

Abstract
During endurance exercise, most (≈75%) of the energy derived from the oxidation of metabolic fuels and ATP hydrolysis of muscle contraction is liberated as heat, the accumulation of which leads to an increase in body temperature. For example, the temperature of exercising muscles can rise to 40°C. Although severe heat injury can be deleterious, several beneficial effects of mild heat stress (HS), such as the improvement of insulin sensitivity in patients with type 2 diabetes, have been reported. However, among all cellular events induced by mild HS from physical activities, the direct effects and mechanisms of mild HS on mitochondrial biogenesis in skeletal muscle are least characterized. AMP-activated protein kinase (AMPK) and sirtuin 1 (SIRT1) are key energy-sensing molecules regulating mitochondrial biogenesis. In C2C12 myotubes, we found that 1 h mild HS at 40°C upregulated both AMPK activity and SIRT1 expression, as well as increased the expression of several mitochondrial biogenesis regulatory genes including peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) and transcription factors involved in mitochondrial biogenesis. In particular, PGC-1α expression was found to be transcriptionally regulated by mild HS. Additionally, after repeated mild HS for 5 days, protein levels of PGC-1α and several mitochondrial oxidative phosphorylation subunits were also upregulated. Repeated mild HS also significantly increased mitochondrial DNA copy number. In conclusion, these data show that mild HS is sufficient to induce mitochondrial biogenesis in C2C12 myotubes. Temperature-induced mitochondrial biogenesis correlates with activation of the AMPK-SIRT1-PGC-1α pathway. Therefore, it is possible that muscle heat production during exercise plays a role in mitochondrial biogenesis.
Keywords: sirtuin, SIRT1, AMPK, PGC-1α, temperature, heat shock proteins, exertion, exercise, mitochondria, mitochondrial reticulum
temperature directly or indirectly affects essentially every aspect of an organism's physiology. It is well known that most species perform best in a fairly narrow temperature range, which is also associated with optimal enzyme functions (40). Mammals produce heat by thermogenesis that involves mechanisms such as shivering and uncoupling protein-induced mitochondrial proton leak (22, 42). Excess heat in mammals is dissipated by evaporative and nonevaporative means (4, 10).
Endurance exercise imposes a combination of stresses such as changes in calcium concentration, mechanical strain, alterations in the ADP-to-ATP ratio, hypoxic stress, and loosening of mitochondrial respiratory control on skeletal muscle (5, 33). Numerous studies have focused on how these stresses contribute to exercise-induced adaptations. Increased heat production is also a byproduct of endurance exercise and can also be counted among the stresses imposed on skeletal muscle by physical exercise. However, the direct effects of mild heat stress (HS)—heating within physiological limits—on mitochondrial biogenesis have not been well characterized in past studies.
Muscle tissue is capable of directly transforming the chemical energy derived from nourishment into mechanical work. Measured by the ratio of work performed to energy cost as calculated from oxygen uptake, the energy conversion efficiency of skeletal muscle is only 20–30% (14, 54). This is because the efficiency of ATP generation is ∼60%, and the efficiency of ATP use is ∼50% (14, 54). During vigorous exercise, the energy cost can increase by 10- to 20-fold compared with the resting metabolic rate (35), and the temperature of exercising muscles can rise to values as high as 40°C, which can negatively affect mitochondrial respiratory control and coupling efficiency (6, 48, 49). The heat generated during endurance exercise must be effectively transported to the skin and dissipated to the environment to prevent heat injury. Although heat stroke is currently the third leading cause of death in athletes in the United States (25), several beneficial effects of mild HS have been reported among several different species. For example, hot tub therapy can improve insulin sensitivity and lower blood glucose levels in patients with type 2 diabetes (24). Both heat treatment at 41 to 41.5°C and overexpression of heat shock protein 72 (HSP72) have been shown to prevent high-fat diet-induced insulin resistance in rodents (11, 17). Finally, mild HS was also observed to extend life span in yeast, worms, and flies (30, 37, 51).
Mild HS raises cellular ATP flux that raises AMP level, which, in turn, activates AMPK (12). AMPK is a key energy sensing molecule that maintains whole body energy homeostasis in response to reduced cellular energy status (19). Activated AMPK switches off biosynthetic pathways to reserve ATP for more essential processes and switches on catabolic pathways to boost ATP production by upregulating oxidative metabolism and mitochondrial biogenesis (19). Recently, an emerging body of evidence suggests that another energy-sensing molecule, SIRT1, works closely with AMPK to control cellular energy expenditure (47). SIRT1, a mammalian class III protein deacetylase, is an important regulator of metabolism, cancer, and life span (34). AMPK and SIRT1 act in cooperation with the master regulator of mitochondrial biogenesis, PGC-1α, to regulate energy homeostasis in response to environmental stimuli and nutritional signals (8, 9, 55). AMPK and SIRT1 directly activate PGC-1α through phosphorylation and deacetylation, respectively (27, 46). AMPK and SIRT1 also increase the expression of mRNAs encoding PGC-1α (2, 27).
Mitochondrial biogenesis entails mitochondrial DNA replication and transcription, as well as the synthesis and import of proteins into the mitochondrial reticulum (29, 31). Because the oxidative phosphorylation system is located in the mitochondrial inner membrane (50), the physiological significance of mitochondrial biogenesis lies in the enhancement of oxidative phosphorylation, which ultimately results in a net ATP gain of more than 15 times the amount of ATP produced by glycolysis (1).
