

Abstract
The mortality rate for acute lung injury (ALI) is reported to be between 35–40%, and there are very few treatment strategies that improve the death rate from this condition. Previous studies have suggested that signaling through the prostaglandin (PG) I2 receptor may protect against bleomycin-induced ALI in mice. We found that mice that overexpress PGI synthase (PGIS) in the airway epithelium were significantly protected against bleomycin-induced mortality and had reduced parenchymal consolidation, apoptosis of lung tissue, and generation of F2-isoprostanes compared with littermate wild-type controls. In addition, we show for the first time in both in vivo and in vitro experiments that PGI2 induced the expression of NADP (H): quinoneoxidoreductase 1 (Nqo 1), an enzyme that prevents the generation of reactive oxygen species. PGI2 induction of Nqo 1 provides a possible novel mechanism by which this prostanoid protects against bleomycin-induced mortality and identifies a potential therapeutic target for human ALI.
Keywords: epithelial cells, lung, prostaglandin I, NAD(P)H: quinoneoxidoreductase 1
acute lung injury (ALI) can be initiated by a variety of stimuli, including trauma, sepsis, and toxic ingestions (25). ALI is characterized by damage to both the lung epithelial and endothelial tissues, resulting in alveolar capillary/epithelial barrier destruction. The consequence of this injury is greater permeability of pulmonary capillary vessels, resulting in leakage of plasma proteins and influx of inflammatory cells into the alveolar spaces, leading to abnormalities of gas exchange (25). Although there have been recent advances in treatment modalities for ALI, the mortality rate for these patients remains exceedingly high at between 35–40% (20). Therefore, determining new approaches to either prevent or minimize the sequelae of ALI are of great importance.
Intra-airway administration of bleomycin has been used by many investigators as a way to model ALI in mice to test therapeutic modalities in preclinical trials (7, 10, 12, 21). The bleomycin model has been used to suggest that prostaglandin I2 (PGI2) may protect against ALI (11, 13). PGI2 is a metabolite of arachidonic acid that is primarily produced by endothelial cells to protect against vascular damage; however, it has been recently shown to have potent anti-inflammatory properties (28, 29). Lovgren and colleagues (11) reported that the inability to signal through the PGI2 receptor IP exacerbated bleomycin-induced lung injury in a murine model. Murakami and colleagues (13) showed that a PGI2 agonist with thromboxane synthase inhibitory activity reduced bleomycin-induced disease in mice; however, the effect of PGI2 by itself in bleomycin-induced acute lung injury is unknown.
We first hypothesized that PGI2 protects mice from bleomycin-induced acute lung injury and tested the hypothesis by using mice that overexpress PGI synthase (PGIS) selectively in the lung epithelium. We found that PGIS overexpression reduced mortality to bleomycin-induced lung injury at three different doses of bleomycin. Intranasal administration of bleomycin first results in injury to and death of airway epithelium through apoptosis and oxidant stress. As PGIS overexpression protected against bleomycin-induced mortality, we further hypothesized that this protective effect is mediated by regulating and increasing epithelial expression of antioxidant enzymes in the acute model of bleomycin-induced lung toxicity.
MATERIALS AND METHODS
Mice.
Pathogen-free 10–14-wk-old female mice were used in all experiments. Transgenic mice that overexpress PGIS (PGIS OE+) were created by using a construct consisting of a human surfactant protein C (SP-C) promoter and full-length rat PGIS cDNA as described previously (6). The SP-C promoter allows targeted gene expression to alveolar and airway epithelial cells (AEC). Transgenic mice were genotyped by performing PCR on genomic DNA isolated from tails as described previously (10). Each line was propagated as heterozygotes. PGIS OE+ mice on a FVB background were always bred with wild-type FVB/N (Jackson Laboratory, Bar Harbor, ME) mice to produce the experimental PGIS OE+ mice as well as the PGIS OE− littermates, which were used as controls in all of the experiments. For all of the experiments, F1-generation mice were used. Cages, bedding, food, and water were sterilized before use. All animal protocols were approved by the IACUC at Vanderbilt University Medical School.
Bleomycin injections.
