

Abstract
Gastrointestinal epithelium faces osmotic stress, both at physiological and pathophysiological conditions. JNK activation is an immediate cellular response to osmotic stress. We investigated the effect of osmotic stress on intestinal epithelial barrier function and delineated the role of JNK2 in osmotic stress-induced tight junction (TJ) regulation in Caco-2 cell monolayers and ileum of Jnk−/− and Jnk2−/− mice. The role of JNK activation in osmotic stress-induced TJ disruption was evaluated using JNK-specific inhibitor and antisense oligonucleotides. Furthermore, the effect of cold restraint stress in vivo on TJ integrity was determined in rats. Osmotic stress disrupted TJs and barrier function in Caco-2 cell monolayers without affecting cell viability. Osmotic stress activated JNK1 and JNK2 and the inhibition of JNK by SP600125 attenuated osmotic stress-induced TJ disruption. TJ disruption and barrier dysfunction by osmotic stress was associated with JNK-dependent remodeling of actin cytoskeleton. Knockdown of JNK2 accelerated TJ assembly and attenuated osmotic stress-induced TJ disruption in Caco-2 cell monolayers. In mouse ileum in vitro, osmotic stress increased paracellular permeability, which was attenuated by SP600125. Osmotic stress disrupted actin cytoskeleton and TJs and increased paracellular permeability in the ileum of wild-type and JNK1−/− mice, but not in JNK2−/− mouse ileum. Cold restraint stress activated JNK in rat ileum and caused JNK-dependent remodeling of actin cytoskeleton and redistribution of occludin and zona occluden-1 from the intercellular junctions. These results reveal the role of JNK2 in the mechanism of osmotic stress-induced TJ disruption in the intestinal epithelium.
Keywords: c-Jun NH2-terminal kinase, occludin, intestine, barrier function, stress
physical, psychological, and chemical stresses are known to alter the gastrointestinal (GI) mucosal homeostasis (3) and play an important role in the pathogenesis of numerous GI diseases, such as functional dyspepsia, irritable bowel syndrome, gastroesophageal reflux disease, peptic ulcer, and inflammatory bowel disease (13). Intestine is lined by low-resistance epithelium that experiences transient mucosal osmotic load during the process of normal digestion (12). Hyperosmolarity is associated with a variety of intestinal inflammatory conditions, such as Crohn's disease (24), ulcerative colitis (19), and necrotizing enterocolitis (5). Hyperosmotic stimulus was shown to induce the production of proinflammatory cytokines and hence contribute to inflammatory reactions in the gut (9, 15).
Epithelial barrier function that prevents the diffusion of toxins, allergens, and pathogens from the GI lumen into the tissue (23) is threatened by various types of stress (11). The epithelial tight junctions (TJs) constitute the major component of gut barrier function, and, therefore, disruption of TJs is the initial factor associated with the pathogenesis of stress-induced inflammatory diseases. The TJ is organized by four types of transmembrane proteins, occludin, claudins, tricellulin, and junctional adhesion molecules, which interact with the scaffold proteins such as zona occludens (ZO)-1, ZO-2, and ZO-3 (8). ZO-1 anchors the transmembrane proteins of TJs to the actin cytoskeleton and interacts with other TJ proteins. TJ proteins interact with numerous signaling proteins, indicating the potential role of intracellular signaling pathways in the regulation of epithelial TJs and barrier function (16).
Nearly 3 decades ago, it was shown that hyperosmotic stress on the apical surface of guinea pig jejunum transiently enhances the barrier function, whereas osmotic stress on both apical and basal surfaces disrupted the barrier function (12). Evidence indicated that transient increase in barrier function by apical osmotic stress was regulated by the actin cytoskeleton, while the mechanism of barrier disruption induced by osmotic stress on both apical and basal surfaces is unclear. c-Jun NH2-terminal kinase (JNK), originally identified as the stress activated protein kinase, is a member of MAPK family of signaling proteins. Various types of stress are known to activate JNK in different cell types (10), and recent studies indicated that JNK may play a role in regulation of TJ integrity in different epithelial cells (4, 7, 14). In Caco-2 cell monolayers, osmotic stress induced activation of JNK, which was suggested to mediate the hyperosmotic stress-induced IL-8 production (9).
In the present study, we investigated the role of JNK activation in osmotic stress-induced TJ regulation in Caco-2 cell monolayers and mouse intestine. The results show that, although apical osmotic stress initially induces a transient increase in resistance, subsequently it disrupts the TJs and barrier function. This effect of osmotic stress is mediated by the activation of JNK2.
METHODS
Reagents.
Cell culture and transfection reagents were purchased from GIBCO-BRL (Grand Island, NY). SP600125 was purchased from EMD Chemicals (San Diego, CA), and FITC-inulin was from Sigma Chemicals (St. Louis, MO). Rabbit polyclonal anti-JNK1/2 and anti-JNK1/2(pT183pY185) [phosphorylated JNK1/2 (p-JNK1/2)] antibodies were purchased from Cell Signaling Solutions (Charlottesville, VA), and mouse monoclonal anti-JNK2 was from Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase (HRP)-conjugated anti-mouse IgG and HRP-conjugated anti-rabbit IgG antibodies were purchased from BD Transduction Laboratories (San Jose, CA). Rabbit polyclonal anti-claudins, anti-ZO-1, anti-occludin antibodies, HRP-conjugated mouse monoclonal anti-occludin, and mouse monoclonal anti-ZO-1 antibodies were purchased from Zymed Laboratories (San Francisco, CA). Cy3-conjugated anti-rabbit IgG, AlexaFluor 488-phalloidin, and AlexaFluor 488-conjugated anti-mouse IgG antibodies were from Molecular Probes (Eugene, OR).
Antisense oligonucleotides.
Antisense oligonucleotides for JNK1 (AS-Jnk1) and JNK2 (AS-Jnk2) were prepared, as described previously (2). Oligonucleotide with scrambled sequence for AS-Jnk1 [missense oligonucleotide (MS-Oligo)] that did not match any sequence for human protein was used as control. These oligonucleotides were custom synthesized in phosphorothioate form by Sigma Genosys (St. Louis, MO).
Cell culture and transfection.