We propose that the beneficial effects of mild HS, such as improved insulin sensitivity, may be associated with mitochondrial biogenesis. We hypothesize that the reduced level of cellular energy induced by mild HS can activate AMPK and SIRT1, both of which further upregulate PGC-1α and the downstream mitochondrial biogenesis program. To test this hypothesis, we used C2C12 myotubes as a model system to study the effect of acute and repeated mild HS on mitochondrial biogenesis by measuring activation of the AMPK-SIRT1-PGC-1α pathway, as well as the levels of transcription factors involved in mitochondrial biogenesis and mitochondrial components such as mitochondrial DNA copy number.
MATERIALS AND METHODS
Cell culture.
Mouse C2C12 myoblast cell line was maintained in DMEM (4.5 g glucose/l) in the presence of 10% fetal bovine serum. Differentiation of C2C12 myoblasts into myotubes was induced with media containing 2% horse serum for 5 days. The cells were grown in a humidified 37°C incubator with 5% CO2 in air.
Mild heat stress exposure.
For mild HS, the culture temperature was set at 40°C. The duration of mild HS was maintained for 1 h for each bout. The frequency was either a single bout on the 5th day of C2C12 myotube differentiation or one bout per day for 5 days since the start of differentiation. After final treatment cells were harvested for subsequent assays.
Total protein extraction.
C2C12 myotubes were washed with 1× PBS, scraped, centrifuged, and resuspended in SDS lysis buffer (50 mM Tris·HCl, pH 6.8, 1% SDS, and 10% glycerol). After brief sonication to break up genomic DNA, an aliquot of the cell lysates was reserved for subsequent protein concentration determinations (BCA assay, Thermo Fisher), and the remainder was supplemented with β-mercaptoethanol.
Western blotting.
Lysates of C2C12 myotubes were electrophoresed, transferred to Hybond-C Extra nitrocellulose membranes (Amersham Biosciences), and probed with antibodies against HSP72 (Enzo), AMPK (Sigma), phosphorylated AMPK (Cell Signaling), SIRT1 (Abcam), Nampt (Bethyl), and PGC-1α (Millipore), as well as with OXPHOS Rodent WB Antibody Cocktail (MitoSciences), which recognizes Complex I subunit NDUFB8, Complex II subunit 30 kDa, Complex III subunit Core 2, Complex IV subunit I, and ATP synthase subunit-α. For normalization purposes, blots were also probed with an antibody to Histone H3 (Upstate). Blots were then developed using Amersham ECL Western Blotting Detection (GE Healthcare). The band intensity was quantified with Photoshop and ImageJ software.
RNA isolation and cDNA synthesis.
Total RNA was isolated from C2C12 myotubes using RiboPure kits (Ambion) according to the manufacturer's instructions. Isolated RNA was reverse transcribed to cDNA using random hexamer primers and SuperScript II reverse transcriptase kit (Invitrogen) according to the manufacturer's instructions.
Real-time quantitative PCR.
Real-time quantitative PCR (qPCR) was performed using 2× HotSybr Real-Time PCR Kit (Mclab) and iQ5 Multicolor Real-Time PCR Detection System (BioRad). Sequences of specific primers are listed in Table 1. The data from qPCR were analyzed with delta-delta threshold cycle (ΔΔCt) method using 18S ribosomal RNA (Rn18s) as the internal control gene. Each ΔCt value was determined by subtracting 18S ribosomal RNA Ct value from the target gene Ct value. The ΔΔCt was calculated by subtracting the ΔCt value of the 37°C control from the ΔCt value of the heat-stressed sample. 2−ΔΔCt represented the average relative amount of mRNA for each target gene.
Table 1.
Primer sequences used for real-time quantitative PCR
| Primers | Strand | Sequence 5′-> 3′ |
|---|---|---|
| Hspa1a | Forward | AGATATGTGGCCTTGAGGACTGTCATTATTTC |
| Reverse | CAAATCACATCAGCGGGGCAGTGCTGAATTG | |
| Hspa1b | Forward | TGCTTGGGCACCGATTACTGTCAAGG |
| Reverse | GGCAGCTAGACTATATGTCTTCCCAGGCTACTG | |
| Sirt1 | Forward | GACGATGACAGAACGTCACAC |
| Reverse | CGAGGATCGGTGCCAATCA | |
| PGC-1α | Forward | AAGTGTGGAACTCTCTGGAACTG |
| Reverse | GGGTTATCTTGGTTGGCTTTATG | |
| PGC-1β | Forward | GGCAGGTTCAACCCCGA |
| Reverse | CTTGCTAACATCACAGAGGATATCTTG | |
| PRC | Forward | CCAAAAAGGATGCCTGCCCTA |
| Reverse | GTAGCCGTGCATGGGAGTG | |
| Nrf1 | Forward | TCT CAC CCT CCA AAC CCA AC |
| Reverse | CCC GAC CTG TGG AAT ACT TG | |
| Nrf2α | Forward | CTCCCGCTACACCGACTAC |
| Reverse | TCTGACCATTGTTTCCTGTTCTG | |
| Tfam | Forward | CAT TTA TGT ATC TGA AAG CTTCC |
| Reverse | CTC TTC CCA AGA CTT CAT TTC | |
| Tfb1m | Forward | AAGATGGCCCTTTCGTTTATGG |
| Reverse | GACTGTGCTGTTTGCTTCCTG | |
| Tfb2m | Forward | CCAAAACCCATCCCGTCAAAT |
| Reverse | AAGGGCTCCAAATGTGGAATAAA | |
| Cycs | Forward | GCAAGCATAAGACTGGACCAAA |
| Reverse | TTGTTGGCATCTGTGTAAGAGAATC | |
| Cox2 | Forward | TGAAGACGTCCTCCACTCATGA |
| Reverse | GCCTGGGATGGCATCAGTT | |
| Cox4 | Forward | ACCAAGCGAATGCTGGACAT |
| Reverse | GGCGGAGAAGCCCTGAA | |
| Glut4 | Forward | TGTGGCCTTCTTTGAGATTGG |
| Reverse | CCCATGCCGACAATGAAGTT | |
| Ldha | Forward | TGCCTACGAGGTGATCAAGCT |
| Reverse | ATGCACCCGCCTAAGGTTCTT | |
| Rn18 s | Forward | CGCCGCTAGAGGTGAAATTCT |
| Reverse | CGAACCTCCGACTTTCGTTCT |
Reporter gene assays.