Bleomycin (Ben Venue Laboratories, Bedford, OH) was administered intranasally at the described doses in a volume of 100 μl after dilution with PBS. The mice were anesthetized with intramuscular ketamine at 40 μg/g and xylazine at 6 μg/g. After being anesthetized, the mice were held upright with the neck fully extended, whereupon they readily inhaled a 100 μl solution of bleomycin placed over their nostrils with a micropipette.
Measurement of 2,3-dinor-6-keto-PGF1α and TxB2 in the urine and the lung.
PGIS OE+ and littermate control PGIS OE− mice were treated with PBS or bleomycin (0.08 U/mouse) intranasally. Twelve to sixteen mice in each treatment group were placed in metabolic cages, four mice per cage, and the urine was collected daily. Each data point represents the concentration of 2,3-dinor-6-keto-PGF1α in the urine collected from one cage. For measurements of 2,3-dinor-6-keto-PGF1α and thromboxane B2 (TxB2) in the lung, mouse lungs were harvested and homogenized with 2 ml RPMI 1640 containing 1 μM indomethacin to inhibit ex vivo production of cyclooxygenase (COX) products. The levels of 2,3-dinor-6-keto-PGF1α in the urine or lung samples and the levels of TxB2 in the lung were measured by a gas chromatographic-mass spectrometric assay as previously described (3).
Measurement of F2-isoprostanes by gas chromatography/negative-ion chemical ionization mass spectrometry.
For measurement of F2-isoprostanes, mouse lungs were perfused with PBS to remove circulating cells and plasma, dissected free from the chest, placed immediately in PBS, and ground in a tissue grinder. Analysis for esterified F2-isoprostanes by gas chromatography/negative-ion chemical ionization mass spectrometry was performed as previously described (4).
Histology and immunohistochemical staining.
Mice were euthanized by CO2 asphyxiation. Lungs were inflated with 10% neutral buffered formalin under a standard inflation pressure of 20 cm water before being dehydrated in 70% ethanol. Lungs were then processed using standard procedures and embedded in paraffin. Sections were cut, mounted on slides, and stained with hematoxylin and eosin to assess the inflammatory infiltrate, and terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) assay was performed to assess for DNA damage and apoptosis. The DeadEnd Colorimetric Detection system (Promega, Madison, WI) was used for TUNEL assay, and the number of positive cells for TUNEL was counted in the whole field of each section under a microscope with ×200 magnification. Slides were scored by a pathologist who was blinded to the groups. Ten sequential, nonoverlapping tissue fields of lung parenchyma were evaluated. Each tissue field was scored using a 0–4 point system (0, no positive cells; 1, ≤1% positive cells; 2, 1–5% positive cells; 3, 5–10% positive cells; and 4, 10–25% positive cells). A mean score for all fields was calculated for each animal.
Mouse lung function measurement.
PGIS OE+ and littermate control PGIS OE− mice were administered either PBS or 0.08 units of bleomycin at day 0. At day 7 after bleomycin administration, the mice were anesthetized, and airway resistance, elastance, and compliance were measured by FlexiVent system (SCIREQ).
Protein measurement.
PGIS OE+ and littermate control PGIS OE− mice were intranasally administered 0.08 units of bleomycin at day 0. Mouse bronchoalveolar lavage (BAL) fluid was harvested at day 0 and at days 1, 3, and 6 after bleomycin injection. Protein concentrations of the BAL fluid were determined by a colorimetric protein assay kit from Bio-Rad (Hercules, CA).
Cell culture.
