

Abstract
Caffeine is sometimes used in cell physiological studies to release internally stored Ca2+. We obtained evidence that caffeine may also act through a different mechanism that has not been previously described and sought to examine this in greater detail. We ruled out a role for phosphodiesterase (PDE) inhibition, since the effect was 1) not reversed by inhibiting PKA or adenylate cyclase; 2) not exacerbated by inhibiting PDE4; and 3) not mimicked by submillimolar caffeine nor theophylline, both of which are sufficient to inhibit PDE. Although caffeine is an agonist of bitter taste receptors, which in turn mediate bronchodilation, its relaxant effect was not mimicked by quinine. After permeabilizing the membrane using β-escin and depleting the internal Ca2+ store using A23187, we found that 10 mM caffeine reversed tone evoked by direct application of Ca2+, suggesting it functionally antagonizes the contractile apparatus. Using a variety of molecular techniques, we found that caffeine did not affect phosphorylation of myosin light chain (MLC) by MLC kinase, actin-filament motility catalyzed by MLC kinase, phosphorylation of CPI-17 by either protein kinase C or RhoA kinase, nor the activity of MLC-phosphatase. However, we did obtain evidence that caffeine decreased actin filament binding to phosphorylated myosin heads and increased the ratio of globular to filamentous actin in precontracted tissues. We conclude that, in addition to its other non-RyR targets, caffeine also interferes with actin function (decreased binding by myosin, possibly with depolymerization), an effect that should be borne in mind in studies using caffeine to probe excitation-contraction coupling in smooth muscle.
Keywords: excitation-contraction coupling, Ca2+ handling, airway smooth muscle, ryanodine, asthma
contraction of smooth muscle (SM) is ultimately dependent on phosphorylation of the regulatory light chain of myosin (MLC20) at Ser19, leading to a marked increase in myosin ATPase activity and cross-bridge cycling with actin. MLCK is widely recognized as the primary effector of this phosphorylation and is activated directly by Ca2+. Phospho-MLC20 (P-MLC20) is then dephosphorylated by myosin light chain phosphatase (MLCP). Contraction can also involve a suppression of tonic MLCP activity, via phosphorylation of MLCP and the MLCP inhibitor protein, CPI-17, in response to the activation of RhoA kinase (ROCK) and PKC pathways, in Ca2+-independent and -dependent fashions, respectively. Furthermore, accumulating evidence suggests that Ca2+-dependent activation of calcineurin causes the actin polymerization mediating agonist-induced canine airway SM (ASM) contraction (51). Thus both MLC20 phosphorylation and actin polymerization are involved in Ca2+-induced SM contraction in response to agonist stimuli.
Given this central role for Ca2+ in contraction, considerable attention has been focused on the regulation of cytosolic concentration of calcium ([Ca2+]i) in SM. [Ca2+]i is elevated by either influx of external Ca2+ through a variety of pathways [voltage-dependent Ca2+ channels (15, 17), nonselective cation channels (12, 20, 21), reverse-mode Na+/Ca2+ exchange (15, 16)] and/or release of Ca2+ sequestered into intracellular organelles, particularly the sarcoplasmic reticulum (SR) (14, 24, 33).
Release of Ca2+ from the SR occurs through two different types of ligand-gated ion channels. One of these is activated by inositol trisphosphate, generated by phospholipase following its activation by plasmalemmal G-protein-coupled receptors for a variety of ligands including acetylcholine (ACh) and serotonin (5-HT) (42). The second type of SR-release channel is often referred to as the ryanodine receptor (RyR) because of its sensitivity to the plant alkaloid ryanodine (42). The endogenous ligand for this receptor is debated, with many proposing it to be cyclic ADP ribose (42). RyR are also activated by cytosolic Ca2+ ions themselves, leading to a phenomenon known as Ca2+-induced Ca2+ release (42). Following discharge of Ca2+ from the SR, a Ca2+-ATPase on the SR membranes is activated and mediates refilling of that internal pool (42).
Because of the importance of the SR in determining [Ca2+]i, many tools have been identified that modulate the activity of those Ca2+ flux pathways. One of the most commonly used among these is caffeine, which at millimolar concentrations increases the Ca2+ sensitivity of the RyR such that they become activated at basal [Ca2+]i (30, 34). For this reason, caffeine is often used to study excitation-contraction coupling in SM, with the intent of inducing a massive release of internally sequestered Ca2+ or possibly even outright depletion of the SR. However, caffeine is also known to mediate other effects at even lower concentrations, including inhibition of phosphodiesterase (PDE) activity and blockade of adenosine receptors (9, 50). In the course of investigating the mechanisms underlying contractile responses to 5-HT in bovine tracheal SM (TSM), we collected evidence that it might also be acting through yet another mechanism, distinct from disruption of Ca2+ handling or PDE activity. Here, we characterize that other mechanism, finding it to comprise a direct inhibition of the contractile apparatus, in part through disruption of actin filaments. Comparisons are made with other molecules that share caffeine's identity as a methylxanthine and PDE inhibitor [theophylline; isobutylmethylxanthine (IBMX)] and as an agonist at bitter taste receptors (quinine).
METHODS
Preparation of isolated tissues.
All experimental procedures were approved by the McMaster University Animal Care Committee, the McMaster University Biosafety Committee, and the St. Joseph's Healthcare Research Ethics Board and conform to the guidelines set out by the Canadian Council on Animal Care. Tracheas were obtained from cows (200–500 kg) euthanized at the local abattoir and transported to the laboratory in ice-cold Krebs buffer (see Solutions and chemicals). Upon arrival at the laboratory, the epithelium was removed, and TSM strips (∼2–3 mm wide, ∼10 mm long) were excised and used immediately or stored at 4°C for use up to 48 h.
Muscle bath technique.
ASM strips were mounted vertically in organ baths using silk suture (Ethicon 4-0) tied to a Grass FT.03 force transducer on one end and to a glass rod that served as an anchor on the other end. These were bathed in Krebs-Ringer buffer containing (in mM) 116 NaCl, 4.2 KCl, 2.5 CaCl2, 1.6 NaH2PO4, 1.2 MgSO4, 22 NaHCO3, 11 d-glucose, bubbled with 95% O2-5% CO2 to maintain pH at 7.4 and maintained at 37°C. Nω-nitro-l-arginine (10−4 M) and indomethacin (10−5 M) were also added to prevent generation of nitric oxide and of cyclooxygenase metabolites of arachidonic acid, respectively. Tissues were passively stretched to impose a preload tension of ∼1 g. Isometric changes in tension were digitized (2 samples per second) and recorded online (DigiMed System Integrator, MicroMed, Louisville, KY) for plotting on the computer. Tissues were equilibrated for 1 h before commencing the experiments, during which time they were challenged with 60 mM KCl three times to assess the functional state of each tissue.
