

Abstract
Calpain activation has been implicated in the disease pathology of Duchenne muscular dystrophy. Inhibition of calpain has been proposed as a promising therapeutic target, which could lessen the protein degradation and prevent progressive fibrosis. At the same time, there are conflicting reports as to whether elevation of calpastatin, an endogenous calpain inhibitor, alters pathology. We compared the effects of pharmacological calpain inhibition in the mdx mouse using leupeptin and a proprietary compound (C101) that linked the inhibitory portion of leupeptin to carnitine (to increase uptake into muscle). Administration of C101 for 4 wk did not improve muscle histology, function, or serum creatine kinase levels in mdx mice. Mdx mice injected daily with leupeptin (36 mg/kg) for 6 mo also failed to show improved muscle function, histology, or creatine kinase levels. Biochemical analysis revealed that leupeptin administration caused an increase in m-calpain autolysis and proteasome activity, yet calpastatin levels were similar between treated and untreated mdx mice. These data demonstrate that pharmacological inhibition of calpain is not a promising intervention for the treatment of Duchenne muscular dystrophy due to the ability of skeletal muscle to counter calpain inhibitors by increasing multiple degradative pathways.
Keywords: protease, skeletal muscle, muscular dystrophy, calpain
duchenne muscular dystrophy (DMD) is an X-linked, progressive muscle-wasting disease caused by the absence of the protein dystrophin (for review, see Ref. 14). Loss of dystrophin increases the susceptibility to muscle fiber damage and membrane rupture in response to normal muscle contractions, which in turn leads to membrane damage and uncontrolled Ca2+ influx (1, 35, 37). A significant consequence of high cytoplasmic Ca2+ levels is the activation of calpains, a group of cysteine proteases, which leads to protein degradation. The continued cycles of muscle degeneration and compensatory regeneration leads to inflammation, and ultimately progressive fibrosis replaces functional muscle. Current pharmaceutical interventions include only prednisone and other corticosteroids that counter inflammation (2, 11, 24). Therefore, an unmet need exists for further drug development to target many of the additional deleterious consequences of the dystrophic process.
Although the ultimate but technically challenging cure for DMD is the restoration of dystrophin, inhibition of the early steps of muscle damage may help to prevent the downstream pathology. One such target may be the calpains, which are activated upon calcium entry. These proteases are responsible for cleavage of larger proteins, such as talin, into polypeptides that are subsequently ubiquitinated and degraded by the proteasome system (30). The increased Ca2+ influx in dystrophin-deficient muscle may result in micro-environments with high Ca2+ concentrations and potentially lead to local calpain activation (19). Elevated calpain activity has been reported on numerous occasions in mdx animals, suggesting that the increased cytosolic Ca2+ is sufficient to induce calpain-induced protein cleavage (1, 18, 31).
Indirect evidence that blocking calpain activity may be beneficial was provided by a previous study showing that two calcium channel blockers (tranilast and diltiazem) reduce serum creatine kinase (CK) activity in mdx mice (20). However, more substantial evidence was demonstrated by the use of leupeptin (LPTN), a nonspecific pharmacological calpain inhibitor, which increased fiber diameter and improved muscle histology (3). Another endogenous modulator of calpain is calpastatin, but there have been mixed results of using increased calpastatin to combat dystrophic pathology, where muscle-specific transgenic expression was shown to either provide histological benefit (32) or not (6), depending on which promoter was utilized. Thus, it remains an open question as to whether or not calpain inhibition is a good therapeutic target for DMD.
While LPTN does effectively inhibit calpain, it is a nonspecific protease inhibitor and can interact with other cysteine and serine proteases, including cathepsins B and L, but not caspases (21, 28). It also can interfere with proteases required for successful completion of the protease cascade involved in blood clotting, making its use in humans tenuous (5). To both decrease the potential toxicity of LPTN and to get better penetration into muscle, a strategy was devised whereby carnitine was covalently linked to LPTN, creating a novel calpain inhibitor known as C101. In theory, the linkage to carnitine affords efficient uptake into muscle cells via the carnitine organic cation transporter found on the sarcolemma. This has the potential to maximize muscle drug content and blood clearance, thus minimizing its potential interference with blood clotting. It was anticipated that greater LPTN activity within the muscle would lead to greater calpain inhibition and concomitant improvement of the mdx phenotype. Specifically, we hypothesized that C101 administration would improve muscle function and histology of mdx mice to a greater extent than was previously reported (3) with LPTN.
MATERIALS AND METHODS
Study Design and Animal Treatments
The experimental protocols in this study were approved by the University of Pennsylvania's Institutional Animal Care and Use Committee. Both C57Bl6 (C57) and mdx mice were utilized. Animals were treated with compound or vehicle for 4 or 26 wk beginning at 2 or 8 wk of age. Treatment compounds included 100 mg/kg C101 or PBS for 4-wk treatment regimens and 36 mg/kg LPTN and PBS for the 26-wk regimen. C101 is a proprietary compound consisting of LPTN bound to carnitine through a linker sequence. Details of each experiment are described below.
Short-term study 1.
Twenty-four mice were obtained from our colonies of mdx (n = 12) and C57Bl6 (n = 12) strains and divided into four matched groups (6/group) receiving PBS or 100 mg/kg of C101. Drug was injected into the intraperitoneal cavity daily beginning in the 8th wk of life and continuing for 4 wk until the animals were killed at 12 wk of age. Serum CK activity, isolated muscle function tests of the extensor digitorum longus (EDL) and soleus muscles, and histological measures on the diaphragm muscles were performed as described below. In addition, three animals in each group were injected with Evan's blue dye (EBD) 24 h prior to death to evaluate muscle fiber integrity.
Short-term study 2.
To follow the same treatment strategy as Badalamente and Stracher (3) mdx animals were injected in the intraperitoneal cavity with either PBS (n = 4) or 100 mg/kg C101 (n = 8) daily for 4 wk, beginning at 2 wk of age and ending at 6 wk of age. These animals were killed, and the gastrocnemius, quadriceps, and diaphragm were removed for histological analysis as described below.
Long-term study.