C2C12 myocytes were transfected with 1 μg of PGL3-PGC-1α 2-kb promoter-firefly luciferase reporter vector (Addgene) (18) and 0.2 μg of CMV Renilla luciferase vector as the control vector in 6-well plates at 90% confluence using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. Cells were left for 5 h with the DNA-Lipofectamine mix. Then the medium was removed and replaced by differentiation medium for 36 h before being treated with mild HS for 1 h. After a 24-h recovery period, the cultures were resuspended into 250 μl of passive lysis buffer from the Dual Luciferase Reporter Assay system (Promega) and 20 μl of the cell lysates were used for determining Renilla and firefly luciferase activities with the Promega kit mentioned above.
Mitochondrial DNA quantification.
Total cellular DNA in C2C12 myotubes was extracted using Blood & Cell Culture DNA Mini Kit (Qiagen) according to the manufacturer's instructions. DNA pellets were resuspended in TE (1 mM EDTA, 10 mM Tris, pH 8.0) prior to qPCR analysis. Real-time qPCR was performed for mitochondrial DNA-encoded cytochrome c oxidase subunit II (Cox2) (primers: forward 5′-GCCGACTAAATCAAGCAACA-3′, reverse 5′-CAATGGGCATAAAGCTATGG-3′) (57) and nucleus-encoded 18S ribosomal RNA genes (primers: forward 5′-TAGAGGGACAAGTGGCGTTC-3′, reverse 5′-CGCTGAGCCAGTCAGTGT-3′) (3). The ratio of Cox2 DNA copies to 18S rRNA represents the relative mitochondrial copy number.
Statistical analysis.
Statistical significance of temperature-induced differences was evaluated by Student's t-test assuming equal variance, and a value of P < 0.05 was considered significant. Results are presented as the mean ± SE of the mean. Statistical analyses were performed using Excel 2007 software (Microsoft).
RESULTS
Mild HS induces cytoprotective heat shock response.
To investigate the direct cellular effects of mild HS on muscle cells, we incubated the C2C12 myotubes at 40°C for 1 h. HSPa1a and HSPa1b are the major stress-inducible genes of the heat shock protein 70-kDa family. They encode proteins with identical amino acid sequences known as HSP72 or HSP70. Although no significant change in HSP72 protein levels was observed immediately after mild HS (Fig. 1A), mRNA levels of Hspa1a and Hspa1b increased by more than 100-fold when measured 1 h after mild HS (data not shown). Two hours and 24 h after mild HS, the mRNA levels were still significantly higher for Hspa1a (3.41-fold, P < 0.05 and 1.82-fold, P < 0.05; Fig. 1C) and for Hspa1b (4.98-fold, P < 0.05 and 1.67-fold, P < 0.05; Fig. 1D). Furthermore, the increase in HSP72 protein levels by 2.55-fold (P < 0.05) was observed 24 h after mild HS (Fig. 1B). Our data demonstrate that mild HS within physiological range is sufficient to induce a cytoprotective heat shock response.
Fig. 1.
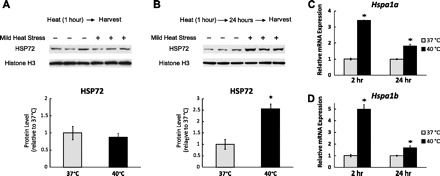
Mild heat stress (HS) activates heat shock response. A: C2C12 myotubes were exposed to 1-h mild HS (40°C) and harvested immediately for Western blotting to determine the heat shock protein (HSP) 72 protein levels. B: C2C12 myotubes were exposed to 1-h mild HS and then returned to the normal culture environment (37°C). After 24 h, cells were harvested for Western blotting to measure the HSP72 protein levels. Results were normalized to Histone H3. Quantification of band intensity is shown below the Western blot. Results are given as the means ± SE from three independent trials. *P < 0.05 compared with the control. C and D: C2C12 myotubes were harvested 2 and 24 h after the above-mentioned mild HS. The mRNA levels of hspa1a and Hspa1b were determined by qPCR and normalized to 18S ribosomal RNA. Results are expressed as means ± SE of triplicate samples in 1 representative experiment from 3 independent trials. *P < 0.05 compared with control.
Mild HS upregulates AMPK activity and SIRT1 expression.
AMPK is a key energy-sensing molecule and is activated when cells undergo energy-depleting stresses. The degree of AMPK T172 phosphorylation (p-AMPK) was used as a measure of AMPK activation (19). It is evident from Fig. 2A that, immediately after mild HS, a significant increase in AMPK T172 phosphorylation (1.6-fold; P < 0.05) was observed while the total AMPK (t-AMPK) protein levels remained unchanged. These results indicate that mild HS rapidly enhances AMPK activity.
Fig. 2.