β-Escin permeabilized muscle strips.
Bovine TSM was excised in the form of strips ∼50–100 μm thick and 3–4 mm long. One end of these was anchored, and the other end was tied to a force transducer (SensoNor AE801; Norway) with single fibers of silk suture, restretched to its in situ length, and bathed in standard Krebs-Ringer buffer (composition given below). After 30–60 min of equilibration, the Krebs-Ringer buffer was replaced with a buffer that mimics the cytosol (i.e., high-millimolar K+ and low free [Ca2+]i) developed by Rodat-Despoix et al. (38), comprised of the following (in mM): 87 KCl, 5.1 MgCl2, 5.2 ATP (disodium salt), 10 creatine phosphate, 2 EGTA, 30 PIPES, pH adjusted to 7.2 by use of KOH. Various pCa were obtained by dilution of a high-Ca2+ form of this cytosolic buffer (one supplemented with 6.5 mM CaCl2 and 10 mM EGTA, estimated to give pCa 5) with appropriate volumes of the nominally Ca2+-free form of this buffer. Tissues were initially bathed in pCa 8 cytosolic buffer, then permeabilized with β-escin (10 μM) for 40 min. Unless noted otherwise, the Ca2+ ionophore A23187 (10−5 M) was also added to this pCa 8 cytosolic buffer to deplete the SR. The permeabilized tissues were then challenged with cytosolic buffers having pCa ranging from 8 to 5 in the presence of various pharmacological tools.
In vitro motility assay.
The in vitro motility assay was used to quantify the velocity of actin propulsion by myosin (νmax) as previously described (5, 26). Myosin was purified from pig stomach antrum (43) and was either phosphorylated (46) in the absence or presence of caffeine (1 and 10 mM), or thiophosphorylated (46) in the absence of caffeine [phosphorylation levels were assessed by phos-tag analysis (44)]. Actin was purified from chicken pectoralis acetone powder (32) and fluorescently labeled with tetramethylrhodamine isothiocyanate (TRITC)-phalloidin (P1951, Sigma-Aldrich Canada) (49). The proteins and buffers were introduced into the flow-through chamber as previously described with the exception that caffeine (or vehicle) was added to the thiophosphorylated myosin and motility buffer before being introduced into the chamber. Measurements were performed at 30°C. Motility was assessed by use of an inverted microscope (IX70, Olympus, Melville, NY) equipped with a high numerical aperture objective (×100 magnification Ach 1.25 numerical aperture, Olympus) and rhodamine epifluorescence. An image-intensified video camera (KP-E500 CCD Camera, Hitachi Kokusai Electric, Woodbury, NY) was used to visualize and record the actin filament movement on computer (Custom Built by Norbec Communication, Montreal, QC, Canada) by using a frame grabber (Pinnacle Studio AV/DV V.9 PCI Card) at 29.94 Hz and an image capturing software (AMCap software V9.20) at 29.94 Hz. The νmax was determined from the total path described by the filaments divided by the elapsed time via our automated version of the National Institutes of Health tracking software (NIH macro in Scion Image 4.02, Scion). The assay was also modified to quantify the number of actin filaments bound by thiophosphorylated myosin. Four washes of actin buffer were added after the labeled actin incubation to wash away unbound actin filaments. No ATP was added to the motility buffer because only binding was assessed. Caffeine (or vehicle) was added to the myosin, to the labeled actin, and to the actin buffer washes. Actin filament binding was assessed by quantifying the number of filaments present in five spots per flow-through chamber throughout the motility surface.
Myosin light chain phosphatase assay.
The recombinant MLCP complex, consisting of the regulatory subunit (MYPT1) and the catalytic subunit (PP1Cδ), was prepared by using a mammalian-cell coexpression system. COS1 cells were transiently transfected for 48 h with a pair of DNA vectors FLAG-metal affinity tag (MAT)-tagged full length human MYPT1 and HA-tagged PP1Cδ using FuGeneHD reagent (Roche). After transfection, the cells expressing recombinant MYPT1 and PP1Cδ were lysed with the lysis buffer containing 20 mM Tris·HCl, pH 8.0, plus 0.5 M NaCl, 0.1 mM EGTA, 5% glycerol, 0.1% Tween-20, 4 mM Pefabloc (Roche), and 0.5 mM Tris[2-carboxyethyl]phosphine (TCEP, Pierce). The clarified cell lysates were subjected to metal affinity purification by use of Talon beads (Invitrogen) and eluted with the buffer including 0.5 M imidazole-HCl, pH 7.0. The eluant, including MYPT1 and PP1Cδ, was used as the MLCP preparation. The MLCP activity was determined from the amount of inorganic phosphate liberated from the substrate, phospho-peptide mimicking MLC20 (3–26) with phospho-Ser19. The amount of released phosphate was determined by using a BIOMOL Green kit (Enzo). The conditions were 25 mM MOPS-NaOH, pH 7.0, plus 0.1 mM EGTA, 0.5 mM TCEP, 4 mM Pefabloc, 1 nM okadaic acid, and caffeine at the indicated concentration. Okadaic acid at low concentration was added to avoid the trace of the activity of PP2A in the MLCP preparation. The reaction was initiated by addition of the substrate and terminated by addition of BIOMOL Green reagent, followed by the measurement of OD 650 nm. The thiophosphorylation of the recombinant MLCP was carried out by preincubating for 30 min at 30°C with ROCK and 1 mM ATPγS, prior to the assay. Mean values ± SE were obtained from triplicate assays in three independent experiments (n = 9).
Kinase assay.
Phosphorylation in the presence of caffeine was assayed by using MLCK purified from chicken gizzard, PKC purified from human red cells (7), and the recombinant human ROCK2 (Millipore). Chicken gizzard MLC20 and recombinant CPI-17 (7) were used as substrates. MLCK, ROCK, and PKC assays were performed using phospho-specific antibodies for phospho-MLC20 Ser19 (Cell Signaling) and phospho-CPI-17 Thr38 as described previously (8).