To evaluate the therapeutic potential of calpain inhibition in ameliorating long-term disease progression, animals were exposed to compound for a period of 6 mo. Animals were divided into four groups as follows: C57Bl6 PBS (n = 6), C57Bl6 LPTN (n = 6), mdx PBS (n = 6), and mdx LPTN (n = 6). Intraperitoneal cavity injections of 36 mg/kg LPTN or PBS started at 2 mo of age and continued daily for 6 mo. We could not conduct long-term C101 studies due to limitations in drug availability. Hence, we chose to use LPTN in our long-term calpain inhibition study. The LPTN dose used in this investigation is identical to the daily dose used in the study by Badalamente and Stracher (3). However, to prevent a potential increase in animal stress, immune response, and injury to the injection site, we elected to use a single daily dose. By 6 mo of age, the diaphragm exhibits clear pathology, including muscle degeneration, central nucleation, and extensive fibrosis (10, 33). Therefore, functional analysis was limited to the diaphragm. Serum CK, histological analysis on the diaphragm muscles, and biochemical measurements on the tibialis anterior muscles were performed.
Muscle Function Tests
Contractile function was performed according to standard techniques (4). Briefly, isolated muscle function was performed on a commercially available apparatus with associated software (Aurora Scientific, Ontario, Canada). Mice were anesthetized with ketamine/xylazine. Muscles were removed and placed in a bath of Ringers solution gas-equilibrated with 95% O2-5% CO2. Sutures were attached to the distal and proximal tendons of the EDL and soleus muscles, and to the central tendon and rib of the diaphragm preparations. Optimum length (Lo) was determined using standard techniques followed by supramaximal stimulation (EDL, 120 Hz, 40 V; Sol, 100 Hz 40 V; diaphragm, 100 Hz, 40 V) to achieve maximum isometric tetanic contractions. Each muscle performed three 500-ms tetanic contractions at Lo with 5 min between each trial. Cross-sectional area and specific tension were calculated using standard equations and constants (7).
Histology
Muscles were dissected, blotted, and weighed and then pinned to Styrofoam and coated in embedding compound (OCT). Muscles were rapidly frozen in melting isopentane (2-methyl butane) and then stored at −80°C until subsequent histological analysis. Ten-micrometer histological sections were cut at −30°C with a Leica CM3000 cryostat (Bannockburn, IL). Hematoxylin and eosin staining was accomplished according to standard techniques. Trichrome staining was done using a kit from Sigma (HT 15). EBD staining was performed on diaphragm sections from EBD-injected animals in accordance with Straub et al., (34) with modifications. Briefly, sections were incubated in PBS for 5 min to remove OCT, acetone for 1 min, and then rinsed in PBS. Sections were dried, coverslips were mounted with Vectashield medium containing 4,6-diamidino-2-phenylindole (DAPI) for nucleus identification, and the slides were sealed with nail polish. Detection conditions were identical for all sections. EBD-positive cells were detected on an eipfluorescence microscope (Leica, Bannockburn, IL) and image analysis software (Openlab, Perkin Elmer) and are reported as percent area of the section.
Immunohistochemistry for laminin was utilized to define fiber area. Tissues were washed in PBS for 10 min, and sections were then circled with a Pap pen to create a well. Blocking was accomplished by covering each section with 5% BSA and incubation for 15 min. Sections were then incubated overnight at 4°C in laminin primary antibody (Laminin Ab-1; Neomarkers, Fremont, CA) at a dilution of 1:100 in 5% BSA. They were then washed three times in PBS and incubated with rabbit Alexa Fluor 488 secondary antibody (Invitrogen) at a dilution of 1:200 in 5% BSA for 1 h in the dark. Sections were washed three times for 10 min in PBS and mounted with Vectashield with DAPI, and the edges were sealed with nail polish. Sections were analyzed for fiber size and the proportion of centrally nucleated fibers.
Biochemical Analyses
Animals were anesthetized and then exsanguinated prior to muscle dissections by cardiac puncture. Blood was allowed to clot for 30 min on ice and was then centrifuged at 4°C for 10 min at 3,000 rpm on a bench-top centrifuge. Supernatant was then removed and stored in liquid nitrogen prior to CK measurements. CK activity was determined on serum by utilizing a kit from Diagnostic Chemicals (cat. no. 310; Oxford, CT) and read in a microplate reader at 340 nm for 30 min. Data are reported as units per liter.
Western blot analysis was done as previously described with minor modifications (29). Muscle was powdered on dry ice and lysed using lysis buffer [50 mM Tris (pH 8.0), 5 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.5% deoxycholic acid, 50 mM DTT with Complete protease inhibitor cocktail (Roche Diagnostic, Indianapolis, IN)] at a 1:10 dilution. Following lysis, whole homogenate was centrifuged at 700 rpm for 10 min to remove large cellular debris. Protein concentration was determined by the method of Biuret. Following dilution to 2.5 mg/ml in reducing buffer, precisely 25-μg protein was loaded into 5–20% gradient gels. Transfer was accomplished by using the I-blot system from Invitrogen (Carlsbad, CA). Primary antibodies were used at 1:1,000 in 1% milk dissolved in Tween-20 Tris-base sodium (TTBS) overnight at 4°C. Primary antibodies were used as follows: calpastatin (cat. no. Ab28254; Abcam), μ-calpain (cat. no. C5611; Sigma), m-calpain (cat. no. Ab81013; Chemicon), and talin (cat. no. T3287; Sigma). Secondary antibodies were incubated for 1 h at room temperature at a 1:2,000 dilution in 1% mild dissolved TTBS. Detection was achieved by enhanced chemiluminescence and X-ray film with subsequent analysis of band intensity.
Zymograms were performed using the technique of Raser (27). Muscle samples were homogenized in lysis buffer at a 1:5 wt/vol ratio, and centrifuged at 12,000 g for 30 min at 4°C. After protein concentration was determined, samples were diluted to 0.5 mg/ml in ×5 sample buffer (20% glycerol, 150 mM Tris·HCl, pH 6.8, 0.75% 2-ME, 0.02% bromophenol blue). Samples were loaded onto a gel pretreated with casein (12% zymogram gel; Bio-Rad). Gels were run in a cold room on ice at 125 V for 3 h. Following electrophoresis, gels were washed with cold water and incubated at room temperature with developing buffer (50 mM Tris·HCl, pH 7.0, 5 mM CaCl2, 0.05% 2-ME) twice for 1 h and a third time, over night. To visualize bands, gels were stained with Coommassie Blue for 1 h, destained, the image captured, and band optical density quantified. Optical density was expressed relative to background intensity.