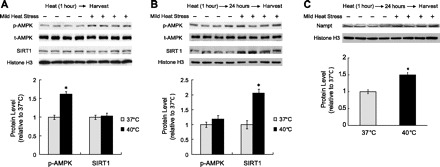
Mild HS upregulates AMPK activity and SIRT1 expression. A: C2C12 myotubes were exposed to 1-h mild HS (40°C) and harvested immediately for Western blotting to determine levels of total and phosphorylated AMPK (t-AMPK and p-AMPK) as well as SIRT1. B and C: C2C12 myotubes were exposed to 1-h mild HS and then returned to the normal culture environment (37°C). After a 24-h recovery period, cells were harvested for Western blotting to measure the expression of abovementioned proteins and Nampt, the rate-limiting enzyme in the NAD+ biosynthesis pathway. Results were normalized to Histone H3. Quantification of band intensity is shown below the Western blot. Results are given as the means ± SE from 4 independent trials.*P < 0.05 compared with the control.
AMPK is known to regulate SIRT1 expression (56). Therefore, we measured SIRT1 expression after mild HS. Our data showed that the SIRT1 protein levels remained unchanged immediately after mild HS (Fig. 2A). However, 24 h after mild HS, the SIRT1 protein levels were upregulated by 2.04-fold (P < 0.05; Fig. 2B). We also found that the Sirt1 mRNA level was upregulated 24 h after mild HS (1.51-fold, P < 0.05; Fig. 3A).
Fig. 3.

Mild HS increases PGC-1α transcription. A: C2C12 myotubes were exposed to mild HS for 1 h and then returned to the normal culture environment. The mRNA levels of Sirt1, PGC-1α, PGC-1β, and PRC were determined by qPCR and normalized to 18S ribosomal RNA. Results are expressed as means ± SE of triplicate samples in one representative experiment from 3 independent trials. *P < 0.05 compared with control. B: C2C12 myoblasts were transfected with PGL3-PGC-1α 2-kb promoter-firefly luciferase reporter and a CMV Renilla luciferase plasmid. After differentiation, the cells were treated with 1-h mild HS and then returned to the normal culture environment. After a 24-h recovery period, cells were harvested for luciferase assay. The data show the ratio of firefly to Renilla luciferase luminescence, which represents the normalized promoter activity of PGC-1α. Results are given as the means ± SE from 5 independent trials. *P < 0.05 compared with the control.
Nicotinamide adenine dinucleotide (NAD+) is required in several important biological functions. In particular, NAD+ is an obligate cofactor for the deacetylase activity of SIRT1 (26). Nicotinamide phosphoribosyltransferase (Nampt) is responsible for the rate-limiting step in the NAD+-biosynthetic pathway in mammals (45). Therefore, we analyzed Nampt expression after mild HS. Our data showed that 24 h after mild HS, Nampt protein levels were significantly upregulated (1.50-fold, P < 0.05; Fig. 2C). In summary, a single bout of mild heat stress results in a rapid upregulation of AMPK activity followed by an increase of Nampt and SIRT1 expression.
Mild heat stress increases PGC-1α transcription.
PGC-1α is a well-studied transcriptional target of both SIRT1 and AMPK (2, 27). In our studies of its expression level after mild HS, no significant change in the mRNA level of PGC-1α was observed 2 h after mild HS while an increase of 1.36-fold (P < 0.05) was seen 24 h later (Fig. 3A). In contrast, the mRNA levels of PGC-1β and PRC, the other two members of PGC-1 family transcription coactivators, remained constant at both 2 h and 24 h after mild HS (Fig. 3A).
To examine whether mild HS upregulates PGC-1α expression at the transcription level, the luciferase assay was used to monitor PGC-1α promoter activity. As shown in Fig. 3B, luciferase activity increased by 3.46-fold (P < 0.05) 24 h after mild HS. In summary, our data indicate that mild HS is sufficient to increase PGC-1α promoter activity and modulate PGC-1α transcription in C2C12 myotubes.
Mild heat stress activates mitochondrial biogenesis program.
To determine whether mitochondrial biogenesis program is upregulated in response to mild heat stress, a series of real-time qPCR assays was conducted to compare the expression of downstream genes of AMPK-SIRT1-PGC-1α pathway. We first analyzed the expression of nuclear respiratory factors 1 and 2 (NRF1 and NRF2), the transcription factors necessary for the coordination of nuclear-mitochondrial gene expression required for mitochondrial biogenesis. The effects of PGC-1α on mitochondrial biogenesis are mediated through its ability to both upregulate expression of NRF1 and NRF2 as well as coactivate both nuclear respiratory factors (55). Similar to PGC-1α, the mRNA levels of both Nrf1 and Nrf2α remained constant 2 h after mild HS while a significant increase was observed after 24 h (1.35-fold, P < 0.05 and 1.46-fold, P < 0.05, respectively; Fig. 4, A and B).
Fig. 4.
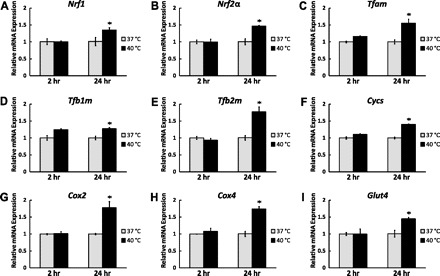
Mild HS activates mitochondrial biogenesis program. C2C12 myotubes were exposed to mild HS for 1 h and then returned to the normal culture environment. Cells were harvested after 2- and 24-h recovery periods. The mRNA levels of nuclear regulatory factors Nrf1 (A), Nrf2α (B), mitochondrial transcription factors Tfam (C), Tfb1m (D), Tfb2m (E), mitochondrial components Cycs (F), Cox2 (G), Cox4 (H), and glucose transporter Glut4 (I) were determined by qPCR and normalized to 18S ribosomal RNA. Results are expressed as means ± SE of triplicate samples in 1 representative experiment from 3 independent trials. *P < 0.05 compared with the control.