Caco-2 cells purchased from ATCC (Rockville, MD) were grown under standard cell culture conditions, as described before (21). Cells (at 70% confluence) were transfected with oligonucleotides using 1 ml antibiotic and serum-free DMEM containing 100 or 250 nM MS-Oligo, AS-Jnk1, or AS-Jnk2 and 3.15 μl of oligofectamine reagent and incubated for 6 h at 37°C. After 24 h, the cells were trypsinized and grown on polycarbonate membranes in transwells (6.5, 12, or 24 mm diameter; Costar, Cambridge, MA), which were used for experiments 12–17 days after seeding.
TJ assembly.
TJ assembly was induced on day 4 posttransfection by the calcium switch method by EGTA treatment (20). The development of barrier function was evaluated by measuring transepithelial electrical resistance (TER) and unidirectional flux of FITC-conjugated inulin, as described before (17, 20, 21).
Osmotic stress.
Cell monolayers in transwells were incubated in DMEM (600 mosM, adjusted by adding mannitol) to induce osmotic stress. Barrier disruption was evaluated by measuring TER and inulin flux. SP600125 (1 μM) was administered 30 min before osmotic load.
Measurement of TER.
TER was measured as described previously (21, 22) using a Millicell-ERS Electrical Resistance System (Millipore, Bedford, MA). TER was calculated as ohms times centimeter squared by multiplying it with the surface area of the monolayer.
Unidirectional flux of inulin.
Cell monolayers were incubated in the presence of FITC-inulin (0.5 mg/ml) in the basal well. At varying times during the experiment, 50 μl of apical medium were withdrawn, and fluorescence was measured using a fluorescence plate reader (BioTEK Instruments, Winooski, VT).
Immunofluorescence microscopy.
Cell monolayers were fixed in 3% paraformaldehyde. Following permeabilization in 0.2% Triton X-100, cell monolayers were blocked in 4% nonfat milk in Tris-buffered saline-Tween (20 mM Tris, pH 7.2, and 150 mM NaCl). They were incubated for 1 h with primary antibodies, followed by 1-h incubation with secondary antibodies and AlexaFluor 488-conjugated anti-mouse IgG and Cy3-conjugated anti-rabbit IgG antibodies. The fluorescence was examined under Zeiss LSM-5 confocal microscope, and images (1 μm x-y sections) were collected using LSM-5-Pascal software. Images were processed using the software Image J (National Institutes of Health) and Adobe Photoshop (Adobe Systems, San Jose, CA). Actin cytoskeleton was stained with AlexaFluor 488-conjugated phalloidin, and images from 1-μm sections (apical to basal) were collected.
Preparation of detergent-insoluble fractions.
Mouse ileal mucosal scrappings were incubated for 15 min with lysis buffer-CS (Tris buffer containing 1.0% Triton X-100, 2 μg/ml leupeptin, 10 μg/ml aprotinin, 10 μg/ml bestatin, 10 μg/ml pepstatin-A, 1 mM vanadate and 1 mM PMSF). Cell lysates were centrifuged at low speed (15,600 g) for 4 min at 4°C to sediment the high-density actin cytoskeleton (detergent-insoluble fraction). Supernatant was used as detergent-soluble fraction. The pellet was suspended in 200 μl of lysis buffer-D and sonicated to homogenize the actin cytoskeleton. Protein contents in different fractions were measured by the bicinchoninic acid method (Pierce Biotechnology, Rockford, IL). Triton-insoluble and triton-soluble fractions were mixed with equal volume of Laemmli's sample buffer (2× concentrated) and heated at 100°C for 5 min.
Immunoblot analysis.
Proteins were separated by SDS-polyacrylamide gel (4–12% gradient) electrophoresis and transferred to nitrocellulose polyvinylidene difluoride membranes. Membranes were blotted for different proteins using specific antibodies in combination with HRP-conjugated anti-mouse IgG or HRP-conjugated anti-rabbit IgG antibodies. The blots were developed using enhanced chemiluminescence method (Amersham, Arlington Heights, IL). The bands were quantitated by densitometric analysis using Image J software (National Institutes of Health).
Animal studies.
All animal experiments were performed according to the protocols that were approved by the institutional animal care and use committee at the University of Tennessee, Memphis, and the University of California, San Francisco. C57BL/6 wild-type (WT), Jnk1+/−, and Jnk2−/− mice were purchased from Jackson Laboratories (Bar Harbor, ME) and bred in our animal care facility. Mice at 6–8 wk of age were used for these studies.
Osmotic stress in vitro.
Strips of ileum from WT, Jnk1+/−, and Jnk2−/− mice were mounted to Ussing chambers and bathed in Krebs Ringer bicarbonate buffer containing 10 mM glucose, as described before (18). Buffer was continuously oxygenated and temperature maintained at 37°C. Osmotic stress was induced by administration of 500 or 700 mosM buffer adjusted with mannitol. Barrier function was evaluated by measuring unidirectional flux of FITC-inulin, and the TJ integrity was assessed by staining the cryosections of tissues for F-actin and ZO-1.
Cold-restrained stress.
Adult male Sprague-Dawley rats were singly housed and maintained on a 12:12-h light-dark cycle with ad libitum access to food and water. Control animals were left in their home cage at room temperature, whereas experimental rats were subjected to cold-restraint stress (CRS) at 4°C in a cylindrical restrainer. In some groups, animals were injected (intraperitoneally) with SP600125 (20 mg/kg body wt) 30 min before initiation of stress. CRS rats were killed 3 h after the onset of stress, along with controls. Ileum was fixed and the cryosections were stained for F-actin, p-JNK, occluding, and ZO-1. Images were collected as described above.
Statistical analyses.
Comparison between two groups was made by Student's t-tests for grouped data. Significance in all tests was set at 95% or greater confidence level.
RESULTS
Osmotic stress disrupts TJs in Caco-2 cell monolayers by a JNK-dependent mechanism.