Murine type II AECs can be difficult to maintain in culture for time periods that allow experimental studies; therefore, type II AECs were isolated from Immortomice. Immortomice, transgenic mice, which have temperature and interferon-γ (IFN-γ)-sensitive expression of the SV40 large T antigen, were obtained from Dr. Robert Whitehead, Vanderbilt University, Nashville, TN. Immortomouse cells proliferate when cultured with IFN-γ at 33°C; however, when cultured without IFN-γ at 37°C, cells lose expression of the SV40 antigen and revert to the primary phenotype. With the use of permissive conditions, Immortomouse type II AECs can be passaged multiple times while retaining type II AEC phenotype (as determined by morphology and SP-C proprotein expression). For these studies, Type II AECs were isolated from three adult 8-wk-old Immortomice using techniques as previously described (23). After isolation, Immortomouse type II AECs were placed in DMEM culture medium with IFN-γ at 10 ng/ml (Sigma, St. Louis, MO) and cultured at 33°C. After two passages, Immortomouse type II AECs were placed in DMEM culture medium without IFN-γ and cultured at 37°C in six-well plates 2 days before being treated with indomethacin (2 μM, Sigma), cicaprost (2 μM, a gift from Dr. M. Huebner at Schering-Plough), and/or bleomycin (0.1 U/ml). Ethanol and water were used as vehicle control treatments for indomethacin and cicaprost, respectively. The treatment of indomethacin was conducted 4 h before adding cicaprost to the culture medium, and bleomycin was added 30 min after cicaprost treatment. The cells were harvested 4 h after cicaprost treatment alone or 4 h after bleomycin treatment and used for total RNA extraction by Qiagen RNeasy Mini kit (Valencia, CA).
PCR.
Real-time quantitative PCR was used to determine the mRNA levels of the PGI2 receptor (IP), NADP (H): quinoneoxidoreductase 1 (Nqo 1), NF-E2-related factor 2 (NRF2), and β-actin. Total cellular RNA of the type II epithelial cells was transcribed to cDNA by using Superscript III reverse transcriptase and oligo dT(16) (Life Technologies, Grand Island, NY) according to the manufacturer's protocols. The cDNA products were used for SYBR Green real-time PCR with gene-specific primers, iTAQ real-time SYBR Green Supermix (Bio-Rad) and iCycler (Bio-Rad). The primers were designed to contain sequences of multiple exons to increase PCR specificity. The primer sequences are as follows: IP, 5′CCGCC AACAG AGACG CCACC AT 3′ (forward) and 5′CGGGC ACACA GGCAA CACAA CCA 3′ (reverse) with a 273 bp amplicon; NRF2, 5′CAGCT ACTCC CAGGT TGCCC ACATT 3′ (forward) and 5′GCCAA ACTTG CTCCA TGTCC TGCTC TAT 3′ (reverse) with a 215 bp amplicon; Nqo 1, 5′TGGCC GAACA CAAGA AGCTG GAA 3′ (forward) and 5′CCCCG TGGAC ACCCT GAAGA GAGT 3′ (reverse) with a 211 bp amplicon; and β-actin, 5′-GACGA TGCTC CCCGG GCTGT A-3′ (forward) and 5′-CGACC AGAGG CATAC AGGGA CAGC-3 (reverse) with a 371 bp amplicon. The thermocycler condition was as follows: stage 1, 95°C for 3 min for one cycle; and stage 2, 95°C for 15 s and 60°C for 45 s for 45 cycles. Standard curves were generated for each gene using increasing amounts of RNA input. Relative levels of gene expression in the samples were extrapolated from the standard curves. β-Actin gene expression was used to normalize the amount of RNA input. The PCR products were analyzed in agarose gels.
BAL and protein measurement.
The animals were given a lethal injection of pentobarbital sodium. BAL was then performed by instilling 800 μl of sterile saline through the tracheostomy tube and then withdrawing the fluid with gentle suction via the syringe. The protein concentration in the BAL fluid was determined by using Bio-Rad Protein Assay according to the manufacturer's instructions.
Statistical analysis.
The P values were calculated by using ANOVA with Bonferroni's posttest (SAS) for multiple-group comparisons and the unpaired Student's t-test for two-group comparisons. Values of P < 0.05 were considered significant.
RESULTS
Bleomycin administration increases production of the stable PGI2 metabolite 2,3-dinor-6-keto-PGF1α in the urine and the lung.