Mitochondrial DNA replication and transcription are coupled events executed by unique enzyme systems. This process is regulated by mitochondrial transcription factors, including TFAM, TFB1M, and TFB2M (13). After nuclear translocation and coactivation by PGC-1α, NRF1 and NRF2 bind and activate the promoters of Tfam, Tfb1m, and Tfb2m (15). Because the mRNA levels of both Nrf1 and Nrf2α were upregulated by mild HS, we subsequently measured the mRNA levels of Tfam, Tfb1m, and Tfb2m. Two hours after mild HS, the mRNA levels of these three mitochondrial transcription factors were not altered. However, 24 h after treatment, a significant increase was observed in the levels of all three transcription factors (1.55-fold, P < 0.05, 1.27-fold, P < 0.05 and 1.77-fold, P < 0.05, respectively; Figs. 4, C–E). In summary, these data demonstrate that the mitochondrial biogenesis program is upregulated by mild heat stress.
Mild heat stress induces gene expression for mitochondrial components.
Because activation of the mitochondrial biogenesis transcriptional cascade should lead to increased expression of mitochondrial components, we analyzed the mRNA levels of mitochondrial oxidative proteins including cytochrome c (Cycs), nuclear-encoded cytochrome c oxidase subunit IV (Cox4), and mitochondrial DNA-encoded respiratory subunit Cox2. No significant changes in the mRNA levels of Cycs, Cox2, and Cox4 were observed 2 h after mild HS while significant increases were seen in all of them 24 h after treatment (1.40-fold, P < 0.05, 1.77-fold, P < 0.05 and 1.74-fold, P < 0.05, respectively; Fig. 4, F–H). In summary, not only the regulatory genes, but also genes of mitochondrial components were upregulated after mild HS.
In addition to the genes involved in mitochondrial biogenesis, we also measured the transcription of glucose transporter Glut4, another target regulated by PGC-1α. No significant change in the mRNA level of Glut4 was observed 2 h after mild HS while an increase of 1.45-fold (P < 0.05) was seen after 24 h (Fig. 4I). At the same time, there was no difference observed in mRNA levels of lactate dehydrogenase isoform A (Ldha) after mild HS after 2 and 24 h (data not shown).
Repeated mild HS upregulates the expression of PGC-1α, oxidative phosphorylation subunits, and mitochondrial DNA copy number.
Our initial studies demonstrated that a single bout of mild HS is capable of activating the expression of a diverse group of genes. To further characterize the adaptive response to mild HS in myotubes, repeated mild HS was used to mimic the repeated HS that occurs in muscle as the result of regular exercise.
C2C12 myotubes were exposed to 1 h of mild HS per day for 5 days and then harvested for subsequent assays. There was no significant difference in SIRT1 protein expression observed between control and HS groups (data not show). However, a significant increase in PGC-1α protein expression was seen (1.58-fold, P < 0.05; Fig. 5). We were also interested in the effect of repeated mild HS on mitochondrial protein levels. As expected, repeated mild HS upregulated the protein levels of four representative oxidative phosphorylation subunits including Complex I subunit NDUFB8 (1.94-fold, P < 0.05), Complex II subunit 30 kDa (1.76-fold, P < 0.05), Complex III subunit Core 2 (1.78-fold, P <0.05), and Complex V, or ATP synthase subunit-α (1.61-fold P < 0.05; Fig. 5).
Fig. 5.
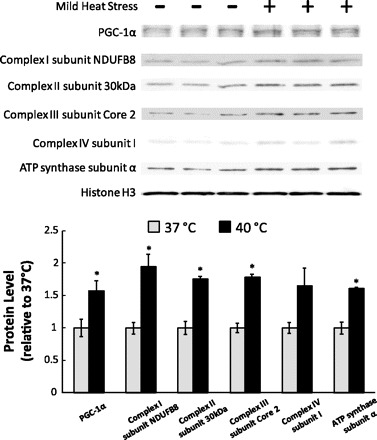
Repeated mild HS upregulates the expression of PGC-1α and oxidative phosphorylation subunits. C2C12 myotubes were exposed to 1-h HS for 5 days. 24 h after the last treatment, cells were harvested and subjected to Western blot analysis. Protein levels were normalized to Histone H3. Quantification of band intensity is shown below the Western blot. Results are given as the means ± SE from 3 independent trials. *P < 0.05 compared with control.
Because mitochondria contain their own DNA encoding 13 proteins, measurements of mitochondria DNA copy number was used to verify changes in mitochondrial biogenesis levels (38). Real-time qPCR analysis showed that repeated mild HS for 5 days upregulated the mitochondrial DNA copy number by 1.68-fold (P < 0.05; Fig. 6). Taken together, repeated mild HS for 5 days resulted in a significant increase in PGC-1α protein expression as well as in mitochondrial components including oxidative phosphorylation proteins and mitochondrial DNA.
Fig. 6.
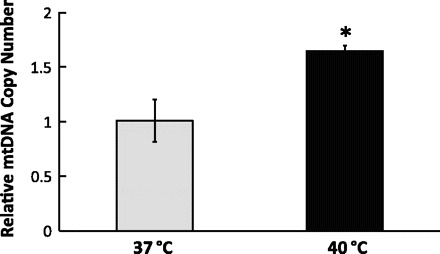
Repeated mild HS increased mitochondrial DNA (mtDNA) copy number. C2C12 myotubes were exposed to 1-h mild HS for 5 days. 24 h after the last treatment, cells were harvested for total DNA extraction. Using qPCR, the relative mitochondrial DNA copy number was determined by the ratio of mitochondrial DNA-encoded Cox2 and nucleus-encoded 18S ribosomal RNA genes. Results are expressed as means ± SE of triplicate samples in 1 representative experiment from 6 independent trials. *P < 0.05 compared with the control.