The chymotrypsin-like activity of the proteasome was determined by the method of Vigouroux (36), with modifications. A frozen section of tibialis anterior was weighed, pulverized on dry ice, and homogenized in a 1:10 wt/vol ratio in a 50 mM Tris·HCl buffer (pH 8.0) containing 1.0 mM EDTA, 1.0 mM EGTA, 1.0 mM DTT, 2.0 mM ATP, 10% glycerol and Complete protease inhibitor cocktail (Roche Diagnostic). The homogenates were centrifuged for 1 h at 100,000 g at 4°C. Protein concentration of the supernatant was determined using the Bradford reaction (Bio-Rad, Hercules, CA) with BSA as a standard. Homogenates were diluted with assay buffer [50.0 mM Tris·HCl (pH 7.5), 40.0 mM KCl, 5.0 mM MgCl2, 2.0 mM ATP, 1.0 mM DTT, 10.0 μg BSA] to normalize protein concentration. In our experimental conditions, components were mixed in a 1:1:2 ratio such that 50-μl homogenate, 50 μl substrate, and 100 μl assay buffer or epoxomicin were in each well. Homogenate contained 10 μg of protein. Substrate (Suc-LLVY-MAC) was dissolved in assay buffer and had a final concentration of 100 μM. Samples were incubated with assay buffer or 100 μM of epoxomicin, in 100 μl assay buffer. After a 1-h incubation at 37°C, measurements were taken with Eex 340 and Eem 465 nm (GENios Pro; TECAN, Mannedorf, Switzerland).
To determine the inhibitor activity of C101, in vitro, 25 μl of dilution buffer (20 mM Tris·HCl, pH 7.5, 1 mM EDTA, 100 mM KCL, 0.1% MCE) containing calpain, 25 μl of dilution buffer containing variable concentrations of C101, and 50 μl of dilution buffer were added to bring the total volume to 100 μl. The reaction was initiated by adding 100 μl of BODIPY-FL-casein (10 μg/ml in dilution buffer + 10 mM Ca2+). Final calpain concentrations were 50, 5, and 2.5 μM. Measurements were made using a Tecan fluorometer, reading the fluorescence at 485-nm excitation and 535-nm emission. Every assay included two blanks. One is a Ca2+ blank (dilution buffer + BODIPY-casein with no calpain or C101) intended to control for nonspecific substrate degradation. The second controls for nonspecific activity for each sample and contains 25 μl of 100 mM EDTA, 25 μl of dilution buffer, 25 μl of calpain solution at the same concentration under investigation, 25 μl of C101 at the same concentration under investigation, and 100 μl of BODIPY-casein. Importantly, the 100 mM EDTA solution was added before the C101 solution to remove Ca2+. Calpain activity is calculated as follows: FU cal = FUsample − (FU Ca blank + FU EDTA blank)/2. Measurements were taken every 5 or 10 min, starting with time 0, and slope was calculated using linear part of the graph. C101 compound levels were determined in serum and in tissues by National Medical Services Labs, Willow Grove, PA.
Statistics
Within each study, groups were compared with an ANOVA (followed by a Bonferroni post hoc test) or t-test, where appropriate. The fiber cross-sectional area was compared using a repeated-measures ANOVA. Statistical significance was defined a priori as P < 0.05. Values are displayed as means ± SE unless otherwise noted.
RESULTS
The goal of this study was to determine whether calpain inhibition could provide morphological and physiological amelioration of pathology in the mdx mouse. A novel compound based on LPTN was developed (C101) to provide efficient exposure of skeletal muscle to this inhibitor and to enable rapid clearance of the inhibitor from the circulation. To confirm that C101 is a calpain inhibitor, an in vitro assay was performed measuring calpain-mediated cleavage of casein in the presence of progressively increasing amounts of C101. Calpain activity was reduced in a dose-dependent fashion confirming that C101 inhibits calpain activity (Fig. 1).
Fig. 1.
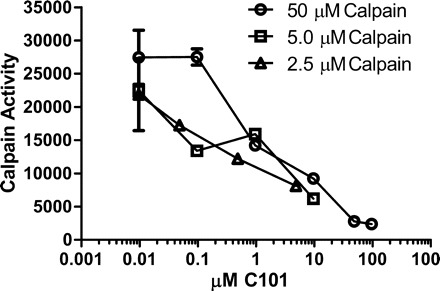
C101 inhibits calpain activity. A range of μM C101 was added to 3 starting concentrations of calpain. In each instance, C101 reduced calpain activity.
To determine whether C101 entered the circulation and skeletal muscle after intraperitoneal injection, C101 content was measured in the serum and tissue lysates. Following a 100-mg/kg injection, peak serum levels occurred by 30 min, with a maximum level of 15,000 ng/ml (equivalent to 29 μM). By 2 h postinjection, blood levels fell to 3 μM, and C101 was undetectable in the circulation by 8 h postinjection. The content of C101 in muscle lysates was determined in a similar manner, but in animals that had received injections for a period of 4 wk. C101 content was ∼8 μM at 1 h postinjection, and 4 μM 24 h following injection, indicating that it is maintained in skeletal muscle. Skeletal muscle is estimated to contain between 0.2 and 0.03 μM calpain (13, 25). Given the levels of calpain and inhibitor in skeletal muscle and data generated in vitro, we expected ∼75% inhibition of calpain with daily C101 administration.
Short-Term Study 1
The first study evaluated C101 treatment for a duration of 4 wk in young adult C57 and mdx mice. Muscle mass, morphology, and function, as well as additional indices of muscle integrity were measured to determine whether C101 administration was beneficial. The body weights of both C57 groups and both mdx groups were similar prior to beginning the experiment and remained so following 4 wk of drug treatment (see Supplemental Table 1. Supplemental data for this article are available online at the American Journal of Physiology–Regulatory, Integrative and Comparative Physiology website.) Overall, mdx animals were larger than C57 animals; however, at the beginning of the experiment only the mdx group was significantly different from both C57 groups, while the mdx 100 mg/kg group was not. Both mdx groups were significantly larger than both C57 groups immediately prior to death. Absolute and relative muscle masses were also similar regardless of drug treatment within animal types; however, mdx animals tended to have larger muscle masses than C57 animals. The relative EDL mass was ∼10% larger in mdx mice treated with 100 mg/kg C101 compared with untreated mdx mice (P < 0.05).