We measured the stable PGI2 metabolite, 2,3-dinor-6-keto-PGF1α in the urine of PGIS OE− and PGIS OE+ mice that were intranasally challenged with either bleomycin (0.08 U) or PBS to determine 1) whether PGI2 might be involved in the response to bleomycin and 2) whether PGIS OE+ mice would produce greater PGI2 in response to bleomycin than PGIS OE− littermates. The results shown in Fig. 1 are for four data points for the day before bleomycin administration (day −1), the combined urine collected for days 2 and 3 after bleomycin administration (days 2-3), the combined urine collected for days 6 and 7 after bleomycin administration (days 6-7), and the combined urine collected for days 8 through 12 after bleomycin administration (days 8-12). The urines were combined for the time points listed above because the bleomycin-treated mice produced less urine than the PBS-treated mice, and there was not sufficient urine for measurement of the PGI2 metabolite on the individual days in the bleomycin-treated animals. The results of the PGI2 metabolite are normalized for mg creatinine to account for any effect of urine concentration that might have occurred in the bleomycin-treated mice. At both day −1 and days 2–3, there was no difference in the urinary concentrations of the PGI2 metabolite in the four groups of mice. However, at days 6–7, the PGI2 metabolite in the PGIS OE+ mice that had been administered intranasal bleomycin (PGIS OE+/bleomycin) was significantly increased (P < 0.05) compared with the other three groups of mice including the PGIS OE− that had been administered intranasal bleomycin (PGIS OE−/bleomycin), the PGIS OE+ mice that had been administered intranasal PBS (PGIS OE+/PBS), or the PGIS OE− mice that had been administered intranasal PBS (PGIS OE−/PBS). At days 8–12, the PGIS OE+/bleomycin mice had ∼25% greater PGI2 metabolite in their urine compared with the PGIS OE−/bleomycin mice although this difference was not statistically significant. At days 8–12, there was an approximate doubling of the PGI2 metabolite in the urine of the bleomycin-treated mice compared with the PBS-treated mice. These results suggest that PGI2 synthesis is modulated by bleomycin administration in the PGIS OE+ mice and that the use of these mice is a valid model to test the effects of increased airway PGI2 on bleomycin induced airway disease.
Fig. 1.
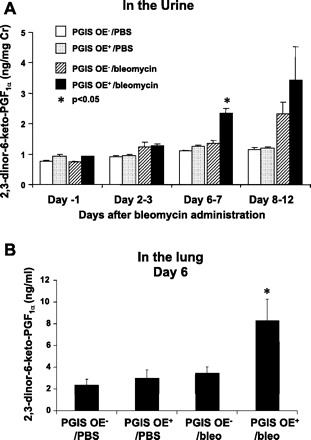
Bleomycin increases production of the levels of 2,3-dinor-6-keto-PGF1α in the urine and the lung. Transgenic mice that overexpress prostaglandin I2 synthase (PGIS OE+) and littermate control PGIS OE− mice were intranasally administered 0.08 U of bleomycin or PBS at day 0. A: 12–16 mice per treatment group were placed in metabolic cages, 4 mice per cage, and the mouse urine was collected daily from day −1 to day 12. B: lungs of bleomycin (bleo)- or PBS-treated mice were harvested at day 6 after bleomycin administration. The levels of 2,3-dinor-6-keto-PGF1α in the urine samples and in the lung homogenate were determined by a gas chromatographic-mass spectrometric assay. Data shown are the means ± SE, n = 3–4; each data point represents a urine sample collected from a cage of 4 mice (A) and n = 4–8 (B). *P < 0.05 vs. all 3 other groups at the same time point.
To further determine whether PGIS overexpression increased local production of PGI2 in the lung, we administered bleomycin or PBS intranasally to PGIS OE+ and PGIS OE− mice and harvested the lungs at 6 days after bleomycin administration. We measured the levels of 2,3-dinor-6-keto-PGF1α in the lung homogenate. We found that bleomycin-treated PGIS OE+ mice had significantly increased levels of 2,3-dinor-6-keto-PGF1α compared with bleomycin-treated PGIS OE− mice, PBS-treated PGIS OE+ mice, or PBS-treated PGIS OE− mice (Fig. 1B), supporting the notion that PGIS overexpression augmented bleomycin-induced PGI2 production. In addition, bleomycin-treated PGIS OE+ mice had decreased levels of TxB2 in the lung (0.024 ± 0.009 ng/g lung tissue, n = 9) compared with bleomycin-treated PGIS OE− mice (0.103 ± 0.028 ng/g lung tissue, n = 7, P < 0.05) at day 3 after bleomycin administration, suggesting a shunting effect of PGH2, the common substrate for prostanoid synthases, toward PGIS pathway.