F-actin-to-G-actin ratio assay.
SM strips were homogenized in F-actin stabilization buffer (50 mM PIPES, pH 6.9, 50 mM NaCl, 5 mM MgCl2, 5 mm EGTA, 5% glycerol, 0.1% Triton X-100, 0.1% Nonidet P-40, 0.1% Tween 20, 0.1% β-mercaptoethanol, 0.0011% anti-foam, 1 mM ATP, 1 μg/ml pepstatin, 1 μg/ml leupeptin, 10 μg/ml benzamidine, 500 μg/ml tosyl arginine methyl ester) provided within a standard commercially available assay kit (Cytoskeleton, Denver, CO). The homogenates were centrifuged at 100,000 g for 60 min at 30°C, then the supernatants (containing G-actin) were collected and kept on ice. The pellets (containing F-actin), on the other hand, were resuspended in ice-cold distilled H2O plus 1 μm cytochalasin D and incubated on ice for 1 h to dissociate the actin filaments into G-actin monomers; the resuspended pellets were gently mixed every 15 min. This cytochalasin-digested mixture was centrifuged at 2,300 g for 2 min at 4°C and the second supernatant was also collected and stored on ice, and 25 μg of protein from both supernatants were subjected to analysis by immunoblot using anti-actin antibody. The total amounts of G-actin and F-actin from the original soluble and insoluble fractions were calculated based on the total protein in each fraction.
Solutions and chemicals.
All chemicals were obtained from Sigma Chemical and prepared as 10 mM stock solutions, either as aqueous solutions (caffeine, KCl, 5-HT, carbachol), ethanol (A23187, nifedipine, ryanodine, quinine, IBMX), or DMSO (cyclopiazonic acid, SQ22536, MDL12330A, IBMX, H89). Aliquots were then added to the muscle baths; the final bath concentration of solvents did not exceed 0.1%, which we have found elsewhere to have little or no effect on mechanical activity.
Data analysis.
Unless indicated otherwise, contractile responses to 5-HT or KCl were expressed as a percentage of the response to bolus addition of 60 mM KCl added during the equilibration period (immediately before onset of the experiment). Reversals of tone in permeabilized tissue strips are expressed as a percentage of the tone evoked by pCa 6.5 buffer. Values are reported as means ± SE; n refers to the number of animals. For the in vitro motility assay, n refers to the number of flow-through chambers. Statistical comparisons were made by Student's t-test or one-way ANOVA; P < 0.05 was considered statistically significant.
RESULTS
Caffeine inhibits excitatory responses in intact bovine TSM strips.
Serotonin evoked contractions in a concentration-dependent fashion (Fig. 1A): these were abolished by pretreatment with 10 mM caffeine, both in the presence and absence of ryanodine (10−5 M) (Fig. 1). This inhibitory effect of caffeine developed within minutes, being half-maximal within 1–2 min, and more than 90% complete within 10 min (Fig. 1B).
Fig. 1.

A: cumulative concentration-response relationships for 5-HT plus/minus cyclopiazonic acid (CPA; 10−5), ryanodine (ryan; 10−5), and/or caffeine (caff; 10 mM) (N > 5 for all). B: tissues were stimulated with a bolus concentration of 5-HT (10−5 M) before and at various times after addition of 10 mM caffeine, as indicated; the mean peak magnitude of the second (postcaffeine) response is plotted here against time after treatment with caffeine (n = 5). C: cumulative concentration-response relationships for 5-HT after pretreating the tissues for 20 min with 0.1, 1, or 10 mM caffeine (n = 5). *Significantly different from control.
If depletion of the SR accounts for the loss of responsiveness to 5-HT, then this effect should be mimicked by cyclopiazonic acid (CPA), which we have previously shown is sufficient to deplete the SR in ASM (13, 18, 19, 23, 24); others have confirmed this property of CPA specifically in bovine TSM (6) and shown complete overlap between ryanodine-sensitive and IP3-sensitive Ca2+ stores (2). However, we show here (Fig. 1A) that 5-HT-evoked contractions are relatively unaffected by pretreatment with CPA plus/minus ryanodine (10−5 M; the latter added to promote Ca2+ release through RyR).
Similarly, if the mechanism of action underlying this inhibitory effect of caffeine is simply through SR depletion, then 1 mM caffeine should also exert some inhibitory effect, since that concentration is also sufficient to sensitize RyR (34). However, this too was not the case (Fig. 1C).
Finally, caffeine-induced depletion of the SR should have little effect on contractions evoked by high millimolar KCl, which in ASM are mediated by promotion of voltage-dependent Ca2+ influx and/or stimulation of RhoA kinase activity (22, 28). However, we show here that caffeine reduces by more than 50% the magnitudes of contractions evoked by all concentrations of KCl (Fig. 2), irrespective of whether the SR has previously already been emptied or not. That is, depletion of the stores using CPA and ryanodine caused a modest left-hand shift in the KCl concentration-response relationship [Fig. 2; which we have previously shown is due to abolition of the superficial buffer barrier (18, 24)], but this prior depletion had no effect on the caffeine-induced reduction of KCl-evoked contractions (Fig. 2). Our observation that caffeine antagonizes KCl-evoked contractions also argues against the possibility that caffeine's effect on 5-HT-evoked contraction is mediated through blockade of 5-HT receptors.
Fig. 2.

Cumulative concentration-response relationships for KCl plus/minus CPA (10−5), ryanodine (10−5), and/or caffeine (10 mM), as indicated; n = 5. *Significantly different from control.
Caffeine acts through inhibition of PDE?