DISCUSSION
This study provides evidence in support of the hypothesis that mild HS is sufficient to induce mitochondrial biogenesis in C2C12 myotubes. Two types of evidence were obtained. First, key molecules of the mitochondrial biogenesis pathway, including energy-sensing AMPK and SIRT1 as well as their downstream targets such as PGC-1α, are activated by mild HS (Figs. 2 and 3). Second, levels of mitochondrial components, including several oxidative phosphorylation proteins and mitochondrial DNA, increase after mild HS (Figs. 4–6).
One of the most prominent mechanisms to rapidly cope with stresses is the heat shock response (21, 36). The heat shock response is almost ubiquitous across all life forms and is induced at temperatures a few degrees above the normal physiological range for most species (42). To cope with stresses, cells need to synthesize several critical heat shock proteins that can be used to restore structures of reversibly damaged proteins while degrading irreversibly damaged proteins as well as synthesizing new proteins (21, 36). All of these events are highly ATP costly processes and force cells into a reduced energy status. Investigators have shown that nondamaging running exercise for 45 min increases muscle temperature from 36.2 to 40°C (41). The authors also showed that the protein level of HSP72 significantly increased 48 h postexercise in muscle. Exercise-induced expression of HSPs in the heart is also one of the mechanisms responsible for the cardioprotective effects of exercise (16, 20). Our results showed that mild HS at 40°C increases immediately the mRNA levels and subsequently the protein levels of HSPa1α and HSPa1β—the major stress-inducible members of the heat shock protein 70-kDa family (Fig. 1). In summary, temperature increase within the physiological range is sufficient to induce the cytoprotective heat shock response.
SIRT1 is well-known for its role in modulating the life span of a number of organisms including yeast, worms, flies, and mice with restricted caloric intake (34). Similarly, mild HS has also long been reported to extend life span in different species (30, 37, 51), although the mechanism is not yet clear. Recently, Westerheide et al. (53) showed that SIRT1 directly deacetylates heat shock factor 1 and thereby regulates the acute heat shock response in mammalian cells. That result may mean that SIRT1 plays a role in heat-induced life span extension. Our results show that SIRT1 protein level significantly increased 24 h after mild HS. To our knowledge, this is the first report showing the direct upregulation of SIRT1 protein expression by heat. Our results may mean that SIRT1 not only functions in the early stages of the heat shock response, but SIRT1 may also provide important long-term functions after HS is removed. Accordingly, upregulation of SIRT1 may play a crucial role in two renowned environmental stressors that extend life span in animals: calorie restriction and mild heat stress.
Based on the evidence of the increase in PGC-1α promoter activity and mRNA level our data show that PGC-1α is transcriptionally regulated by mild HS. Interestingly, PGC-1α levels have been found to be greatly induced in muscle and brown fat by cold exposure (44). The common effect of cold exposure and excess heat on cells is an increase in cellular energy requirement. Based on previous findings and those of the current study, there is clear evidence that PGC-1α plays a crucial role in response to temperature fluctuations in cells, and PGC-1α is widely considered to be the master regulator of mitochondrial biogenesis. As well, it has been shown that another member of the sirtuin family of protein deacetylases—SIRT3 is a downstream target of PGC-1α (32). SIRT3 is localized in mitochondria and regulates mitochondrial respiration capacity, oxidative stress, and insulin resistance in skeletal muscle (28). It will be interesting to test whether SIRT3 expression level and activity are also upregulated by mild HS in the future.
Glucose uptake is a rate-limiting step in muscle glucose metabolism (52), and both gain-of-function and loss-of-function mutations have shown that GLUT4 is essential in the maintenance of normal glucose homeostasis (7, 58). As well, in skeletal muscle, Glut4 is one of the targets regulated by PGC-1α (39). In our studies, Glut4 transcription was significantly upregulated by mild HS, possibly due to an increase in PGG-1α expression. Thus results of our study may reveal an additional factor that, among others (11, 17), contributes to the heat-induced improvement of glucose tolerance and insulin sensitivity.
Multiple lines of evidence have shown that endurance exercise training results in an increase in skeletal muscle mitochondrial biogenesis (23). The physiological significance of mitochondrial biogenesis is the enhancement of cellular oxidative phosphorylation, which improves endurance performance at the organismal level. We found that the levels of several oxidative phosphorylation proteins and mitochondrial DNA copy number significantly increased after repeated mild HS. Because we show that in C2C12 myotubes mild HS increases markers of mitochondrial biogenesis similar to what occurs in regular endurance training in vivo, it is possible that muscle heat production during exercise plays a role in mitochondrial biogenesis.
Mitochondrial biogenesis is a vital and exciting area of cell biology. Lack of normal mitochondrial function has a negative impact in insulin resistance and aging, among others (43). In this report, we show that mitochondrial biogenesis is upregulated in response to mild HS in C2C12 myotubes. The activation of AMPK-SIRT1-PGC-1α pathway and its downstream targets is correlated with the temperature-induced changes in mitochondrial biogenesis. One limitation of our report is that C2C12 myotube culture system may not entirely reflect what occurs in vivo. Our findings need to be confirmed in animal models to see whether the temperature-induced effects are reproducible in vivo. Future studies are also needed to test the hypothesis of a causal link between AMPK-SIRT1-PGC1α activation and temperature-induced mitochondrial biogenesis in muscle. If so, then, exercise-induced changes in muscle temperature may have the effect of raising the background levels of metabolic gene expression, upon which exercise-induced changes in metabolites, ions, and other known affectors of mitochondrial biogenesis operate. The results presented here may suggest the possibility of developing treatments for conditions caused by mitochondrial deficit using heat modalities when the ability for exercise is impaired.