Mice that overexpress PGIS are protected against bleomycin-induced weight loss.
On the basis of the data shown above that the stable PGI2 metabolite was increased after bleomycin administration and previous studies that suggested that signaling through IP might protect against bleomycin-induced weight loss (11, 13), we hypothesized that PGI2 protects against bleomycin-induced disease. To test this hypothesis, we weighed both PGIS OE+ and PGIS OE− mice after bleomycin administration. After 9 days following bleomycin administration, PGIS OE+ mice (n = 13) lost significantly less weight (Fig. 2, P < 0.05) than PGIS OE− mice (n = 10). The mice were weighed to day 9 because mortality began to occur in the PGIS OE− mice.
Fig. 2.
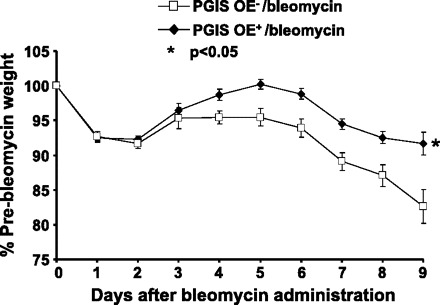
PGIS overexpression protects mice against bleomycin-induced weight loss. PGIS OE+ (n = 13) and PGIS OE− (n = 10) mice were intranasally administered 0.08 U of bleomycin at day 0. The mouse body weights were monitored daily from days 0 to 9. Data shown are the means ± SE. *P < 0.05 vs. PGIS OE−/bleomycin.
Mice that overexpress PGIS are protected against bleomycin-induced mortality.
On the basis of the finding that the PGIS OE+ were protected from weight loss following airway administration of bleomycin compared with PGIS OE− mice, we hypothesized that PGI2 was protective against bleomycin-induced mortality. We performed dose-response experiments in which either 0.08 U, 0.16 U, or 0.32 U of bleomycin were administered intranasally. At the 0.08-U dose (Fig. 3A), there was a trend (P = 0.057) for an increase in the proportion surviving at day 13 after bleomycin administration in the PGIS OE+ mice (18 of 19 mice surviving) compared with the PGIS OE− littermates (7 of 10 mice surviving). At the 0.16-U dose (Fig. 3B), there was a significant increase (P = 0.0054) in the proportion surviving at day 13 after bleomycin administration in the PGIS OE+ mice (4 of 6 mice surviving) compared with the PGIS OE− littermates (0 of 6 mice surviving). At the 0.32-U dose (Fig. 3C), there was a trend (P = 0.15) for an increase in the proportion surviving at day 13 after bleomycin administration in the PGIS OE+ mice (2 of 6 mice surviving) compared with the PGIS OE− littermates (0 of 6 mice surviving). Therefore, we found that PGIS OE significantly protected against bleomycin-induced mortality at the 0.16-U dose, and there was a trend for protection against bleomycin-induced mortality at the 0.08- and 0.32-U doses.
Fig. 3.
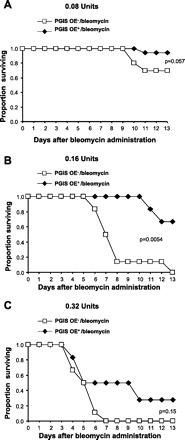
PGIS overexpression protects mice against bleomycin-induced mortality. PGIS OE+ mice and littermate control PGIS OE− mice were intranasally administered 0.08 U (A), 0.16 U (B), or 0.32 U (C) of bleomycin at day 0. The Kaplan-Meier Survival Curve shows mouse survival rate; n = 19 PGIS OE+ mice and 10 PGIS OE− mice (A) and n = 6 mice (B and C).
Mice that overexpress PGIS have reduced lung consolidation.