Osmotic stress was induced in Caco-2 cell monolayers by applying hyperosmotic buffer on the apical surface (600 mosM). Osmotic stress rapidly reduced TER (Fig. 1A) and increased inulin permeability (Fig. 1B). Under these conditions, osmotic stress did not induce cytotoxicity as assessed by a lack of lactate dehydrogenase release (Fig. 1C) and preservation of cell viability (Fig. 1D). Osmotic stress induced a rapid increase in the levels of p-JNK1 and p-JNK2 (Fig. 2A); JNK1 phosphorylation was transient, while JNK2 phosphorylation was sustained at least for 2 h. p-JNK was distributed predominantly in the intracellular compartment of osmotic stress-treated cells (Fig. 2B). The band above the JNK2 band appears to be a nonspecific band. Pretreatment of cell monolayers with SP600125, a widely used JNK inhibitor, significantly attenuated osmotic stress-induced increase in inulin permeability (Fig. 2C) and redistribution of occludin and ZO-1 from the junctions (Fig. 2D). The osmolarity of 600 mosM was selected on the basis of similar conditions used in the previous study (12). We also evaluated the effects of 400, 500, and 600 mosM buffers and found that the effect on the barrier function was dose dependent (data not shown). Similar hyperosmolarity induced by sodium chloride instead of mannitol also showed a similar effect on the barrier function (data not shown).
Fig. 1.
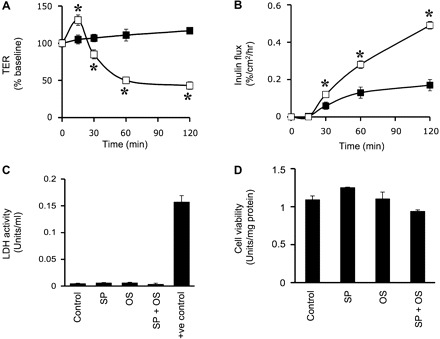
Osmotic stress (OS) disrupts tight junctions (TJs) in Caco-2 cell monolayers. A and B: Caco-2 cell monolayers were incubated with (□) or without (■) hyperosmotic buffer (600 mosM). Transepithelial electrical resistance (TER; A) and inulin permeability (B) were measured. Values are means ± SE (n = 6). *Significantly (P < 0.05) different from corresponding control values. C and D: Caco-2 cell monolayers were incubated with or without SP600125 (SP) for 1 h followed by exposure to OS for 1 h. C: aliquots of apical medium were assayed for lactate dehydrogenase (LDH) activity. D: the cells were assayed for cytotoxicity by WST-1 assay. Values are means ± SE (n = 6). Cells incubated with 0.1% Triton X-100 were used as a positive control.
Fig. 2.
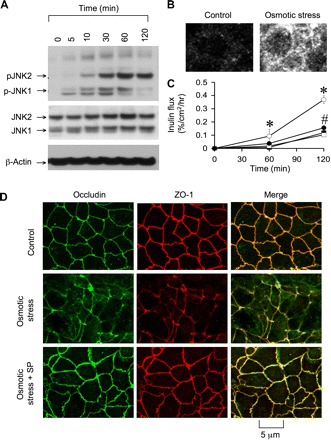
c-Jun NH2-terminal kinase (JNK) activation mediates OS-induced TJ disruption. A: at varying times after incubation with hyperosmotic buffer (600 mosM), cell extracts were immunoblotted for different proteins. B: cell monolayers after 60-min incubation, with or without OS, were fixed and stained for phosphorylated JNK (p-JNK) by immunofluorescence method. C: cell monolayers were incubated with (solid symbols) or without (open symbols) SP (1 μM) for 1 h, followed by incubation with (circles) or without (squares) hyperosmotic buffer. Inulin permeability was measured. Values are means ± SE (n = 6). *Significantly (P < 0.05) different from corresponding control values. #Significantly (P < 0.05) different from corresponding values for OS group. D: cell monolayers, untreated and SP treated, were incubated with or without (control) OS for 1 h. Cell monolayers were fixed and stained for occludin and zona occluden (ZO)-1.
Knockdown of JNK promotes TJ assembly and prevents osmotic stress-induced TJ disruption.
Transfection of Caco-2 cells with JNK-specific antisense oligonucleotides, AS-Jnk1 and AS-Jnk2, reduced the levels of JNK1 and JNK2 (Fig. 3A); AS-Jnk2 selectively knocked down JNK2 without significant influence on JNK1 level, whereas AS-Jnk1 reduced both JNK1 and JNK2. Although BLAST analysis confirmed that AS-Jnk1 sequence is specific for JNK1, it reduced the levels of both JNK1 and JNK2. This was also observed in the original study (2). Furthermore, we designed two additional antisense oligos for other target sequences, but failed to specifically knock down JNK1. It is likely that JNK1 expression may indirectly regulate the expression of JNK2 at posttranscriptional level. AS-Jnk1- and AS-Jnk2-transfected cell monolayers showed significantly higher TER (Fig. 3B) and lower inulin permeability (Fig. 3C). Significantly high levels of ZO-1 and occludin were localized at the intercellular junctions in AS-Jnk1- and AS-Jnk2-transfected cell monolayers (Fig. 3D); AS-Jnk2 was much more efficient than AS-Jnk1 in this effect.
Fig. 3.
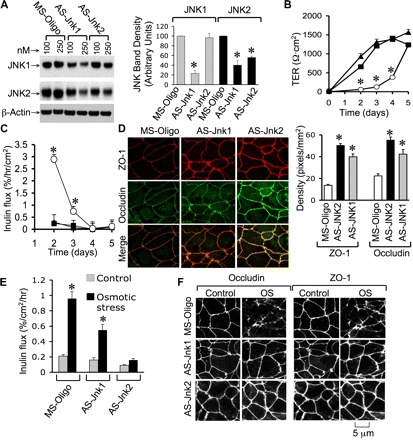
Reduced expression of JNK1/2 enhances TJ integrity and attenuates OS-induced TJ disruption. A, left: Caco-2 cells were transfected with 100 or 250 nM missense oligonucleotides (MS-Oligo) or antisense oligonucleotides (AS-Jnk1 and AS-Jnk2). Right: proteins extracted on day 3 after transfection were immunoblotted for different proteins and quantitated by densitometric analysis. B and C: cells transfected with MS-Oligo (○), AS-Jnk1 (■), or AS-Jnk2 (▲) was seeded on transwells. TER (B) and inulin permeability (C) were measured at varying times. Values are means ± SE (n = 6). *Significantly (P < 0.05) different from corresponding values for AS-Jnk1 and AS-Jnk2 groups. D, left: cell monolayers were fixed and stained for occludin and ZO-1 by immunofluorescence method. Right: densitometric measurement of junctional fluorescence for occludin and ZO-1. Values are means ± SE (n = 3; each value is an average of 6 measurements in different regions of the monolayer). *Significantly (P < 0.05) different from corresponding MS-Oligo values. E: Caco-2 cells transfected with MS-Oligo, AS-Jnk1, or AS-Jnk2 were incubated with (solid bars) or without (shaded bars) hyperosmotic buffer. Inulin permeability was measured at 1 h. Values are means ± SE (n = 6). *Significantly (P < 0.05) different from corresponding control values. F: after 1-h incubation, with or without OS, cell monolayers (transfected with different oligos) were fixed and stained for occludin and ZO-1.