Altogether, then, the data suggest that mere depletion of the SR does not account for the inhibitory effect of millimolar caffeine on contractions in ASM. Being a methylxanthine, caffeine at concentrations 100-fold lower than those used here can also inhibit PDE activity (50), which might functionally antagonize the excitatory responses. IBMX (10−4 M), another methylxanthine with well-documented inhibitory action of PDE activity, fully reproduced the effect of caffeine (Fig. 3A). However, theophylline, a methylxanthine that has been used clinically to reverse bronchoconstriction through its ability to inhibit PDE activity (35, 37), when applied at a concentration of 100 μM [sufficient to fully inhibit PDE (35)], had no effect against KCl-evoked contractions (Fig. 3A). Similarly, 1 mM caffeine [which is also sufficient to inhibit PDE activity (50)] applied for 30 or 120 min did not reproduce this inhibitory effect (Fig. 3B), and 10 mM caffeine applied for 120 min did not have any greater effect than that at 10 min (Fig. 3C). Also not consistent with the interpretation that caffeine inhibits contraction by causing accumulation of cyclic nucleotides was our finding that its inhibitory effect was not augmented by rolipram [10−5 M; selective inhibitor of PDE4, the cAMP-dependent subtype that is prevalent in bovine ASM; (39)], nor reduced by the adenylate cyclase inhibitors SQ22536 or MDL 12330A (10−5 M) (Fig. 3D). Finally, we tested the effect of the protein kinase A inhibitor H89 (3 × 10−5 M) (Fig. 3D) on this effect of caffeine. Given that H89 is also a powerful inhibitor of ROCK (3), which in turn modulates the contractile response to KCl (22, 28, 40), we compared the effect of H89 in tissues that had concurrently been pretreated with or without the ROCK-selective inhibitor Y27632 (3): H89 did not remove the caffeine-induced suppression (Fig. 3D). Altogether, these various lines of evidence rule out the interpretation that this caffeine-induced suppression that we describe here is owing to inhibition of PDE, accumulation of cAMP and activation of PKA.
Fig. 3.
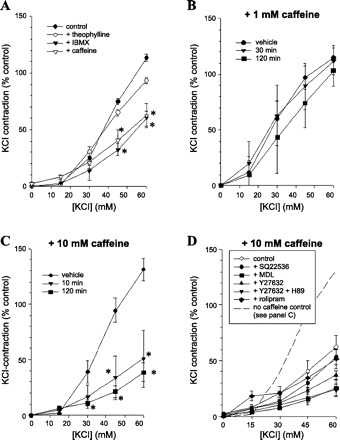
A: cumulative concentration-response relationships for KCl after pretreating tissues for 30 min with vehicle, caffeine (10 mM), theophylline (10−4), or IBMX (10−4), as indicated; n = 4. *Significantly different from control. Cumulative concentration-response relationships for KCl after incubations for various durations (as indicated) with 1 mM caffeine (B) or 10 mM caffeine (C); n = 5 for both. *Significantly different from control. D: cumulative concentration-response relationships for KCl after pretreating the tissues for 20 min with caffeine plus/minus either one of the adenylate cyclase inhibitors SQ22536 (10−5) or MDL12330A (10−5), the PDE4-selective inhibitor rolipram (10−5), the ROCK-inhibitor Y27632 (10−5), Y27632 plus the ROCK/PKA-inhibitor H89 (both 10−5), or vehicle, as indicated; n = 5 for all. There was no statistically significant reversal of the caffeine-induced effect in response to SQ22536, MDL12330A, H89 (+/−Y27632), nor any significant augmentation of this caffeine-induced inhibition in response to rolipram.
Caffeine acts as a direct functional antagonist?
Next, we explored the possibility that caffeine was acting as a direct functional antagonist by using the permeabilized muscle strip approach. First, to confirm that our preparation allowed direct access to the contractile apparatus, we compared responses to pCa 6.5 conditions before and after membrane permeabilization, as well as their sensitivity to voltage-dependent Ca2+ channel blockers. TSM strips did not exhibit a contractile response when challenged with pCa 6.5 cytosolic buffer prior to permeabilization with β-escin (Fig. 4A); likewise, permeabilization with β-escin in pCa 8 cytosolic buffer did not lead to an increase in tone (in fact, relaxation was often seen during permeabilization; Fig. 4A). However, once permeabilized by β-escin, challenge with pCa 6.5 cytosolic buffer (approximates [Ca2+]i during maximal agonist-evoked excitation) evoked a substantial and sustained contractile response (Fig. 4, A and B) that was unaffected by subsequent application of nifedipine to block voltage-dependent Ca2+ channels (Fig. 4B). Altogether, these observations confirm that the tone evoked in β-escin-permeabilized tissues by pCa 6.5 cytosolic buffer is due to direct stimulation of the contractile apparatus by free Ca2+.
Fig. 4.

A: original recordings from tissue challenged with pCa 6.5 buffer before and after permeabilization, evoking a contractile response only in the latter case. Subsequent application of IBMX (10−3) evoked a further increase in tone, whereas caffeine (10 mM) evoked a small and brief contraction but then completely reversed all tone. B: in a second tissue permeabilized by β-escin and treated with A23187 to release internal Ca2+ (as evidenced by the increase in tone) before constricting with pCa 6.5 buffer, nifedipine (10−6 M) had no effect but caffeine evoked only a reversal of tone. C: mean reversals of tone evoked by caffeine, IBMX, quinine, or theophylline applied at concentrations indicated. *Significantly different from the mean response evoked by 10 mM caffeine. Italicized numerals in parentheses indicate value of n in each case.
In bovine TSM strips permeabilized and then constricted using pCa 6.5 cytosolic buffer as described above, subsequent challenge with 10 mM caffeine led to a transient spike-like increase in tone, followed by a precipitous abolition of all tone (Fig. 4, A–C). The initial small contraction appears to be mediated by release of internal Ca2+, since it was occluded by pretreatment of the tissues using A23187 (10−5 M; e.g., compare Fig. 4, A and B). One millimolar caffeine evoked only a small drop in pCa 6.5 tone (Fig. 4C). The relaxant effect of caffeine is not mediated by inhibition of PDE, since the nonselective PDE inhibitor IBMX (10−3 M) exerted the opposite effect on pCa 6.5 tone (Fig. 4C). Theophylline at a concentration of 100 μM [sufficient to fully inhibit PDE (35)] caused no statistically significant reversal of pCa 6.5 tone (Fig. 4C); 1 and 10 mM theophylline; however, completely reversed pCa 6.5 tone. Finally, caffeine is also an agonist of bitter taste receptors (27), which have been shown to be bronchodilators (4): here, we found quinine, another bitter tastant, to be considerably and statistically significantly less effective than caffeine in reversing the contractile response to pCa 6.5 when applied at 1 mM or at 10 mM (Fig. 4C).
Caffeine disrupts the contractile apparatus.