Muscle function was assessed in the EDL and soleus muscles (Table 1). There were no significant changes in force generation or CSA in response to C101 treatment in muscles from either C57 or mdx mice. Comparisons between muscles from mdx mice and those from C57 mice showed that CSA measurements were consistently larger, and specific tension was consistently lower in the mdx groups.
Table 1.
Muscle function following 28 days of vehicle or C101 administration in mdx and C57 mice
| EDL Tet, mN | EDL CSA, mm2 | EDL ST, N/cm2 | Sol Tet, mN | Sol CSA, mm2 | Sol ST, N/cm2 | |
|---|---|---|---|---|---|---|
| C57 control | 393 ± 2 | 1.66 ± 0.05 | 22.5 ± 0.5 | 208 ± 7 | 1.06 ± 0.02 | 21.5 ± 0.5 |
| C57 100 mg/kg | 393 ± 2 | 1.65 ± 0.04 | 23.0 ± 0.7 | 220 ± 5 | 0.97 ± 0.04 | 22.2 ± 0.4 |
| mdx control | 383 ± 15 | 2.15 ± 0.06*† | 19 ± 0.4*† | 229 ± 18 | 1.31 ± 0.07*† | 18.8 ± 0.9† |
| mdx 100 mg/kg | 393 ± 8 | 2.18 ± 0.05*† | 18.5 ± 0.6*† | 238 ± 5 | 1.25 ± 0.04*† | 19 ± 0.9† |
All values are means ± SE. Eight-week-old mice were given daily injections of PBS or 100 mg/kg C101 for 4 wk. For both muscles tested, tetanic force (Tet) was similar between groups. For the extensor digitorum longus (EDL), both mdx groups had a larger cross-sectional area (CSA) and smaller specific tension (ST) when compared with both C57 groups. For the soleus (Sol), both mdx groups had a larger CSA when compared with both C57 groups and a smaller ST when compared with the C57 100 mg/kg group.
Significant difference when compared with C57 (P < 0.05);
significant difference when compared with C57 100 mg/kg (P < 0.05).
Muscle integrity was assessed by determining serum CK activity (Supplemental Fig. 1). CK is an enzyme specific to muscle cells, which when damaged, will leak CK into the interstitial space and the blood. Increasing serum CK activity is indicative of increased muscle cell membrane damage. CK was dramatically increased in mdx animals compared with C57, but C101 treatment did not affect the elevated serum CK in mdx mice. Therefore, C101 did not protect the sarcolemma from damage.
In the diaphragm, subjective observation following hematoxylin and eosin staining showed C57 and C57 100 mg/kg muscles were indistinguishable upon microscopic inspection (Supplemental Fig. 2). Muscles from mdx and mdx 100 mg/kg animals were also indistinguishable by microscopic inspection. The two mouse types, however, differed greatly in their appearance compared with each other. In mdx animals, greatly increased numbers of infiltrating immune cells, as indicated by increased numbers of nonskeletal muscle nuclei, were observed. In addition, areas of damage or necrosis were obvious in mdx animals. Finally, centralized nuclei, a feature of regenerating skeletal muscle, were common in muscles from mdx animals. By contrast, in C57 animals, there were far fewer infiltrating nuclei, and the detection of damage, necrosis, and centralized nuclei was rare, all indications of healthy skeletal muscle.
EBD absorbance was used as an additional quantitative measure of necrosis. EBD is excluded by fibers that are viable and absorbed by necrotic fibers (Fig. 2, A–D). It is observed visually as brightly lit fibers on a dull background when excited and measured quantitatively as a percentage of the total section area. C101 failed to reduce necrosis in mdx muscles (Fig. 2E). In addition, C101 failed to reduce the background emission signal in mdx animals indicating a similar basal absorbance, likely through small lesions in the sarcolemma (Fig. 2F).
Fig. 2.

A–D: magnification, ×20. Representative pictures of sections from EBD-treated animals: C57 control (A), C57 100 mg·kg−1·day−1 C101 (B), mdx (C), and mdx 100 mg·kg−1·day−1 C101 (D). These sections were also stained with DAPI, a nuclear stain that appears as blue. Note that the red background is far more visible in C and D (mdx) than the A and B (C57). In addition, in C and D areas of intense color can be seen indicating areas of necrosis. The % necrotic area (E) in each section was significantly increased in mdx animals compared with C57 animals. Threshold intensity was set at 100 intensity units (0–255) to identify Evan's blue dye (EBD)-positive cells. Intensity below this threshold was excluded from the sum of the necrotic area. This total area was then made relative to the section area. The background intensity (F) was increased in mdx animals, indicating a higher level of EBD absorbance into the cells, likely through sarcolemmal lesions. Median intensity units were quantified from each section. Note that regardless of group, median intensity was below the threshold of 100 intensity units used to identify the % necrotic area, indicating that the higher background intensity observed in mdx animals did not contribute to the increased necrotic area. *Significantly different from C57; †significantly different from C57 100 mg·kg−1·day−1 C101.
Short-Term Study 2
The second study evaluated C101 treatment for a duration of 4 wk spanning the weaning transition from 2 to 6 wk of age to follow the same treatment strategy as Badalamente and Stracher (3). Body and muscle weights were similar between groups (Supplemental Table 2). Inspection of the hematoxylin and eosin-stained muscles did not reveal a difference in tissue morphology between C101 and PBS-treated mdx animals in the gastrocnemius or diaphragm (Supplemental Fig. 3). Diaphragm sections were assessed for fiber area distribution and central nucleation because previous reports demonstrated a reduction in fiber area variability and central nucleation by calpain inhibition (Fig. 3; 23). However, there was no effect of C101 treatment on fiber area distribution or the proportion of fibers with central nuclei (data not shown).
Fig. 3.
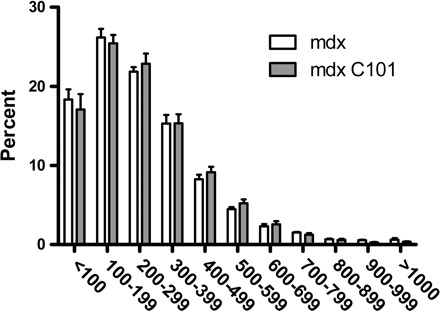
Fiber area distribution in the diaphragm following 4 wk of intraperitoneal injection of PBS or C101.