GRANTS
This research was supported by Henry Summer Research Fellowship to C-TL and NIH R01 AR050459 to GAB, and a gift from CytoSport, Inc.
DISCLOSURES
GAB has a financial interest in CytoSport, Inc. Otherwise, no conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: C.-T.L. and G.A.B. conception and design of research; C.-T.L. performed experiments; C.-T.L. and G.A.B. analyzed data; C.-T.L. and G.A.B. interpreted results of experiments; C.-T.L. prepared Figs.; C.-T.L. drafted manuscript; C.-T.L. and G.A.B. edited and revised manuscript; C.-T.L. and G.A.B. approved final version of manuscript.
REFERENCES
- 1. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. New York: Garland Science, 2002 [Google Scholar]
- 2. Amat R, Planavila A, Chen SL, Iglesias R, Giralt M, Villarroya F. SIRT1 controls the transcription of the peroxisome proliferator-activated receptor-gamma Co-activator-1alpha (PGC-1alpha) gene in skeletal muscle through the PGC-1alpha autoregulatory loop and interaction with MyoD. J Biol Chem 284: 21872–21880, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bai RK, Perng CL, Hsu CH, Wong LJ. Quantitative PCR analysis of mitochondrial DNA content in patients with mitochondrial disease. Ann NY Acad Sci 1011: 304–309, 2004 [DOI] [PubMed] [Google Scholar]
- 4. Brooks GA, Fahey TD, White TP, Baldwin KM. Exercise Physiology: Human Bioenergetics and Its Applications. New York: McGraw-Hill, 2005 [Google Scholar]
- 5. Brooks GA, Hittelman KJ, Faulkner JA, Beyer RE. Temperature, skeletal muscle mitochondrial functions, and oxygen debt. Am J Physiol 220: 1053–1059, 1971 [DOI] [PubMed] [Google Scholar]
- 6. Brooks GA, Hittelman KJ, Faulkner JA, Beyer RE. Tissue temperatures and whole-animal oxygen consumption after exercise. Am J Physiol 221: 427–431, 1971 [DOI] [PubMed] [Google Scholar]
- 7. Brozinick JT, Jr, McCoid SC, Reynolds TH, Nardone NA, Hargrove DM, Stevenson RW, Cushman SW, Gibbs EM. GLUT4 overexpression in db/db mice dose-dependently ameliorates diabetes but is not a lifelong cure. Diabetes 50: 593–600, 2001 [DOI] [PubMed] [Google Scholar]
- 8. Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab 11: 213–219, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carrier DR, Kapoor AK, Kimura T, Nickels MK, Satwanti Scott EC, So JK, Trinkaus E. The energetic paradox of human running and hominid evolution [and comments and reply]. Curr Anthropol 25: 483–495, 1984 [Google Scholar]
- 11. Chung J, Nguyen AK, Henstridge DC, Holmes AG, Chan MH, Mesa JL, Lancaster GI, Southgate RJ, Bruce CR, Duffy SJ, Horvath I, Mestril R, Watt MJ, Hooper PL, Kingwell BA, Vigh L, Hevener A, Febbraio MA. HSP72 protects against obesity-induced insulin resistance. Proc Natl Acad Sci USA 105: 1739–1744, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Corton JM, Gillespie JG, Hardie DG. Role of the AMP-activated protein kinase in the cellular stress response. Curr Biol 4: 315–324, 1994 [DOI] [PubMed] [Google Scholar]
- 13. Falkenberg M, Larsson NG, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem 76: 679–699, 2007 [DOI] [PubMed] [Google Scholar]
- 14. Gaesser GA, Brooks GA. Muscular efficiency during steady-rate exercise: effects of speed and work rate. J Appl Physiol 38: 1132–1139, 1975 [DOI] [PubMed] [Google Scholar]
- 15. Gleyzer N, Vercauteren K, Scarpulla RC. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol Cell Biol 25: 1354–1366, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Golbidi S, Laher I. Molecular mechanisms in exercise-induced cardioprotection. Cardiol Res Pract 2011: 972807, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gupte AA, Bomhoff GL, Swerdlow RH, Geiger PC. Heat treatment improves glucose tolerance and prevents skeletal muscle insulin resistance in rats fed a high-fat diet. Diabetes 58: 567–578, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc Natl Acad Sci USA 100: 7111–7116, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hardie DG, Sakamoto K. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology (Bethesda) 21: 48–60, 2006 [DOI] [PubMed] [Google Scholar]
- 20. Harris MB, Starnes JW. Effects of body temperature during exercise training on myocardial adaptations. Am J Physiol Heart Circ Physiol 280: H2271–H2280, 2001 [DOI] [PubMed] [Google Scholar]
- 21. Hendrick JP, Hartl FU. Molecular chaperone functions of heat-shock proteins. Annu Rev Biochem 62: 349–384, 1993 [DOI] [PubMed] [Google Scholar]
- 22. Hochachka PW, Somero GN. Biochemical Adaptation: Mechanism and Process in Physiological Evolution. Cambridge, MA: Oxford University Press, 2002 [Google Scholar]
- 23. Holloszy JO, Coyle EF. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J Appl Physiol 56: 831–838, 1984 [DOI] [PubMed] [Google Scholar]
- 24. Hooper PL. Hot-tub therapy for type 2 diabetes mellitus. N Engl J Med 341: 924–925, 1999 [DOI] [PubMed] [Google Scholar]
- 25. Howe AS, Boden BP. Heat-related illness in athletes. Am J Sports Med 35: 1384–1395, 2007 [DOI] [PubMed] [Google Scholar]
- 26. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403: 795–800, 2000 [DOI] [PubMed] [Google Scholar]