Altogether, the data presented above indicate that the inhibitory effect of caffeine and theophylline are downstream of [Ca2+]i elevation. We therefore used a variety of assays to probe for any potential effect of caffeine on the various molecular events that determine the interactions between actin and myosin.
First, we used a standard commercially available ELISA kit to compare levels of filamentous and monomeric actin, to determine whether caffeine promoted the depolymerization of actin filaments. Tissues were challenged with carbachol for 20 min, then challenged with either 10 mM caffeine or vehicle (Krebs buffer) for another 20 min before flash freezing and processing for F-actin vs. G-actin. In this way, we found the F-actin-to-G-actin ratio in caffeine-treated tissues to be 66 ± 22% of that in control tissues (4 assays using tissues from 8 animals; P < 0.05).
A standard in vitro motility assay was used to quantify actin-myosin interaction directly. We found that the νmax was not significantly altered when the myosin had been phosphorylated in the presence of caffeine vs. control (Fig. 5A; ○). When caffeine was added to the thiophosphorylated myosin and the motility buffer, νmax was not significantly different from control either (Fig. 5A, ▲; P = 0.09), suggesting that caffeine does not alter the cross-bridge cycling. On the other hand, we found that the number of actin filaments bound by thiophosphorylated myosin in the assay (in which there is no cell membrane to act as a barrier to caffeine) was statistically significantly reduced by 1 and 10 mM caffeine compared with control (Fig. 5B; P < 0.05).
Fig. 5.

A: velocity of actin propulsion by myosin (νmax) when 1) myosin is phosphorylated in the presence of caffeine (or vehicle) (i.e., prior to introduction onto the motility surface; ○) and 2) caffeine (or vehicle) is added to the already thiophosphorylated myosin as well as the final assay buffer containing ATP, to ensure that the caffeine is not flushed out of the motility assay during velocity measurements (▲); n = 3 for both assays. B: number of actin filaments bound to the motility surface when caffeine (or vehicle) was added to the thiophosphorylated myosin, the labeled actin, and the actin buffer used to wash away the unattached actin filaments (no ATP was present in these conditions); n = 5.
Using the phospho-immunoblotting approach, we compared phosphorylation of MLC20 by MLCK (Fig. 6A, top; in the presence of Ca2+/calmodulin), of CPI-17 by ROCK (Fig. 6A, middle), or of CPI-17 by PKC (Fig. 6A, bottom) in the presence vs. absence of caffeine (0, 0.1 or 1.0 mM). We found phosphorylation of MLC20 or of CPI-17 to be unaffected by caffeine, suggesting that the latter does not inhibit MLCK, and further suggesting that it does not affect MLCP through inhibition of ROCK or PKC.
Fig. 6.
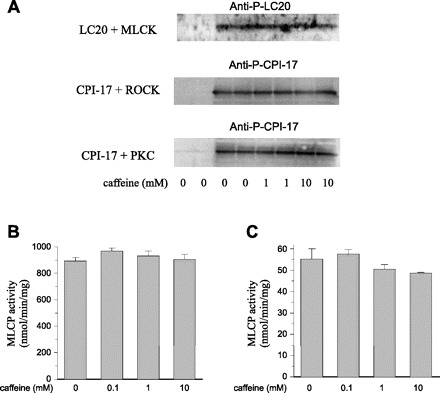
A: kinase assays were performed with regulatory light chain of myosin (MLC20; LC20) plus Ca2+/calmodulin-MLCK (MLCK) (top), or recombinant CPI-17 plus ROCK (middle) or PKC (bottom), and the reaction products were subjected to immunoblotting using the antibodies indicated. Myosin light chain phosphatase (MLCP) assay was performed as described in materials and methods. The specific activity (nmol released phosphate·min−1·mg−1 of MLCP) was determined by using untreated (B) or thiophosphorylated MLCP (C) incubated with phospho-MLC20 peptide in the presence of caffeine indicated. Of note, thiophosphorylation of MLCP reduced the specific activity 16-fold. Mean value ± SE was obtained from triplicate assays in 3 independent experiments (n = 9).
Finally, we further tested whether caffeine directly activates MLCP using the recombinant MLCP complex. Unphosphorylated MLCP (Fig. 6B) was slightly activated in the presence of 0.1 mM caffeine (n = 9, P < 0.05), but not affected at the higher concentrations. MLCP is inactivated when the regulatory subunit MYPT1 is phosphorylated at Thr696 or Thr853. Nonetheless, the inactivation of MLCP by ROCK-mediated thiophosphorylation (Fig. 6C) was not affected by the addition of caffeine (0.1 to 10 mM). We found no effect of caffeine on P-MLC20 dephosphorylation in either case, suggesting caffeine does not suppress contractions by enhancing MLCP activity.
DISCUSSION
In this study, we examined a wide variety of possible mechanisms by which 10 mM caffeine might inhibit contractions in ASM. Our group and others have elsewhere shown that it can release internally sequestered Ca2+ from the SR when applied at millimolar concentrations (18, 19, 23) and can inhibit PDE activity as well as block adenosine receptors at 10- to 100-fold lower concentrations (9, 50). Here, we identified another mechanism by which it can interfere with excitation-contraction coupling when applied at millimolar concentrations. This suppression was not due to suppression of PDE activity (with consequent accumulation of cyclic nucleotides and activation of PKA), since it was not prevented by inhibiting adenylate cyclase (SQ22536 or MDL12330A) or PKA (H89), nor exacerbated by inhibiting PDE4 (rolipram) nor mimicked by other interventions that are known to inhibit PDE (theophylline, submicromolar caffeine). Instead, it seems to involve a direct functional antagonism of the contractile apparatus itself, which manifests within minutes of exposure to caffeine (Figs. 1B and 4). That is, after permeabilizing the membrane using β-escin and depleting the internal Ca2+ store using A23187, it rapidly reversed tone evoked by direct application of Ca2+. As such, caffeine must be somehow interfering downstream of Ca2+-mediated activation of the contractile apparatus. We dissected the latter using a variety of molecular techniques, finding caffeine to have no effect on phosphorylation of MLC20 by MLCK, actin filament motility when using myosin phosphorylated in the presence of caffeine, actin filament motility catalyzed by MLCK, phosphorylation of CPI-17 by either PKC or ROCK, nor the activity of MLCP.