Transfection of cells with AS-Jnk1 or AS-Jnk2 significantly attenuated osmotic stress-induced inulin permeability (Fig. 3E). AS-Jnk2 almost abolished inulin permeability, whereas the effect of AS-Jnk1 partially attenuated osmotic stress-induced permeability. Protection of barrier function from osmotic stress was accompanied by the preservation of junctional distribution of occludin and ZO-1 (Fig. 3F). The reduction in JNK levels in Fig. 3A corresponds to the data presented in Fig. 3, B–D, which clearly demonstrates an equal effect of AS-Jnk1 and AS-Jnk2 on barrier function. On the other hand, the data presented in Fig. 3, E and F, are from separate experiments, in which knockdown of JNK2 was greater in AS-Jnk2-transfected cells compared with AS-Jnk1-transfected cells. Accordingly, AS-Jnk2 prevented osmotic stress-induced barrier dysfunction more completely than AS-Jnk1.
Knockdown of JNK accelerates calcium-induced TJ assembly.
Transfection with AS-Jnk1 or AS-Jnk2 accelerated the development of TER (Fig. 1A) and barrier-to-inulin permeability (Fig. 4B) during calcium-mediated TJ assembly. Accelerated development of barrier function was associated with enhanced junctional organization of occludin and ZO-1 (Fig. 4C). Similarly, long-term treatment of Caco-2 cells with SP600125 for 2 days starting 24 h after seeding also resulted in enhanced junctional distribution of occludin and ZO-1 (Fig. 4, D and E) and acceleration of calcium-induced development of barrier function (Fig. 4, F and G). Knockdown of JNK by antisense oligos significantly increased the levels of ZO-1 and claudin-4 (Cldn-4), while the levels of other TJ proteins were unaffected (Fig. 5, A and B). Similarly, long-term treatment with SP600125 also increased the levels of ZO-1 and Cldn-4 (Fig. 5, C and D).
Fig. 4.
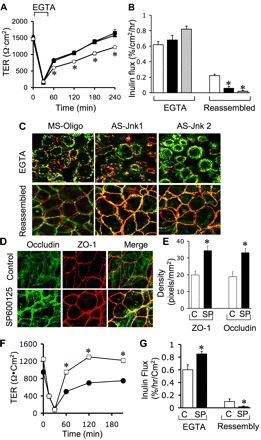
Knockdown or inhibition of JNK accelerates TJ assembly in Caco-2 cell monolayers. A and B: Caco-2 cells transfected with MS-Oligo (○, open bar), AS-Jnk1 (■, solid bar), and AS-Jnk2 (▲, shaded bar). TJ assembly was induced by calcium switch method. A: TER was measured at varying times. B: inulin permeability was measured during the EGTA treatment and at 4 h after calcium replacement. Values in different panels are means ± SE (n = 6). *Significantly (P < 0.05) different from corresponding values for MS-Oligo-transfected cells. C: Caco-2 cells were fixed after EGTA treatment or at 4 h after calcium replacement and stained for occludin and ZO-1. D: Caco-2 cells were incubated with or without SP (1 μM; on day 2 postseeding), and on day 3 postseeding cell monolayers were fixed and stained for occludin and ZO-1. E: the density of fluorescence at the intercellular junctions was evaluated using the software, Image J. Values are means ± SE (n = 6). *Values for SP-treated cells are significantly (P < 0.05) different from corresponding values for control monolayers. F and G: TJ assembly in control (● or C) and SP-treated (□ or SP) cells was evaluated by measuring TER (F) and inulin permeability (G). Values are means ± SE (n = 6). *Values for SP-treated cells (□ or SP) are significantly (P < 0.05) different from corresponding values for control cells (● or C).
Fig. 5.
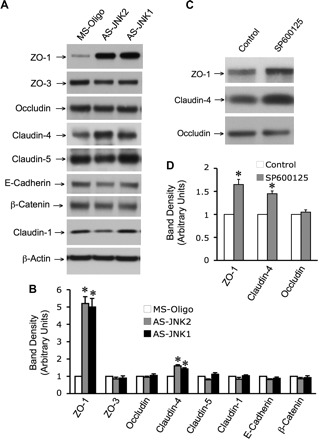
JNK regulates the levels of TJ proteins. Caco-2 cells were transfected with MS-Oligo or AS-Jnk1 and AS-Jnk2. Proteins extracted in 3 days after transfection were immunoblotted for TJ and adherens junction proteins (A), and the band densities were evaluated by using Image J software (B). Values are means ± SE (n = 3). *Values for AS-Jnk1 (solid bars) or AS-Jnk2-tranfected cells (shaded bars) are significantly (P < 0.05) different from corresponding values for MS-Oligo-transfected cells (open bars). C: Caco-2 cells were incubated with SP (1 μM) for 24 h, and the proteins were extracted were immunoblotted for TJ proteins. D: the band densities were evaluated by using Image J software. Values are means ± SE (n = 3). *Values for SP-treated cells (shaded bars) are significantly (P < 0.05) different from corresponding values for untreated cells (open bars) (P < 0.05).
Osmotic stress induces remodeling of actin cytoskeleton by a JNK-dependent mechanism.
A recent study suggested that JNK may regulate the organization of actin ring and TJ integrity in T84 cell monolayers (14). Therefore, we determined the effect of osmotic stress on the organization of actin cytoskeleton in Caco-2 cell monolayers. In the control cell monolayers, actin cytoskeleton is organized differently at the apical, middle, and the basal parts of the cell (Fig. 6). Osmotic stress induced reorganization of apical microvilli, middle actomyosin ring, and basal stress fibers. The effect of osmotic stress on the actomyosin and cortical actin network was much more pronounced than on the actin at the apical and basal parts of the cell. Pretreatment of cell monolayers with SP600125 attenuated the osmotic stress-induced remodeling of actin cytoskeleton.