Long-Term Study
To determine whether there was cumulative benefit of calpain inhibition that cannot be demonstrated in short-term treatment regimens, we evaluated calpain inhibition for a period of 6 mo using LPTN. LPTN (36 mg/kg) or PBS was injected intraperitoneally daily for 6 mo into C57 and mdx mice. Following this course of injections, body weights and muscle masses were similar within animal types. Overall, mdx muscles were larger than C57 muscles both absolutely and relatively, but did not differ according to injection type (Supplemental Table 3). The diaphragm was the muscle of focus for the 6-mo treatment group because in mdx animals of this age there is progressive fibrosis and necrosis that mimics the pathology associated with DMD, while mouse limb muscles show very little functional decline beyond 3 mo (10, 33). Specific tension (Fig. 4) and tetanic force (data not shown) from the diaphragm were greatly reduced in mdx animals compared with C57. LPTN administration failed to improve muscle force in mdx animals.
Fig. 4.

Specific tension in the diaphragm following 6 mo of daily injections of PBS or 36 mg/kg ip leupeptin (LPTN). C57 animals produced significantly more specific tension than mdx animals. Leupeptin had no effect. *Significantly different from C57 (P < 0.05); †significantly different from C57 LPTN (P < 0.05).
Sections of diaphragm were frozen for histological sectioning and staining. In similar fashion to earlier results found in short-term studies 1 and 2, hematoxylin and eosin staining revealed significant immune cell infiltration, apparent areas of necrosis, and centralized nuclei in sections taken from mdx animals (Supplemental Fig. 4). By contrast, sections from C57 animals appeared healthy. Furthermore, trichrome staining indicated significant fibrosis in mdx animals that was not apparent in C57 animals (Fig. 5). LPTN did not appear to be of benefit in either staining condition. Consistent with the histological findings of increased degeneration, serum CK measures showed that activity in mdx animals was not corrected following 6 mo of LPTN administration (Supplemental Fig. 5). Following laminin staining, the fiber cross-sectional area was determined (Supplemental Fig. 6). Dystrophic animals have a much higher proportion of small fibers, likely indicating regenerating or necrotic fibers, compared with C57 animals. This abnormal fiber area distribution was unchanged with LPTN treatment.
Fig. 5.
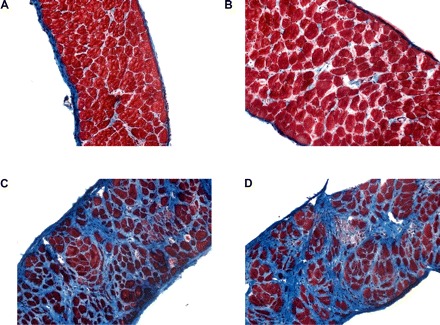
Representative micrographs (magnification, ×20) of trichrome-stained diaphragms from C57 (A–B) and mdx (C–D) animals. Animals were treated for 6 mo with daily injections of PBS (A, C) or 36 mg/kg LPTN (B, D).
We next attempted to determine why LPTN failed to rescue mdx animals. Talin is a calpain substrate, and its cleavage can be used as an indicator of calpain activity. Following 6 mo of LPTN administration, calpain activity was similar within groups (Fig. 6A). Intact talin was similar between all groups; however, the calpain cleavage product was increased nearly threefold in mdx animals compared with C57 animals. Proportional talin cleavage was not changed by administration of LPTN. Proteasome activity was also measured in muscles. Activity increased in an animal type and LPTN-dependent manner, where C57 animals had the lowest activity and mdx LPTN had the highest activity (Fig. 6B).
Fig. 6.
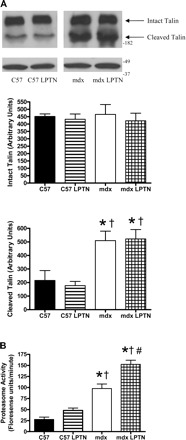
Protease activities following 6 mo of daily PBS or 36 mg/kg LPTN injections. A: representative Western blot showing intact and cleaved talin (top) and corresponding actin blot as a loading control (bottom). Intact talin is similar between groups; however, there is a dramatic increase in cleavage in mdx animals. B: proteasome activity was increased in both sets of mdx animals compared with both C57 groups. Furthermore, LPTN caused an additional increase in proteasome activity in mdx animals. Data are means ± SE. *Significantly different from C57 (P < 0.05); †significantly different from C57 LPTN (P < 0.05); #significantly different from mdx (P < 0.05).
Because calpain activity was maintained in the mdx LPTN group following the long-term addition of 36 mg·kg−1·day−1 of LPTN, we postulated that calpains must be activated at a higher rate in treated mdx animals compared with untreated mdx animals to keep calpain activity constant. Immunoblotting for m-calpain revealed that mdx animals treated with LPTN increased autolysed m-calpain, indicating an elevation in active calpain (Fig. 7A) (12, 19). Thus, it appears that calpain activation was increased in response to exogenous calpain inhibition, maintaining calpain activity. Additionally, zymograms demonstrated increased m-calpain expression in mdx animals compared with C57 animals, independent of LPTN treatment (Fig. 8). Autolysis and activation of μ-calpain did not change in response to drug treatment (Fig. 7B). Zymography, however, indicates a compensatory response to LPTN in both C57 and mdx animals suggesting that μ-calpains may also be involved in a compensatory response (Fig. 8). Calpastatin is an endogenous modulator of calpain activity, and so levels of this protein were measured as a possible source for the maintenance of calpain activity. There was an apparent difference in the expression of calpastatin between C57 animals and mdx animals, but calpastatin expression was similar between the mdx group and the mdx LPTN group (Supplemental Fig. 7).
Fig. 7.
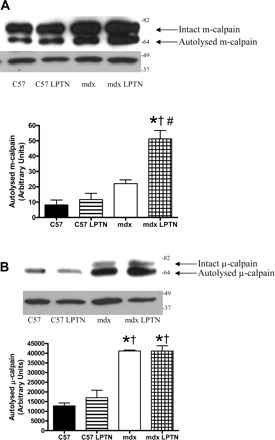
Representative Western blots and corresponding graphical representations of the optical densities from m-calpain (A; actin controls shown) from animals treated with LPTN or PBS for 6 mo. The μ-calpain activation remained unchanged by LPTN (B; shown with actin controls). Data are means ± SE. *Significantly different from C57 (P < 0.05); †significantly different from C57 LPTN (P < 0.05); #significantly different from mdx (P < 0.05).
Fig. 8.