However, two observations suggest to us that caffeine exerted its effect through actin. First, it increased the ratio of globular to filamentous actin in TSM tissues that had been stimulated with the cholinergic agonist carbachol. Second, it dramatically reduced the binding of actin filaments by thiophosphorylated myosin in the in vitro motility assay. We acknowledge that the latter assay used purified skeletal actin with smooth muscle myosin, as our group (5, 26) and others have done many times in the past. This is done to simplify the assay: actin is otherwise accompanied by many regulatory proteins (calponin, caldesmon, tropomyosin, SM22), each exhibiting a variety of isoforms and phosphorylation states. Nonetheless, this approach provides an invaluable starting point from which to begin to understand the function and regulation of the contractile apparatus. Thus, from the data so obtained, it seems that caffeine dramatically reduces actin binding by myosin and/or promotes thin filament depolymerization (i.e., decreases the F- to G-actin ratio). Recent studies suggest that ASM contraction is also regulated through actin polymerization. Several groups have already established that disruption of actin filaments using latrunculin can severely impair contraction of ASM (1, 29). Zhao et al. (51) reported that ACh-triggered Ca2+ transients activate the Ca2+-calmodulin-dependent phosphatase calcineurin, which dephosphorylates ADF/cofilin and promotes actin polymerization in parallel with TSM contraction. Actin polymerization also occurs in arterial SM in response to α-adrenergic stimulation and leads to vasoconstriction (25). We presume that caffeine-induced actin depolymerization disrupts cross-bridge formation in the intact and permeabilized SM tissues, leading to a profound relaxation, without affecting myosin motor function per se.
Others have examined the effect of caffeine on the contractile apparatus in other muscle preparations and found this agent to inhibit MLCK itself in chicken gizzard SM (31) or to interfere with actin-myosin cross-bridge cycling in dog cardiac muscle (36). Neither of those two studies assessed actin filament polymerization state nor its binding to myosin filaments, although the second study did find actomyosin Mg2+-ATPase activity to be decreased by 20 mM caffeine (36). Theophylline (another methylxanthine, like caffeine) inhibited actin polymerization in guinea pig peritoneal macrophages, although that study linked the change to elevation of cAMP levels (11). Yet another group found caffeine inhibits the activation of JNK and thereby decreases the phosphorylation of c-Jun in cultured cerebellar granule neurons, but apparently through a mechanism that does not involve RyR (48). All of these effects of caffeine on non-RyR targets should be kept in mind in studies that use millimolar concentrations of caffeine to probe excitation-contraction coupling in SM.
Our finding that caffeine reversed KCl-evoked contractions led us to conclude that it is acting through some mechanism other than depletion of the internal Ca2+ pool, since our previous studies have shown that KCl contractions are augmented under conditions in which the internal Ca2+ store is depleted by cyclopiazonic acid plus/minus ryanodine, a phenomenon involving disruption of the “superficial buffer barrier” (18, 24). In contrast, Perez and Sanderson (33) have reported that KCl-evoked contractions are determined by the frequency of Ca2+ waves that KCl triggers from the SR. Both laboratories have shown that the KCl-evoked mechanical responses are not blocked by atropine (22, 33), ruling out any role for KCl-induced depolarization of nerve endings and consequent release of ACh (which both laboratories agree would elicit release of internal Ca2+). Both laboratories also agree that KCl promotes voltage-dependent Ca2+ influx, which is then shunted to the SR by the internal Ca2+ pump (18, 33) and that when the SR becomes overloaded it releases its store via RyR (33, 45). The only disagreement, then, is whether the KCl-evoked contractions are actually dependent on Ca2+ release from the SR and, as such, whether a caffeine sensitivity of KCl contractions indicates a mechanism involving depletion of the store (their view) or some non-SR-dependent mechanism (our view). Our group (22, 28) and several others (10, 40, 41, 47) have shown that depolarizing stimuli like KCl can activate the RhoA/ROCK signaling cascade and thereby sensitize the contractile apparatus to even small bursts of Ca2+ from an overloaded and decanting SR (which would be abolished by depletion of the store), but this does not mean that the KCl contraction is actually dependent on store-derived Ca2+.
These findings may be useful in developing therapeutic agents for the treatment of asthma. Although it is unrealistic to propose applying or delivering millimolar concentrations of caffeine to the airways, a better understanding of how this agent exerts its inhibitory effect(s) may set the stage for a more potent and clinically safe therapeutic tool. Theophylline (a methylxanthine like caffeine) and PDE inhibitors (of which IBMX is a nonselective congener) have been used in the treatment of asthma, and caffeine can act through that same mechanism that they share in common (50), but that pathway does not explain the phenomenon we describe here. It is interesting that caffeine is also an agonist for bitter taste receptors, which have recently been shown to elicit bronchodilatory actions through a mechanism that does not involve the cyclic nucleotide signaling cascade (4). We wondered whether caffeine might also be acting through those receptors but did not find quinine, a potent bitter tastant, to fully mimic the inhibition of KCl-evoked contractions.
In conclusion, we find that millimolar concentrations of caffeine can reverse ASM contraction in part through disruption of actin filaments. This mechanism joins several other ones through which caffeine can exert its actions, including inhibition of adenosine receptors (9), PDE activity (50), JNK (48), MLCK (31), and actin-myosin cross-bridge cycling (36), as well as sensitization of RyR (30, 34), and future studies should keep these in mind when using millimolar caffeine.
GRANTS
These studies were supported by operating funds provided by the Canadian Institutes of Health Research (MOP, L. J. Janssen; MOP, A-M. Lauzon), the National Institutes of Health (R01 DK088905; M. Eto), and Natural Science and Engineering Council of Canada (RGPIN; A-M. Lauzon).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHORSHIP CONTRIBUTIONS
Participated in research design: Janssen, Lauzon, Eto. Conducted experiments: Tazzeo, Bates, Roman, Khasnis. Contributed new reagents or analytic tools: not applicable. Performed data analysis: Tazzeo, Bates, Lauzon, Khasnis, Eto, Janssen. Wrote or contributed to the writing of the manuscript: Tazzeo, Bates, Lauzon, Eto, Janssen.
ACKNOWLEDGMENTS
The authors thank Dr. W. Gerthoffer for valuable discussion of these data.