Because we found that PGIS OE significantly protected against bleomycin-induced mortality at the 0.16-U dose and there was a trend for protection against bleomycin-induced mortality at the 0.08- and 0.32-U doses, we hypothesized that PGIS OE reduces the inflammation that results from airway bleomycin administration. Both PGIS OE+ and PGIS OE− mice were administered 0.08 U of bleomycin intranasally, and the lungs were harvested 6 and 14 days later. Lung sections were reviewed by a pathologist blinded to the genotype of the mice. Scoring of the percent consolidation in the lung sections revealed significantly decreased consolidation in the PGIS OE+ mice compared with the PGIS OE− mice at both 6 days (n = 5 per group; P < 0.05) and 14 days (n = 6 per group; P < 0.05) after bleomycin (Fig. 4). Therefore, PGIS overexpression reduces lung inflammation following bleomycin administration.
Fig. 4.
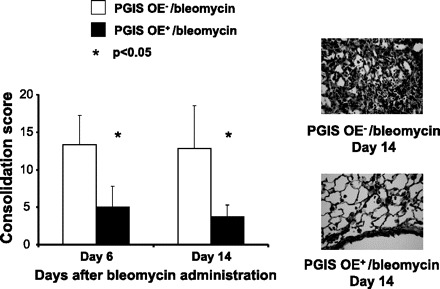
PGIS overexpression decreases bleomycin-induced lung consolidation. PGIS OE+ mice and littermate control PGIS OE− mice were intranasally administered 0.08 U of bleomycin at day 0. Mouse lungs were harvested at days 6 and 14. The lung sections of the paraffin-embedded tissue were stained with hematoxylin and eosin. The lung tissue consolidation was scored by a pathologist who was blinded to the sample identifications. Representative pathological images of the lung sections are shown. Data shown are the means ± SE, n = 5–6 mice. *P < 0.05 vs. PGIS OE−/bleomycin at the same time point.
Mice that overexpress PGIS have reduced bleomycin-induced apoptosis in the lungs.
Nana-Sinkam and colleagues (15) have previously reported that PGI2 prevents pulmonary endothelial cell apoptosis in response to cigarette smoke-induced injury (15). We therefore hypothesized that PGIS OE protects against bleomycin-induced apoptosis. We tested this hypothesis by performing TUNEL staining on the lungs of mice harvested 6 and 14 days after 0.08 U of intranasal bleomycin administration. Lung sections were reviewed by a pathologist blinded to the genotype of the mice. Scoring of the percent of TUNEL+ cells in the lung sections revealed significantly decreased TUNEL staining in the PGIS OE+ mice compared with the PGIS OE− mice at both 6 days (n = 5 per group; P < 0.05) and 14 days (n = 6 per group; P < 0.05) after bleomycin administration (Fig. 5). The TUNEL-positive cells appeared to be resident and recruited inflammatory cells as well as lung structural cells. Therefore, we found that PGIS OE+ mice were protected against bleomycin-induced apoptosis compared with their nontransgenic littermate controls.
Fig. 5.
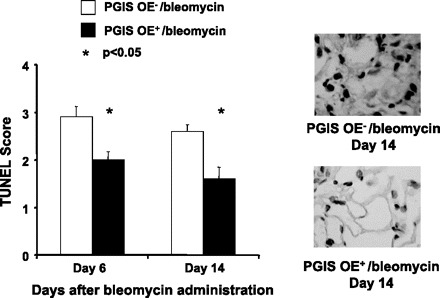
PGIS overexpression decreases bleomycin-induced apoptosis of pulmonary cells. PGIS OE+ mice and littermate control PGIS OE− mice were intranasally administered 0.08 U of bleomycin at day 0. Mouse lungs were harvested at days 6 and 14. The lung sections of the paraffin-embedded tissue were subjected to terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) assay with the DeadEnd Colorimetric Detection System. The TUNEL-positive cells in the lung section were counted and scored by a pathologist who was blinded to the groups. Representative TUNEL assay images are shown, and TUNEL+ cells were stained brown. Data shown are the mean ± SE, n = 5–6 mice. *P < 0.05 vs. PGIS OE−/bleomycin at the same time point.
PGIS OE does not affect lung function 7 days after bleomycin administration.
We measured airway mechanics in PGIS OE+ and PGIS OE− mice 7 days after intranasal challenge with either bleomycin (0.08 U) or PBS. There was no significant difference in the airway resistance, elastance, and compliance between PGIS OE+ mice and PGIS OE− mice (data not shown), indicating that PGIS overexpression did not alter the airway mechanics at day 7 after bleomycin administration.