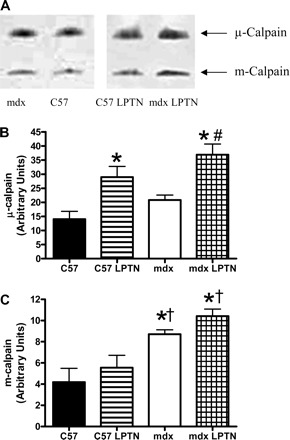
Representative casein zymograms (A) and corresponding graphical representations of optical densities from μ-calpain (B) and m-calpain (C) from animals treated with LPTN or PBS for 6 mo. Data are means ± SE. *Significantly different from C57 (P < 0.05); †significantly different from C57 LPTN (P < 0.05); #significantly different from mdx (P < 0.05).
DISCUSSION
This investigation tested the hypothesis that calpain inhibition ameliorates the pathology associated with dystrophin-deficiency in the mdx mouse. The calpain inhibitors, C101, an LPTN-based compound (LPTN bound to carnitine through a linker sequence), or LPTN itself were examined. Collectively, these compounds were tested in three independent investigations. In short-term study 1, where 8-wk-old animals were injected with C101 for 4 wk, C101 failed to improve muscle function or histology. This was also the case in short-term study 2, where 2-wk-old animals were injected with C101 for 4 wk. We tested LPTN itself for 6 mo (long-term study) where we showed that LPTN failed to improve muscle function and histology in the diaphragms of mdx mice. Furthermore, we demonstrate the basis for this failure to rescue includes heightened compensatory activation of calpain, as well as an increased proteasome activity. These results suggest that any pharmaceutical intervention targeting calpain directly is likely to fail to improve dystrophic pathology because of the endogenous regulation of this and other degradative pathways.
Our results are at odds with the previous work in which LPTN supplementation was demonstrated to ameliorate mdx pathology (3). In that investigation, LPTN was administered by intramuscular injection twice daily at 12 mg/kg and 18 mg/kg from 2 wk to 6 wk of age. These doses were sufficient to reduce calpain activity to control or near control levels. Furthermore, they returned muscle fiber CSA to control levels and greatly improved muscle histology. However, administration of C101 over the same time did not normalize fiber size nor reduce central nucleation even though the levels of exposure were sufficient to reduce calpain activity by 75%.
Two other groups have reported that drugs targeting calpain activity are of benefit to mdx animals. Following 4 wk of treatment (3–7 wks), Lescop et al. (23) demonstrated that the variability of the fiber area distribution was significantly reduced. Additionally, a report from Burdi et al. (9) showed that 4–6 wk (4–5 wks to 8–10 wks) of calpain inhibition improved grip strength and Cl− conductance, reduced TGF-β1, and lowered CK activity; however, it failed to improve histological measures in mdx mice.
Closer examination of the previous calpain inhibition/dystrophic skeletal muscle work reveals inconsistent outcomes. In the work by Badalamente and Stracher (3), calpain inhibition increased fiber areas in the gastrocnemius, soleus, tibialis anterior, and diaphragm. In a related measure, Lescop et al. (23) report a reduction in the variability of the cross-sectional area. Furthermore, Badalamente and Stracher (3) report histological improvements (TA), while no such claim is made by Burdi et al. (gastrocnemius) (9). In fact, the latter study provides additional detail and reveals that the percent necrotic area and percent fibers with centralized nuclei are similar in mdx animals and calpain inhibitor-treated animals (9). It appears that the conclusions of the Badalamente and Stracher study (3) could have been biased by an under-sampling of the muscle cross sections in their histological assessments.
The data from the short-term study 2 and the long-term study are in good agreement with that of Lescop et al. (23) in that the diaphragmatic cross-sectional area was not increased; however, our data did not indicate a reduction in the variability of fiber cross-sectional area. Furthermore, all three of our investigations (short-term study 1, short-term study 2, long-term study) were in good agreement with Burdi et al. (9) in that calpain inhibition failed to improve muscle histology. Muscle function, however, was not improved in our studies as it was in the report from Burdi et al. (9). This could be due to several factors. The first is that we measured muscle function ex vivo while Burdi et al. (9) used performance-based testing. It should be noted that the inhibitor used by Burdi et al. (9) also contained antioxidant activity, which could have provided the improved muscle function, while the calpain inhibitory activity did not. Recent data from our lab indicate that catalase overexpression increased muscle function in 4- and 6-wk-old mdx animals (J. T. Selsby, unpublished observations). Additionally, others have reached similar conclusions regarding the use of antioxidants in dystrophin-deficient muscle (8, 15, 16, 38).
To understand why LPTN failed to improve muscle function and histology in the mdx mouse, we assessed possible compensatory cellular responses to LPTN. Talin cleavage is a measure of in vivo calpain activity, while zymograms are an indicator of maximal potential calpain activity or content, although zymograms lack the sensitivity to detect small changes. Calpain autolysis is an approximate indicator of calpain activation. Activated calpain can be inhibited by LPTN. Initially we found that talin cleavage was similar between treated and untreated animals, indicating that despite treating animals for 6 mo with a calpain inhibitor, total in vivo calpain activity was unchanged. As all phenotypic data indicated that the drug intervention was not effective, this was not overly surprising. We reasoned that total calpain activity could be maintained despite an exogenous inhibitor, if calpain activation were increased.
We observed that long-term exposure to an environment with increased LPTN (i.e., calpain inhibition) in mdx animals caused a selective increase in the autolysis of m-calpain. This increase in autolysis is predictive of increased in vivo m-calpain activation and activity (12, 19). Because proportional talin cleavage is similar between treated and untreated mdx animals, the combination of increased exogenous calpain inhibitor combined with increased m-calpain autolysis and activation appears to result in the maintenance of calpain activity at a constant set point. The m-calpain zymograms indicate that expression of calpain is similar between LPTN-treated and control mdx mice. Despite similar expression of m-calpain, autolysis is higher in LPTN-treated mdx mice compared with control mdx mice, again, indicating the maintenance or defense of a calpain activity set point in dystrophin-deficient muscle.
The similarity in μ-calpain autolysis in LPTN-treated animals (both C57 and mdx) compared with control animals suggests that μ-calpain is not involved in the compensatory response. Conversely, increased maximal μ-calpain activity is increased (zymograms) in response to LPTN treatment in both C57 and mdx animals, allowing the possibility that μ-calpain may be involved in this adaptation. This indicates that even healthy cells may respond to long-term calpain inhibition by an increase in calpain content. This is further suggestive of a calpain activity set point, as calpain activity and autolysis were similar in both C57 groups despite an increase in μ-calpain content in the C57 LPTN group compared with the control C57 group.