REFERENCES
- 1. An SS, Laudadio RE, Lai J, Rogers RA, Fredberg JJ. Stiffness changes in cultured airway smooth muscle cells. Am J Physiol Cell Physiol 283: C792–C801, 2002 [DOI] [PubMed] [Google Scholar]
- 2. Bergner A, Sanderson MJ. Acetylcholine-induced calcium signaling and contraction of airway smooth muscle cells in lung slices. J Gen Physiol 119: 187–198, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95–105, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deshpande DA, Wang WC, McIlmoyle EL, Robinett KS, Schillinger RM, An SS, Sham JS, Liggett SB. Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nat Med 16: 1299–1304, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dyrda P, Tazzeo T, Doharris L, Nilius B, Roman HN, Lauzon AM, Aziz T, Lukic D, Janssen LJ. Acute response of airway muscle to extreme temperature includes disruption of actin-myosin interaction. Am J Respir Cell Mol Biol 44: 213–221, 2011 [DOI] [PubMed] [Google Scholar]
- 6. Ethier MF, Yamaguchi H, Madison JM. Effects of cyclopiazonic acid on cytosolic calcium in bovine airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 281: L126–L133, 2001 [DOI] [PubMed] [Google Scholar]
- 7. Eto M, Kitazawa T, Matsuzawa F, Aikawa S, Kirkbride JA, Isozumi N, Nishimura Y, Brautigan DL, Ohki SY. Phosphorylation-induced conformational switching of CPI-17 produces a potent myosin phosphatase inhibitor. Structure 15: 1591–1602, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eto M, Kitazawa T, Yazawa M, Mukai H, Ono Y, Brautigan DL. Histamine-induced vasoconstriction involves phosphorylation of a specific inhibitor protein for myosin phosphatase by protein kinase C alpha and delta isoforms. J Biol Chem 276: 29072–29078, 2001 [DOI] [PubMed] [Google Scholar]
- 9. Fredholm BB, Persson CG. Xanthine derivatives as adenosine receptor antagonists. Eur J Pharmacol 81: 673–676, 1982 [DOI] [PubMed] [Google Scholar]
- 10. Ghisdal P, Vandenberg G, Morel N. Rho-dependent kinase is involved in agonist-activated calcium entry in rat arteries. J Physiol 551: 855–867, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hamachi T, Hirata M, Koga T. Effect of cAMP-elevating drugs on Ca2+ efflux and actin polymerization in peritoneal macrophages stimulated with N-formyl chemotactic peptide. Biochim Biophys Acta 804: 230–236, 1984 [DOI] [PubMed] [Google Scholar]
- 12. Helli PB, Janssen LJ. Properties of a store-operated non-selective cation channel in airway smooth muscle. Eur Respir J 32: 1529–1539, 2008 [DOI] [PubMed] [Google Scholar]
- 13. Helli PB, Pertens E, Janssen LJ. Cyclopiazonic acid activates a Ca2+-permeable, non-selective cation conductance in porcine and bovine tracheal smooth muscle. J Appl Physiol 99: 1759–1768, 2005 [DOI] [PubMed] [Google Scholar]
- 14. Hirota S, Helli P, Janssen LJ. Ionic mechanisms and Ca2+ handling in airway smooth muscle. Eur Respir J 30: 1400–1419, 2007 [DOI] [PubMed] [Google Scholar]
- 15. Hirota S, Janssen LJ. Store-refilling involves both L-type calcium channels and reverse-mode sodium-calcium exchange in airway smooth muscle. Eur Respir J 30: 269–278, 2007 [DOI] [PubMed] [Google Scholar]
- 16. Hirota S, Pertens E, Janssen LJ. The reverse mode of the Na+/Ca2+ exchanger provides a source of Ca2+ for store refilling following agonist-induced Ca2+ mobilization. Am J Physiol Lung Cell Mol Physiol 292: L438–L447, 2007 [DOI] [PubMed] [Google Scholar]
- 17. Janssen LJ. T-type and L-type Ca2+ currents in canine bronchial smooth muscle: characterization and physiological roles. Am J Physiol Cell Physiol 272: C1757–C1765, 1997 [DOI] [PubMed] [Google Scholar]
- 18. Janssen LJ, Betti PA, Netherton SJ, Walters DK. Superficial buffer barrier and preferentially directed release of Ca2+ in canine airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 276: L744–L753, 1999 [DOI] [PubMed] [Google Scholar]
- 19. Janssen LJ, Sims SM. Emptying and refilling of Ca2+ store in tracheal myocytes as indicated by ACh-evoked currents and contraction. Am J Physiol Cell Physiol 265: C877–C886, 1993 [DOI] [PubMed] [Google Scholar]
- 20. Janssen LJ, Sims SM. Histamine activates Cl− and K+ currents in guinea-pig tracheal myocytes: convergence with muscarinic signalling pathway. J Physiol 465: 661–677, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Janssen LJ, Sims SM. Substance P activates Cl− and K+ conductances in guinea-pig tracheal smooth muscle cells. Can J Physiol Pharmacol 72: 705–710, 1994 [DOI] [PubMed] [Google Scholar]
- 22. Janssen LJ, Tazzeo T, Zuo J, Pertens E, Keshavjee S. KCl evokes contraction of airway smooth muscle via activation of RhoA and Rho-kinase. Am J Physiol Lung Cell Mol Physiol 287: L852–L858, 2004 [DOI] [PubMed] [Google Scholar]
- 23. Janssen LJ, Walters DK, Wattie J. Regulation of [Ca2+]i in canine airway smooth muscle by Ca2+-ATPase and Na+/Ca2+ exchange mechanisms. Am J Physiol Lung Cell Mol Physiol 273: L322–L330, 1997 [DOI] [PubMed] [Google Scholar]
- 24. Janssen LJ, Wattie J, Lu-Chao H, Tazzeo T. Muscarinic excitation-contraction coupling mechanisms in tracheal and bronchial smooth muscles. J Appl Physiol 91: 1142–1151, 2001 [DOI] [PubMed] [Google Scholar]