Mice that overexpress PGIS do not have a reduction in the BAL fluid protein concentration following bleomycin administration.
Because we found that PGIS OE+ mice were protected against bleomycin-induced apoptosis compared with their nontransgenic littermate controls, we hypothesized that there would be a reduction in the permeability of pulmonary capillary vessels in PGIS OE+ mice after bleomycin administration and a decrease in the resulting leakage of plasma proteins. To test this hypothesis, we performed BAL at four different time points: baseline (no bleomycin administration), and 1, 3, and 6 days after bleomycin administration. Contrary to our hypothesis, we did not find that PGIS OE status affected the protein concentration of the BAL fluid after bleomycin injury (data not shown).
Mice that overexpress PGIS have reduced F2-isoprostanes in the lungs 3 days after bleomycin administration.
One mechanism by which bleomycin causes epithelial cell death is the induction of oxidant stress (16). Isoprostane formation is a dependable marker of oxidant injury both in vivo and in vitro, and these mediators also are biologically active and may regulate oxidant injury (5). Because we found that PGIS OE+ mice were protected against bleomycin-induced apoptosis compared with their nontransgenic littermate controls, we hypothesized that PGIS OE would result in decreased formation of F2-isoprostanes in the lungs. To test this hypothesis, we euthanized PGIS OE+ and PGIS OE− mice and measured F2-isoprostanes in the lung at three different time points: 3, 6, and 10 days after bleomycin administration. We found that there was a significant reduction in F2-isoprostanes in the lungs of PGIS OE+ mice 3 days after bleomycin administration (P < 0.05) compared with PGIS OE− mice (Fig. 6). However, we did not find a difference in the F2-isoprostane levels between the PGIS OE+/bleomycin and PGIS OE−/bleomycin mice at 6 and 10 days after bleomycin. These results suggest that PGIS OE regulates oxidative stress early after bleomycin administration, thereby possibly explaining how PGIS OE protects against bleomycin-induced mortality.
Fig. 6.
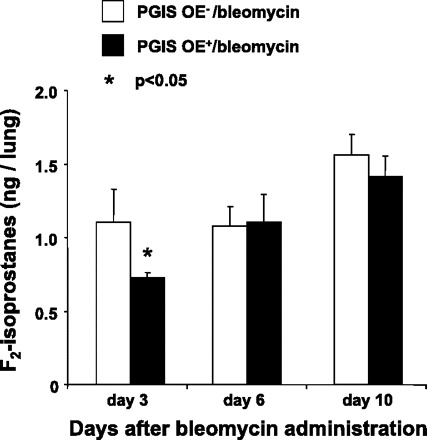
PGIS-overexpressing mice have reduced levels of F2-isoprostanes in the lung. PGIS OE+ mice and littermate control PGIS OE− mice were intranasally administered 0.08 U of bleomycin at day 0. The levels of F2-isoprostanes in the ground lung homogenate were determined by a gas chromatographic-negative-ion chemical ionization mass spectrometric assay. Data shown are the means ± SE, n = 5–6 mice, *P < 0.05 vs. PGIS OE−/bleomycin at the same time point.
Mice that overexpress PGIS have increased Nqo 1 expression in the lungs at baseline and after bleomycin administration.
Given that the PGIS OE+ mice had decreased F2-isoprostane levels in the lungs 3 days after bleomycin administration and that F2-isoprostane formation is a dependable measure of oxidant injury, we hypothesized that PGI2 modulates genes that regulate oxidative stress. To test this hypothesis, we performed real-time PCR for genes that regulate oxidant stress at baseline and 3, 6, and 11 days after bleomycin administration. We found that there was no difference between the PGIS OE+ and PGIS OE− groups in the mRNA expression of NRF2 (data not shown). However, there was a significant increase in mRNA expression of Nqo 1 gene at baseline (P < 0.05) and at days 3 (P < 0.05) and 11 (P < 0.001) after bleomycin administration (Fig. 7). These results suggest that PGIS OE regulates expression of Nqo 1, a detoxification enzyme that plays a critical role in protecting cells against chemically induced oxidative stress and cytotoxicity (9, 19).
Fig. 7.