Alternatively, cells could maintain calpain activity by decreasing expression of the endogenous inhibitor calpastatin. While there are obvious differences between dystrophin-deficient animals and C57 animals, LPTN administration did not cause a change in calpastatin expression or cleavage products. Previously, researchers have found similar banding patterns in skeletal muscle (17, 22, 26, 32). There also have been mixed reports of the efficacy of calpain inhibition using transgenic overexpression of calpastatin in the mdx mouse (5,21). As with the pharmacological studies, the reason for the discrepancy is unclear, since the age of the animals examined were similar in the two studies. It is possible that due to the use of different promoters, the study in which benefit was observed had higher levels of calpastatin expression, which may have overwhelmed the ability of the muscle to increase calpain activity.
Like calpain activity, proteasome activity was increased in mdx mice compared with C57 demonstrating increased proteolysis in dystrophic muscle compared with control. Interestingly, in vitro, maximal 26S proteasome activity was increased with LPTN administration in mdx mice indicating that the proteasome also undergoes a compensatory change in expression and/or function. However, given that calpain activity was similar between groups and calpain cleavage products become proteasome substrates, it is unclear why the proteasome is upregulated in this context. Speculatively, long-term LPTN administration either may directly or indirectly inhibit proteasome activity. Hence, in similar fashion as calpains, proteasomes may upregulate to maintain activity at some set point. Alternatively, long-term LPTN administration may have caused a compensatory increase in calpain activity too subtle to detect in this investigation resulting in increased production of cleavage product/proteasome substrate thus driving increased proteasome expression.
Aside from demonstrating that calpain inhibition is not an effective therapeutic strategy for dystrophic muscle, one positive aspect of the study was the validation of the carnitine delivery system. Covalent linkage of the active portion of LPTN to carnitine resulted in the rapid delivery of the drug to the muscle and caused no pathology in C57 animals. Thus, while delivery of calpain inhibitory activity by this approach was ineffective in dystrophic animals, the same strategy could be used to target other peptides or small molecules to muscle for therapeutic purposes.
Perspectives and Significance
C101, a LPTN-based drug and calpain inhibitor, failed to improve dystrophic pathology in mdx mice. Additionally, long-term administration of LPTN failed to reduce dystrophic pathology. We have determined that this failure is likely due to a compensatory mechanism, wherein increased activation of m-calpain, and potentially increased activation of μ-calpain, leads to maintenance of calpain activity in the face of the exogenous calpain inhibitor. While it is possible lower doses, shorter durations, or alternating drug dosing with periods of drug withdrawal (or some combination) could prove to be more effective, based on the evidence presented here we believe that it is unlikely that directly targeting calpain inhibition can provide therapeutic relief for Duchenne muscular dystrophy.
GRANTS
This work was supported by Wellstone Muscular Dystrophy Cooperative Research Center Grant U54-AR052646, National Institute of Arthritis and Musculoskeletal and Skin Diseases grants F32-AR-055005-01 and AR053461, and a Parent Project Muscular Dystrophy postdoctoral fellowship.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
Supplementary Material
ACKNOWLEDGMENTS
Present address of J. Selsby: Department of Animal Science, Iowa State University, Ames, IA 50011.
REFERENCES
- 1. Alderton JM, Steinhardt RA. Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J Biol Chem 275: 9452–9460, 2000. [DOI] [PubMed] [Google Scholar]
- 2. Angelini C. The role of corticosteroids in muscular dystrophy: a critical appraisal. Muscle Nerve 36: 424–435, 2007. [DOI] [PubMed] [Google Scholar]
- 3. Badalamente MA, Stracher A. Delay of muscle degeneration and necrosis in mdx mice by calpain inhibition. Muscle Nerve 23: 106–111, 2000. [DOI] [PubMed] [Google Scholar]
- 4. Barton ER, Morris L, Kawana M, Bish LT, Toursel T. Systemic administration of l-arginine benefits mdx skeletal muscle function. Muscle Nerve 32: 751–760, 2005. [DOI] [PubMed] [Google Scholar]
- 5. Brass LF, Shattil SJ. Inhibition of thrombin-induced platelet activation by leupeptin. Implications for the participation of calpain in the initiation of platelet activation. J Biol Chem 263: 5210–5216, 1988. [PubMed] [Google Scholar]
- 6. Briguet A, Erb M, Courdier-Fruh I, Barzaghi P, Santos G, Herzner H, Lescop C, Siendt H, Henneboehle M, Weyermann P, Magyar JP, Dubach-Powell J, Metz G, Meier T. Effect of calpain and proteasome inhibition on Ca2+-dependent proteolysis and muscle histopathology in the mdx mouse. FASEB J 22: 4190–4200, 2008. [DOI] [PubMed] [Google Scholar]
- 7. Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol 404: 71–82, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buetler TM, Renard M, Offord EA, Schneider H, Ruegg UT. Green tea extract decreases muscle necrosis in mdx mice and protects against reactive oxygen species. Am J Clin Nutr 75: 749–753, 2002. [DOI] [PubMed] [Google Scholar]
- 9. Burdi R, Didonna MP, Pignol B, Nico B, Mangieri D, Rolland JF, Camerino C, Zallone A, Ferro P, Andreetta F, Confalonieri P, De Luca A. First evaluation of the potential effectiveness in muscular dystrophy of a novel chimeric compound, BN 82270, acting as calpain-inhibitor and anti-oxidant. Neuromuscul Disord 16: 237–248, 2006. [DOI] [PubMed] [Google Scholar]
- 10. Chamberlain JS, Metzger J, Reyes M, Townsend D, Faulkner JA. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J 21: 2195–2204, 2007. [DOI] [PubMed] [Google Scholar]