Fig. 6.
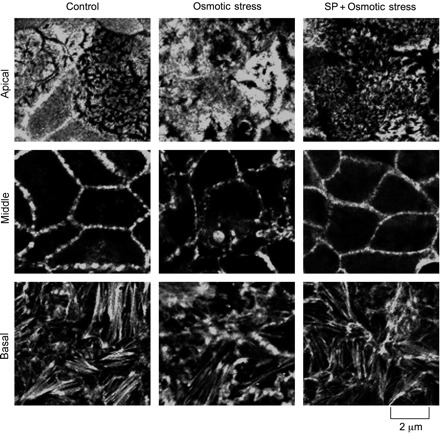
OS induces remodeling of actin cytoskeleton by a JNK-dependent mechanism. Caco-2 cell monolayers were pretreated with or without SP (1 μM), followed by exposure to hyperosmotic buffer for 30 min. Fixed cell monolayers were stained for F-actin using AlexaFluor-488-labeled phalloidin. Images from 2-μm sections at the top, middle, and bottom of the cells are presented.
Selective activation of JNK1 by EGF does not disrupt TJs.
EGF is known to activate MAP kinase signaling of various types of cells (1). Therefore, we investigated the effect of EGF on JNK activation and its influence on the epithelial barrier function. Administration of EGF to Caco-2 cell monolayers induced a rapid, but transient, increase in p-JNK1, with a peak level achieved at 5 min (Fig. 7A). EGF-induced JNK1 activation was cleared by 30 min. EGF did not induce phosphorylation of JNK2, indicating a selective activation of JNK1 by EGF. Total JNK1 and JNK2 levels were unaffected by EGF treatment. Under these conditions, however, EGF failed to increase inulin permeability; it rather slightly reduced the inulin flux (Fig. 7B), an effect that was unaffected by SP600125. This information, along with the observation that AS-Jnk2 is more effective in preventing osmotic stress-induced barrier disruption, suggested that JNK2 is the primary signaling element associated with osmotic stress-induced TJ disruption. Therefore, further studies were conducted in the ileum of Jnk1−/− and Jnk2−/− mice.
Fig. 7.
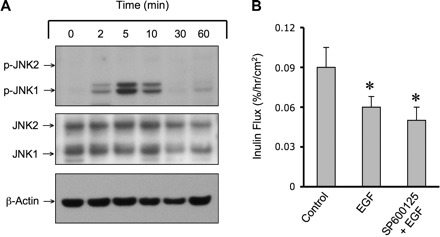
Selective activation of JNK1 by EGF failed to disrupt barrier function. A: Caco-2 cell monolayers at different days postseeding were incubated with EGF (30 nM) for varying times. Protein extracts were immunoblotted for p-JNK, JNK, and β-actin. B: Caco-2 cell monolayers were incubated with or without SP (1 μM), followed by incubation with or without EGF (30 nM) for 60 min. Inulin permeability was measured. Values are means ± SE (n = 4). *Values are significantly (P < 0.05) different from corresponding control values.
Osmotic stress disrupts TJs in mouse ileum by JNK2-dependent mechanism.
To further confirm the specific role of JNK2 in TJ regulation and to establish the physiological relevance of the observation made in Caco-2 cell monolayers, studies were conducted using Jnk1−/− and Jnk2−/− mice. In WT mouse ileum, osmotic stress increased inulin permeability, which was significantly attenuated by SP600125 (Fig. 8A). Osmotic stress-induced inulin permeability in the ileum of Jnk1−/− mice was similar to that in the ileum of WT mice, while osmotic stress failed to increase inulin permeability in Jnk2−/− mouse ileum (Fig. 8B). Immunoblot analysis (Fig. 8C) and immunofluorescence detection method (Fig. 8D) showed that osmotic stress increased p-JNK level in the ileum of WT and Jnk1−/− mice, with only minimal appearance of p-JNK in the ileum of Jnk2−/− mice. In the ileum of WT and Jnk1−/−, osmotic stress induced reorganization of actin cytoskeleton (Fig. 8E), but only minimal modification of actin cytoskeletal organization was seen in osmotic stress-treated ileum of Jnk2−/− mice.
Fig. 8.
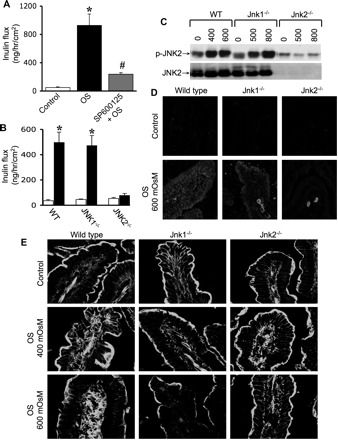
OS induces JNK2-dependent barrier dysfunction in mouse ileum. A: mouse ileal sheets were mounted to Ussing chambers and preincubated with or without SP, followed by administration of hyperosmotic buffer (OS). FITC-inulin transport across the tissue was measured at 1 h. Values are means ± SE (n = 5). *Values are significantly (P < 0.05) different from corresponding control values. #Value is significantly (P < 0.05) different from the value for OS group. B: ileal sheets from wild-type (WT), Jnk1−/−, and Jnk2−/− mice were mounted to Ussing chambers and incubated with (solid bars) or without (open bars) hyperosmotic buffer. FITC-inulin transport across the tissue was measured at 1-h incubation. Values are means ± SE (n = 5). *Values are significantly (P < 0.05) different from corresponding control values. C and D: ileal sheets from WT, Jnk1−/−, and Jnk2−/− mice were mounted to Ussing chambers and incubated with or without hyperosmotic buffer. C: mucosal extracts were immunoblotted for p-JNK and JNK2. D: cryosections of untreated and OS-treated tissues were stained for p-JNK. E: cryosections of untreated and OS-treated tissues were fixed and stained for F-actin using AlexFluor 488-labeled phalloidin.