- 25. Kim HR, Graceffa P, Ferron F, Gallant C, Boczkowska M, Dominguez R, Morgan KG. Actin polymerization in differentiated vascular smooth muscle cells requires vasodilator-stimulated phosphoprotein. Am J Physiol Cell Physiol 298: C559–C571, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leguillette R, Gil FR, Zitouni N, Lajoie-Kadoch S, Sobieszek A, Lauzon AM. (+)Insert smooth muscle myosin heavy chain (SM-B) isoform expression in human tissues. Am J Physiol Cell Physiol 289: C1277–C1285, 2005 [DOI] [PubMed] [Google Scholar]
- 27. Lindemann B. Receptors and transduction in taste. Nature 413: 219–225, 2001 [DOI] [PubMed] [Google Scholar]
- 28. Liu C, Zuo J, Pertens E, Helli PB, Janssen LJ. Regulation of Rho/ROCK signaling in airway smooth muscle by membrane potential and [Ca2+]i. Am J Physiol Lung Cell Mol Physiol 289: L574–L582, 2005 [DOI] [PubMed] [Google Scholar]
- 29. Mehta D, Gunst SJ. Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J Physiol 519: 829–840, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meissner G. Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium 35: 621–628, 2004 [DOI] [PubMed] [Google Scholar]
- 31. Ozaki H, Kasai H, Hori M, Sato K, Ishihara H, Karaki H. Direct inhibition of chicken gizzard smooth muscle contractile apparatus by caffeine. Naunyn Schmiedebergs Arch Pharmacol 341: 262–267, 1990 [DOI] [PubMed] [Google Scholar]
- 32. Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol 85: 164–181, 1982 [DOI] [PubMed] [Google Scholar]
- 33. Perez JF, Sanderson MJ. The frequency of calcium oscillations induced by 5-HT, ACH, and KCl determine the contraction of smooth muscle cells of intrapulmonary bronchioles. J Gen Physiol 125: 535–553, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pessah IN, Stambuk RA, Casida JE. Ca2+-activated ryanodine binding: mechanisms of sensitivity and intensity modulation by Mg2+, caffeine, and adenine nucleotides. Mol Pharmacol 31: 232–238, 1987 [PubMed] [Google Scholar]
- 35. Polson JB, Krzanowski JJ, Goldman AL, Szentivanyi A. Inhibition of human pulmonary phosphodiesterase activity by therapeutic levels of theophylline. Clin Exp Pharmacol Physiol 5: 535–539, 1978 [DOI] [PubMed] [Google Scholar]
- 36. Powers FM, Solaro RJ. Caffeine alters cardiac myofilament activity and regulation independently of Ca2+ binding to troponin C. Am J Physiol Cell Physiol 268: C1348–C1353, 1995 [DOI] [PubMed] [Google Scholar]
- 37. Rabe KF, Magnussen H, Dent G. Theophylline and selective PDE inhibitors as bronchodilators and smooth muscle relaxants. Eur Respir J 8: 637–642, 1995 [PubMed] [Google Scholar]
- 38. Rodat-Despoix L, Aires V, Ducret T, Marthan R, Savineau JP, Rousseau E, Guibert C. Signalling pathways involved in the contractile response to 5-HT in the human pulmonary artery. Eur Respir J 34: 1338–1347, 2009 [DOI] [PubMed] [Google Scholar]
- 39. Rousseau E, Dostie J, Taoudi-Benchekroun M, Cadieux A, Beaudry C, Lugnier C. Specific cyclic nucleotide phosphodiesterase inhibitors differently modulate contractile kinetics in airway smooth muscle. Can J Physiol Pharmacol 73: 1784–1794, 1995 [DOI] [PubMed] [Google Scholar]
- 40. Sakamoto K, Hori M, Izumi M, Oka T, Kohama K, Ozaki H, Karaki H. Inhibition of high K+-induced contraction by the ROCK inhibitor Y-27632 in vascular smooth muscle: possible involvement of ROCK in a signal transduction pathway. J Pharmacol Sci 92: 56–69, 2003 [DOI] [PubMed] [Google Scholar]
- 41. Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y, Takuwa Y. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ Res 93: 548–556, 2003 [DOI] [PubMed] [Google Scholar]
- 42. Sanders KM. Invited Review: Mechanisms of calcium handling in smooth muscles. J Appl Physiol 91: 1438–1449, 2001 [DOI] [PubMed] [Google Scholar]
- 43. Sobieszek A, Bremel RD. Preparation and properties of vertebrate smooth-muscle myofibrils and actomyosin. Eur J Biochem 55: 49–60, 1975 [DOI] [PubMed] [Google Scholar]
- 44. Takeya K, Loutzenhiser K, Shiraishi M, Loutzenhiser R, Walsh MP. A highly sensitive technique to measure myosin regulatory light chain phosphorylation: the first quantification in renal arterioles. Am J Physiol Renal Physiol 294: F1487–F1492, 2008 [DOI] [PubMed] [Google Scholar]
- 45. Tazzeo T, Zhang Y, Keshavjee S, Janssen LJ. Ryanodine receptors decant internal Ca2+ store in human and bovine airway smooth muscle. Eur Respir J 32: 275–284, 2008 [DOI] [PubMed] [Google Scholar]
- 46. Trybus KM, Lowey S. Mechanism of smooth muscle myosin phosphorylation. J Biol Chem 260: 15988–15995, 1985 [PubMed] [Google Scholar]
- 47. Urban NH, Berg KM, Ratz PH. K+ depolarization induces RhoA kinase translocation to caveolae and Ca2+ sensitization of arterial muscle. Am J Physiol Cell Physiol 285: C1377–C1385, 2003 [DOI] [PubMed] [Google Scholar]
- 48. Wang WY, Li MT, Pi RB, Qiu PX, Su XW, Lin SZ, Yan GM. Antagonistic action of caffeine against LY294002-induced apoptosis in cerebellar granule neurons. Acta Pharmacol Sin 21: 35–40, 2000 [PubMed] [Google Scholar]
- 49. Warshaw DM, Desrosiers JM, Work SS, Trybus KM. Mechanical interaction of smooth muscle crossbridges modulates actin filament velocity in vitro. Prog Clin Biol Res 327: 815–826, 1990 [PubMed] [Google Scholar]
- 50. Wells JN, Kramer GL. Phosphodiesterase inhibitors as tools in cyclic nucleotide research: a precautionary comment. Mol Cell Endocrinol 23: 1–9, 1981 [DOI] [PubMed] [Google Scholar]
- 51. Zhao R, Du L, Huang Y, Wu Y, Gunst SJ. Actin depolymerization factor/cofilin activation regulates actin polymerization and tension development in canine tracheal smooth muscle. J Biol Chem 283: 36522–36531, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]