- 11. Cohen L, Morgan J, Bozyk ME. Variable effects of corticosteroid treatment of serum enzyme activities in Duchenne's muscular dystrophy. Res Commun Chem Pathol Pharmacol 17: 529–538, 1977. [PubMed] [Google Scholar]
- 12. Cong J, Goll DE, Peterson AM, Kapprell HP. The role of autolysis in activity of the Ca2+-dependent proteinases (μ-calpain and m-calpain). J Biol Chem 264: 10096–10103, 1989. [PubMed] [Google Scholar]
- 13. Dayton WR, Goll DE, Zeece MG, Robson RM, Reville WJ. A Ca2+-activated protease possibly involved in myofibrillar protein turnover. Purification from porcine muscle. Biochemistry 15: 2150–2158, 1976. [DOI] [PubMed] [Google Scholar]
- 14. Deconinck N, Dan B. Pathophysiology of duchenne muscular dystrophy: current hypotheses. Pediatr Neurol 36: 1–7, 2007. [DOI] [PubMed] [Google Scholar]
- 15. Dorchies OM, Wagner S, Buetler TM, Ruegg UT. Protection of dystrophic muscle cells with polyphenols from green tea correlates with improved glutathione balance and increased expression of 67LR, a receptor for (−)-epigallocatechin gallate. Biofactors 35: 279–294, 2009. [DOI] [PubMed] [Google Scholar]
- 16. Dorchies OM, Wagner S, Vuadens O, Waldhauser K, Buetler TM, Kucera P, Ruegg UT. Green tea extract and its major polyphenol (−)-epigallocatechin gallate improve muscle function in a mouse model for Duchenne muscular dystrophy. Am J Physiol Cell Physiol 290: C616–C625, 2006. [DOI] [PubMed] [Google Scholar]
- 17. Doumit ME, Koohmaraie M. Immunoblot analysis of calpastatin degradation: evidence for cleavage by calpain in postmortem muscle. J Anim Sci 77: 1467–1473, 1999. [DOI] [PubMed] [Google Scholar]
- 18. Gailly P, De Backer F, Van Schoor M, Gillis JM. In situ measurements of calpain activity in isolated muscle fibres from normal and dystrophin-lacking, mdx mice. J Physiol 582: 1261–1275. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev 83: 731–801, 2003. [DOI] [PubMed] [Google Scholar]
- 20. Iwata Y, Katanosaka Y, Shijun Z, Kobayashi Y, Hanada H, Shigekawa M, Wakabayashi S. Protective effects of Ca2+ handling drugs against abnormal Ca2+ homeostasis and cell damage in myopathic skeletal muscle cells. Biochem Pharmacol 70: 740–751, 2005. [DOI] [PubMed] [Google Scholar]
- 21. Kaushal GP, Singh AB, Shah SV. Identification of gene family of caspases in rat kidney and altered expression in ischemia-reperfusion injury. Am J Physiol Renal Physiol 274: F587–F595, 1998. [DOI] [PubMed] [Google Scholar]
- 22. Kent MP, Spencer MJ, Koohmaraie M. Postmortem proteolysis is reduced in transgenic mice overexpressing calpastatin. J Anim Sci 82: 794–801, 2004. [DOI] [PubMed] [Google Scholar]
- 23. Lescop C, Herzner H, Siendt H, Bolliger R, Hennebohle M, Weyermann P, Briguet A, Courdier-Fruh I, Erb M, Foster M, Meier T, Magyar JP, vonSprecher A. Novel cell-penetrating α-keto-amide calpain inhibitors as potential treatment for muscular dystrophy. Bioorg Med Chem Lett 15: 5176–5181, 2005. [DOI] [PubMed] [Google Scholar]
- 24. Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev 1: CD003725, 2008. [DOI] [PubMed] [Google Scholar]
- 25. Murphy RM, Verburg E, Lamb GD. Ca2+ activation of diffusible and bound pools of μ-calpain in rat skeletal muscle. J Physiol 576: 595–612, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parr T, Jewell KK, Sensky PL, Brameld JM, Bardsley RG, Buttery PJ. Expression of calpastatin isoforms in muscle and functionality of multiple calpastatin promoters. Arch Biochem Biophys 427: 8–15, 2004. [DOI] [PubMed] [Google Scholar]
- 27. Raser KJ, Posner A, Wang KK. Casein zymography: a method to study μ-calpain, m-calpain, and their inhibitory agents. Arch Biochem Biophys 319: 211–216, 1995. [DOI] [PubMed] [Google Scholar]
- 28. Ruff RL, Secrist D. Inhibitors of prostaglandin synthesis or cathepsin B prevent muscle wasting due to sepsis in the rat. J Clin Invest 73: 1483–1486, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Selsby JT, Dodd SL. Heat treatment reduces oxidative stress and protects muscle mass during immobilization. Am J Physiol Regul Integr Comp Physiol 289: R134–R139, 2005. [DOI] [PubMed] [Google Scholar]
- 30. Smith IJ, Dodd SL. Calpain activation causes a proteasome-dependent increase in protein degradation and inhibits the Akt signalling pathway in rat diaphragm muscle. Exp Physiol 92: 561–573, 2007. [DOI] [PubMed] [Google Scholar]
- 31. Spencer MJ, Croall DE, Tidball JG. Calpains are activated in necrotic fibers from mdx dystrophic mice. J Biol Chem 270: 10909–10914, 1995. [DOI] [PubMed] [Google Scholar]
- 32. Spencer MJ, Mellgren RL. Overexpression of a calpastatin transgene in mdx muscle reduces dystrophic pathology. Hum Mol Genet 11: 2645–2655, 2002. [DOI] [PubMed] [Google Scholar]
- 33. Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, Narusawa M, Leferovich JM, Sladky JT, Kelly AM. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352: 536–539, 1991. [DOI] [PubMed] [Google Scholar]
- 34. Straub V, Rafael JA, Chamberlain JS, Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol 139: 375–385, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Van debrouck C, Martin D, Colson-Van Schoor M, Debaix H, Gailly P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J Cell Biol 158: 1089–1096, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vigouroux S, Furukawa Y, Farout L, SJK, Briand M, Briand Y. Peptidase activities of the 20/26S proteasome and a novel protease in human brain. J Neurochem 84: 392–396, 2003. [DOI] [PubMed] [Google Scholar]
- 37. Wang X, Weisleder N, Collet C, Zhou J, Chu Y, Hirata Y, Zhao X, Pan Z, Brotto M, Cheng H, Ma J. Uncontrolled calcium sparks act as a dystrophic signal for mammalian skeletal muscle. Nat Cell Biol 7: 525–530, 2005. [DOI] [PubMed] [Google Scholar]
- 38. Whitehead NP, Pham C, Gervasio OL, Allen DG. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J Physiol 586: 2003–2014, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.