Immunofluorescence localization of occludin and ZO-1 showed that occludin and ZO-1 are colocalized at the intercellular junctions of epithelial cells in the ileum of WT, Jnk1−/−, and Jnk2−/− mice, and osmotic stress induced redistribution of occludin and ZO-1 from the intercellular junctions in the ileum of WT and Jnk1−/− mice, but not in the ileum of Jnk2−/− mice (Fig. 9A). Previous studies showed that, in the intact epithelium, the TJ proteins, such as occludin and ZO-1, are bound to the actin cytoskeleton and can be recovered in the detergent-insoluble fractions of the cell (17). Therefore, we analyzed the levels of occludin and Cldn-4 in the Triton-insoluble fractions in control and osmotic stress-treated mouse ileum. Osmotic stress induced a dramatic reduction in the levels of Triton-insoluble fractions of occludin and Cldn-4 in the ileum of WT and Jnk1−/− mice, but the Triton-insoluble fractions of both occludin and Cldn-4 were unaffected by osmotic stress in the ileum of Jnk1−/− mice (Fig. 9B).
Fig. 9.
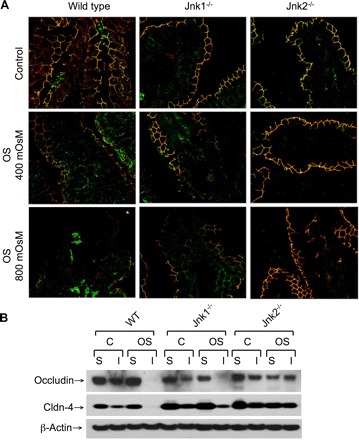
OS disrupts TJs in mouse ileum by a JNK2-dependent mechanism. A: ileal sheets from WT, Jnk1−/−, and Jnk2−/− mice were mounted to Ussing chambers and incubated with or without hyperosmotic buffer. Cryosections of untreated and OS-treated tissues were costained for occludin (green) and ZO-1 (red). B: triton-soluble (S) and triton-insoluble (I) fractions were prepared from WT, Jnk1−/−, and Jnk2−/− mouse ileal strips that were incubated with (OS) or without (C) hyperosmotic buffer and immunoblotted for TJ proteins.
CRS disrupts TJs in rat ileum by a JNK-dependent mechanism.
To determine the role of JNK activity in stress-induced TJ disruption in vivo, we analyzed the effect of CRS on TJ integrity and evaluated the effect of SP600125 on CRS-induced TJ disruption. CRS increased p-JNK levels in rat ileum, which was associated with the disorganized actin cytoskeleton (Fig. 10A). Confocal microscopy for occludin and ZO-1 revealed that CRS induced a redistribution of junctional occludin and ZO-1 in both villi and crypts (Fig. 10B). Administration of SP600125 attenuated CRS-induced redistribution of TJ proteins.
Fig. 10.
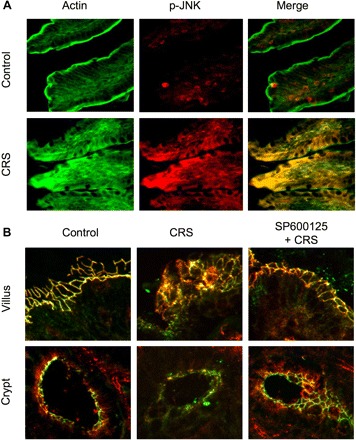
Cold restraint stress (CRS) disrupts TJs in rat ileum by a JNK-dependent mechanism. A: adult male rats were injected with SP 30 min before CRS. Cryosections of ileum after 3 h of CRS were stained for F-actin (green) and p-JNK (red). B: cryosections of ileum were stained for occludin (green) and ZO-1 (red).
DISCUSSION
Stress plays an important role in the pathogenesis of numerous GI diseases (13). Osmotic stress is one type of stress that the intestine faces under physiological (12) and pathophysiological (5, 19, 24) conditions. The mechanism associated with stress-induced perturbation of intestinal mucosal homeostasis is not well understood. Activation of JNK is one of the initial responses to stress (10), and recent studies indicated that JNK may play a role in the epithelial TJ regulation (4, 7, 14). Here we present evidence to demonstrate that JNK2 plays a role in osmotic stress-induced loss of TJ integrity in Caco-2 cell monolayers and mouse ileum.
Previous study demonstrated that exposure of guinea pig ileum in vitro to osmotic stress on both mucosal and serosal surfaces leads to disruption of epithelial barrier function, while osmotic load up to 600 mosM on the apical surface alone induced a transient increase in the barrier function (12). Our present study shows that apical osmotic load up to 600 mosM induces a transient increase in the resistance of Caco-2 cell monolayers, with a peak resistance achieved at 15 min. However, continued exposure to osmotic stress leads to reduction in resistance and increase in inulin permeability without altering cell viability. Therefore, as suggested by Madara (12), initial transient increase in barrier function may relate to the mucosal defense against transient changes in osmolarity during the normal process of digestion and absorption. However, if the osmotic stress is sustained, as in pathophysiological conditions, it may lead to disruption of barrier function. This is supported by the previous observations that hyperosmotic stress in the intestine induces the expression of proinflammatory cytokines (9, 15). The present study shows that osmotic stress disrupts TJs without inducing cell death.
It is important to understand the mechanism of osmotic stress-induced TJ disruption in the intestinal epithelium. Activation of JNK is well known as one of the early signals activated by various types of stress in various cells, including the intestinal epithelial cells. Recent studies indicated that JNK may play a role in the regulation of TJ integrity in cultured epithelial monolayers (4, 7, 14, 26). The attenuation of osmotic stress-induced TJ disruption by SP600125 in the present study revealed the role of JNK activity in osmotic stress-induced TJ disruption. Rapid increase in the levels of p-JNK1/2 indicated that osmotic stress activates JNK1 and JNK2. Osmotic stress-induced TJ disruption in Caco-2 cell monolayers was almost abolished by AS-Jnk2, indicating the potential role of JNK2 in osmotic stress-induced TJ disruption. AS-Jnk1 produced a partial reduction in osmotic stress-induced barrier disruption. The observation that AS-Jnk1 reduced the levels of both JNK1 and JNK2 questioned whether the effect of AS-Jnk1 was caused by reduction of JNK2, rather than reduced JNK1. JNK1 was selectively activated in Caco-2 cells by EGF. EGF, however, failed to disrupt the barrier function, suggesting that JNK1 is not involved in TJ regulation under the present experimental conditions. A recent study indicated that JNK1 regulates TJs in T84 cell monolayers (14). This is in contrast to our current observation that JNK2 rather than JNK1 is responsible for the regulation of TJ integrity in Caco-2 cell monolayers. T84 cells are poorly differentiated cells, whereas Caco-2 cells are highly differentiated enterocyte-like cells. JNK2 may play a major role in differentiated enterocytes, while JNK1 may be important in undifferentiated cells.