- 27. Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA 104: 12017–12022, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, Verdin EM, Kahn CR. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci USA 108: 14608–14613, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joseph AM, Pilegaard H, Litvintsev A, Leick L, Hood DA. Control of gene expression and mitochondrial biogenesis in the muscular adaptation to endurance exercise. Essays Biochem 42: 13–29, 2006 [DOI] [PubMed] [Google Scholar]
- 30. Khazaeli AA, Tatar M, Pletcher SD, Curtsinger JW. Heat-induced longevity extension in Drosophila. I Heat treatment, mortality, and thermotolerance. J Gerontol A Biol Sci Med Sci 52: B48–B52, 1997 [DOI] [PubMed] [Google Scholar]
- 31. Kirkwood SP, Munn EA, Brooks GA. Mitochondrial reticulum in limb skeletal muscle. Am J Physiol Cell Physiol 251: C395–C402, 1986 [DOI] [PubMed] [Google Scholar]
- 32. Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, Fang F, Chang Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLos One 5: e11707, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Koulmann N, Bigard AX. Interaction between signalling pathways involved in skeletal muscle responses to endurance exercise. Pflügers Arch 452: 125–139, 2006 [DOI] [PubMed] [Google Scholar]
- 34. Kwon HS, Ott M. The ups and downs of SIRT1. Trends Biochem Sci 33: 517–525, 2008 [DOI] [PubMed] [Google Scholar]
- 35. Lim CL, Byrne C, Lee JK. Human thermoregulation and measurement of body temperature in exercise and clinical settings. Ann Acad Med Singapore 37: 347–353, 2008 [PubMed] [Google Scholar]
- 36. Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet 22: 631–677, 1988 [DOI] [PubMed] [Google Scholar]
- 37. Lithgow GJ, White TM, Melov S, Johnson TE. Thermotolerance and extended life-span conferred by single-gene mutations and induced by thermal stress. Proc Natl Acad Sci USA 92: 7540–7544, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Medeiros DM. Assessing mitochondria biogenesis. Methods 46: 288–294, 2008 [DOI] [PubMed] [Google Scholar]
- 39. Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Lehman JJ, Kelly DP, Spiegelman BM. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci USA 98: 3820–3825, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Molles MC. Ecology: Concepts and Applications. New York: McGraw-Hill, 2009 [Google Scholar]
- 41. Morton JP, MacLaren DP, Cable NT, Bongers T, Griffiths RD, Campbell IT, Evans L, Kayani A, McArdle A, Drust B. Time course and differential responses of the major heat shock protein families in human skeletal muscle following acute nondamaging treadmill exercise. J Appl Physiol 101: 176–182, 2006 [DOI] [PubMed] [Google Scholar]
- 42. Moyes CD, Schulte PM. Principles of Animal Physiology. Cambridge, UK: Pearson, 2006 [Google Scholar]
- 43. Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300: 1140–1142, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92: 829–839, 1998 [DOI] [PubMed] [Google Scholar]
- 45. Revollo JR, Grimm AA, Imai S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol 23: 164–170, 2007 [DOI] [PubMed] [Google Scholar]
- 46. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434: 113–118, 2005 [DOI] [PubMed] [Google Scholar]
- 47. Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, Ido Y. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab 298: E751–E760, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Saltin B, Gagge AP, Bergh U, Stolwijk JA. Body temperatures and sweating during exhaustive exercise. J Appl Physiol 32: 635–643, 1972 [DOI] [PubMed] [Google Scholar]
- 49. Saltin B, Gagge AP, Stolwijk JA. Muscle temperature during submaximal exercise in man. J Appl Physiol 25: 679–688, 1968 [DOI] [PubMed] [Google Scholar]
- 50. Saraste M. Oxidative phosphorylation at the fin de siecle. Science 283: 1488–1493, 1999 [DOI] [PubMed] [Google Scholar]
- 51. Shama S, Lai CY, Antoniazzi JM, Jiang JC, Jazwinski SM. Heat stress-induced life span extension in yeast. Exp Cell Res 245: 379–388, 1998 [DOI] [PubMed] [Google Scholar]
- 52. Wallberg-Henriksson H, Zierath JR. GLUT4: a key player regulating glucose homeostasis? Insights from transgenic and knockout mice (review). Mol Membr Biol 18: 205–211, 2001 [DOI] [PubMed] [Google Scholar]
- 53. Westerheide SD, Anckar J, Stevens SM, Jr., Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323: 1063–1066, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Whipp BJ, Wasserman K. Efficiency of muscular work. J Appl Physiol 26: 644–648, 1969 [DOI] [PubMed] [Google Scholar]
- 55. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124, 1999 [DOI] [PubMed] [Google Scholar]
- 56. Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem 285: 19051–19059, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yoon JC, Ng A, Kim BH, Bianco A, Xavier RJ, Elledge SJ. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev 24: 1507–1518, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB, Wojtaszewski JF, Hirshman MF, Virkamaki A, Goodyear LJ, Kahn CR, Kahn BB. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med 6: 924–928, 2000 [DOI] [PubMed] [Google Scholar]