The present study shows that knockdown of JNK in Caco-2 cell monolayers accelerates the development of barrier function postseeding. The barrier function and junctional assembly of occludin and ZO-1 in nonspecific oligo-transfected cell monolayers reached maximum on day 4–5 postseeding, while, in JNK knockdown cells, maximal barrier function was achieved on day 2–3. These data suggest that reduced expression of JNK accelerates the assembly of TJs. We observed a selective and transient activation of JNK2 on day 2 after seeding, which may have delayed the TJ assembly. However, the barrier function eventually reached the maximal level and was undistinguishable from the missense oligo-transfected cell monolayers. Similar to the observation made in T84 cells (14), JNK knockdown accelerated calcium-induced TJ assembly in Caco-2 cells. However, JNK2-specific anitsense oligo was more effective than AS-Jnk1 that reduced the level of both JNK1 and JNK2. This observation is different from the previous study showing the role of JNK1 in TJ disruption in T84 cells (14). Results demonstrate that JNK activity destabilizes TJs in Caco-2 cell monolayers. The role of JNK activity in destabilization of TJs was further confirmed by the observation that a long-term treatment with SP600125, the JNK-selective inhibitor, also accelerates de novo TJ assembly and calcium-induced reassembly.
One mechanism associated with TJ regulation involves alteration of the expression of TJ proteins. Conflicting results have been reported regarding the role of JNK in TJ protein expression. While JNK enhanced the expression of Cldn-4 (25) and ZO-1 (26), it decreased occludin level in isolated alveolar epithelium (6). Our present study shows that knockdown or inhibition of JNK leads to increase in the levels of ZO-1 and Cldn-4. However, evidence suggests that the changes in protein level are related to changes in proteolysis rather than changes in protein expression. A recent study showed that JNK1 prevents the formation of contractile F-actin ring in calcium-depleted T84 cells (14). Our study shows that osmotic stress induces reorganization of actin cytoskeleton in Caco-2 cell monolayers. While the apical microvilli and basal stress fibers are disorganized, the actin ring in the middle part of the cell is dramatically disrupted by osmotic stress. The osmotic stress-induced reorganization of actin cytoskeleton in the midcell region was attenuated by SP600125, suggesting the role of JNK in osmotic stress-induced actin reorganization. These results indicate that JNK-dependent remodeling of actin cytoskeleton plays an important role in osmotic stress-induced TJ disruption and barrier dysfunction.
Although AS-Jnk2 specifically knocked down JNK2, AS-Jnk1 was not very selective on JNK1. The absence of TJ disruption by selective activation of JNK1 by EGF suggested that JNK1 may not play a role in TJ disruption in Caco-2 cells. To further distinguish the roles of JNK1 and JNK2 in osmotic stress-induced TJ disruption, we conducted studies in the ileum of WT, Jnk1−/−, and Jnk2−/− mice. Exposure of WT mouse ileum to hyperosmotic stress in vitro resulted in a significant increase in inulin permeability, and the osmotic stress-induced permeability was attenuated by SP600125. These data indicate the role of JNK activity in osmotic stress-induced barrier disruption in mouse ileum, confirming the observation made in Caco-2 cells. Osmotic stress induced barrier dysfunction in the ileum of WT and Jnk1−/− mice, but the TJ integrity was unaffected in Jnk2−/− mouse ileum. These data confirm that JNK2, rather than JNK1, is responsible for osmotic stress-induced TJ disruption in the intestinal epithelium. JNK2 activation was observed in the osmotic stress-treated ileum of WT and Jnk1−/− mice, while very little p-JNK was detectable in osmotic stress-treated ileum of Jnk2−/− mice. Osmotic stress induced reorganization of the actin cytoskeleton in WT and Jnk1−/− mouse ileum, but not in Jnk1−/− mouse ileum. Redistribution of occludin and ZO-1 from the intercellular junctions indicated that osmotic stress disrupts TJs in the ileum of WT and Jnk1−/− mice. Such a redistribution of occludin and ZO-1 was absent in osmotic stress-treated ileum of Jnk2−/− mice. This was further supported by the observation that loss of detergent-insoluble fraction of occludin and Cldn-4 was absent in osmotic stress-treated ileum of Jnk2−/− mice.
The physiological relevance of JNK activity in the stress-induced TJ disruption in intestinal epithelium was further confirmed by the analysis of stress-induced TJ disruption in the rat ileum in vivo. CRS in rats resulted in activation of JNK in the ileal mucosa, which was associated with reorganization of the actin cytoskeleton and redistribution of occludin and ZO-1 from the intercellular junctions. CRS-induced redistribution of occludin and ZO-1 was attenuated by SP600125 administration, indicating that JNK activity is involved in stress-induced intestinal epithelial TJ disruption in vivo. The in vivo study may involve multiple factors, including those related to indirect influences of other cells on epithelial cells. Nevertheless, the present observation made in CRS rats indicates that JNK-dependent TJ regulation exists in an in vivo condition. Further studies are needed to delineate the role of different cell types involved in the stress-induced barrier disruption.
In conclusion, the present study shows that JNK2 plays an important role in the osmotic stress-induced TJ disruption and barrier dysfunction in the intestinal epithelium. This study raises the question of whether JNK activation induced by various other types of stress also affect the TJs of intestine, and whether JNK-mediated barrier disruption is involved in the pathogenesis of GI diseases. Further studies are required to address the effects of different isoforms of JNK in different epithelial monolayers and the mechanisms involved in JNK-induced TJ disruption.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
ACKNOWLEDGMENTS
We thank Ankur Seth, Julia Hoy, and Min Liao for excellent technical assistance.
G. Samak is an overseas associate fellow of the Department of Biotechnology, Govt. of India.
REFERENCES
- 1. Agell N, Bachs O, Rocamora N, Villalonga P. Modulation of the Ras/Raf/MEK/ERK pathway by Ca(2+), and calmodulin. Cell Signal 14: 649–654, 2002 [DOI] [PubMed] [Google Scholar]
- 2. Bost F, McKay R, Bost M, Potapova O, Dean NM, Mercola D. The Jun kinase 2 isoform is preferentially required for epidermal growth factor-induced transformation of human A549 lung carcinoma cells. Mol Cell Biol 19: 1938–1949, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buret AG. How stress induces intestinal hypersensitivity. Am J Pathol 168: 3–5, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carrozzino F, Pugnale P, Feraille E, Montesano R. Inhibition of basal p38 or JNK activity enhances epithelial barrier function through differential modulation of claudin expression. Am J Physiol Cell Physiol 297: C775–C787, 2009 [DOI] [PubMed] [Google Scholar]
- 5. Cheromcha DP, Hyman PE. Neonatal necrotizing enterocolitis. Inflammatory bowel disease of the newborn. Dig Dis Sci 33: 78S–84S, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cohen TS, Gray Lawrence G, Khasgiwala A, Margulies SS. MAPK activation modulates permeability of isolated rat alveolar epithelial cell monolayers following cyclic stretch. PLoS One 5: e10385, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Walle JV, Sergent T, Piront N, Toussaint O, Schneider YJ, Larondelle Y. Deoxynivalenol affects in vitro intestinal epithelial cell barrier integrity through inhibition of protein synthesis. Toxicol Appl Pharmacol 245: 291–298, 2010 [DOI] [PubMed] [Google Scholar]
- 8. Gonzalez-Mariscal L, Betanzos A, Avila-Flores A. MAGUK proteins: structure and role in the tight junction. Semin Cell Dev Biol 11: 315–324, 2000 [DOI] [PubMed] [Google Scholar]
- 9. Hubert A, Cauliez B, Chedeville A, Husson A, Lavoinne A. Osmotic stress, a proinflammatory signal in Caco-2 cells. Biochimie 86: 533–541, 2004 [DOI] [PubMed] [Google Scholar]
- 10. Lin A. Activation of the JNK signaling pathway: breaking the brake on apoptosis. Bioessays 25: 17–24, 2003 [DOI] [PubMed] [Google Scholar]
- 11. Lutgendorff F, Akkermans LM, Soderholm JD. The role of microbiota and probiotics in stress-induced gastro-intestinal damage. Curr Mol Med 8: 282–298, 2008 [DOI] [PubMed] [Google Scholar]
- 12. Madara JL. Increases in guinea pig small intestinal transepithelial resistance induced by osmotic loads are accompanied by rapid alterations in absorptive-cell tight-junction structure. J Cell Biol 97: 125–136, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mawdsley JE, Rampton DS. Psychological stress in IBD: new insights into pathogenic and therapeutic implications. Gut 54: 1481–1491, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Naydenov NG, Hopkins AM, Ivanov AI. c-Jun N-terminal kinase mediates disassembly of apical junctions in model intestinal epithelia. Cell Cycle 8: 2110–2121, 2009 [DOI] [PubMed] [Google Scholar]
- 15. Nemeth ZH, Deitch EA, Szabo C, Hasko G. Hyperosmotic stress induces nuclear factor-kappaB activation and interleukin-8 production in human intestinal epithelial cells. Am J Pathol 161: 987–996, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rao R. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front Biosci 13: 7210–7226, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rao RK, Basuroy S, Rao VU, Karnaky KJ, Jr, Gupta A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J 368: 471–481, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rao RK, Riviere PJ, Pascaud X, Junien JL, Porreca F. Tonic regulation of mouse ileal ion transport by nitric oxide. J Pharmacol Exp Ther 269: 626–631, 1994 [PubMed] [Google Scholar]
- 19. Schilli R, Breuer RI, Klein F, Dunn K, Gnaedinger A, Bernstein J, Paige M, Kaufman M. Comparison of the composition of faecal fluid in Crohn's disease and ulcerative colitis. Gut 23: 326–332, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seth A, Sheth P, Elias BC, Rao R. Protein phosphatases 2A and 1 interact with occludin and negatively regulate the assembly of tight junctions in the CACO-2 cell monolayer. J Biol Chem 282: 11487–11498, 2007 [DOI] [PubMed] [Google Scholar]
- 21. Suzuki T, Elias BC, Seth A, Shen L, Turner JR, Giorgianni F, Desiderio D, Guntaka R, Rao R. PKC eta regulates occludin phosphorylation and epithelial tight junction integrity. Proc Natl Acad Sci U S A 106: 61–66, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suzuki T, Seth A, Rao R. Role of phospholipase Cgamma-induced activation of protein kinase Cepsilon (PKCepsilon) and PKCbetaI in epidermal growth factor-mediated protection of tight junctions from acetaldehyde in Caco-2 cell monolayers. J Biol Chem 283: 3574–3583, 2008 [DOI] [PubMed] [Google Scholar]
- 23. Van Itallie CM, Anderson JM. The molecular physiology of tight junction pores. Physiology (Bethesda) 19: 331–338, 2004 [DOI] [PubMed] [Google Scholar]
- 24. Vernia P, Gnaedinger A, Hauck W, Breuer RI. Organic anions and the diarrhea of inflammatory bowel disease. Dig Dis Sci 33: 1353–1358, 1988 [DOI] [PubMed] [Google Scholar]
- 25. Wray C, Mao Y, Pan J, Chandrasena A, Piasta F, Frank JA. Claudin-4 augments alveolar epithelial barrier function and is induced in acute lung injury. Am J Physiol Lung Cell Mol Physiol 297: L219–L227, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yanai R, Ko JA, Nomi N, Morishige N, Chikama T, Hattori A, Hozumi K, Nomizu M, Nishida T. Upregulation of ZO-1 in cultured human corneal epithelial cells by a peptide (PHSRN) corresponding to the second cell-binding site of fibronectin. Invest Ophthalmol Vis Sci 50: 2757–2764, 2009 [DOI] [PubMed] [Google Scholar]