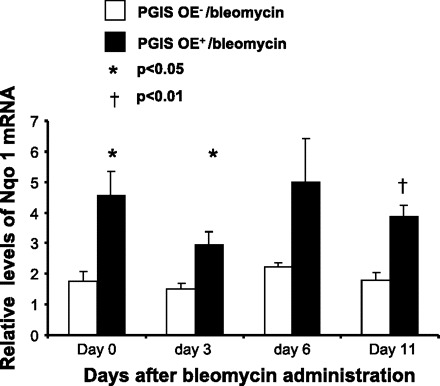
PGIS overexpression increases NADP (H): quinoneoxidoreductase 1 (Nqo 1) expression in the lung at baseline and after bleomycin administration. PGIS OE+ mice and littermate control PGIS OE− mice were intranasally administered 0.08 U of bleomycin at day 0. Mouse lungs were harvested at days 0, 3, 6 and 11. Total RNA was extracted from the lung tissue. The mRNA levels of Nqo 1 in the lung were determined by real-time PCR. Data shown are the means ± SE of 3 separate experiments, n = 4 mice (day 0) and 18–26 mice (days 3-11). *P < 0.05 and †P < 0.01 vs. PGIS OE−/bleomycin at the same time point.
A PGI2 analog, cicaprost, regulates Nqo 1 expression in type II cells in vitro.
On the basis of evidence that PGIS OE increased Nqo 1 expression in vivo, we hypothesized that a PGI2 analog increases Nqo 1 mRNA expression in pulmonary Type II epithelial cells in vitro that were isolated from Immortomice. To determine whether these Type II cells would respond to a PGI2 analog, PCR was performed to determine whether the Type II cells expressed the predominant PGI2 receptor IP. As shown in Fig. 8A, these Type II cells expressed IP. We then sought to determine whether PGI2 regulated Nqo 1 mRNA expression by treating the cells with the stable PGI2 analog cicaprost or its vehicle water. We found that cicaprost increased Nqo 1 mRNA levels approximately twofold compared with vehicle-treated cells (Fig. 8B). We next investigated whether endogenous prostanoid production regulated Nqo 1 mRNA expression by treating the cells with either the COX inhibitor indomethacin or its vehicle ethanol. We found that COX inhibition did not alter Nqo 1 expression in this Type II cell culture (Fig. 8C). Treatment of the Type II cells with indomethacin and bleomycin did not alter Nqo 1 mRNA expression either. However, Type II cells exposed to bleomycin and treated with cicaprost and indomethacin had a twofold increase of Nqo 1 mRNA expression compared with cells exposed to bleomycin and treated with the vehicle for cicaprost (water) and indomethacin. These in vitro results support the in vivo findings found in the PGIS OE mice and suggest that the PGI2 analog cicaprost increases Nqo 1 expression in alveolar Type II cells.
Fig. 8.
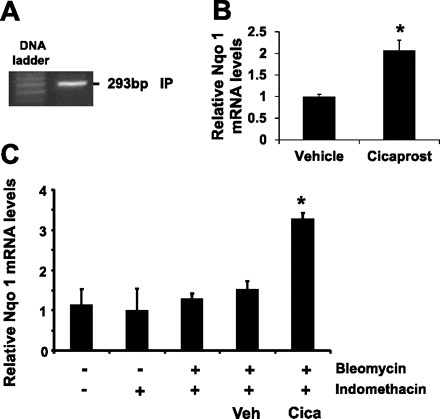
The PGI2 analog cicaprost regulates Nqo 1 expression in Immortomice Type II epithelial cells in vitro. A: mRNA levels of PGI2 IP receptor in the pulmonary Type II cells of Immortomice were determined by real-time PCR, and the PCR products were analyzed on an agarose gel. B: Type II cells were cultured and treated with cicaprost (2 μM) or vehicle (water). C: Type II cells were treated with indomethacin (2 μM) or vehicle solution at 4 h before treating with either cicaprost (2 μM) or vehicle (water). Bleomycin (0.1 U/ml) was added to the culture 30 min after cicaprost or vehicle treatment. The cells were harvested 4 h after cicaprost (B) or 4 h after bleomycin treatment (C), and the levels of Nqo 1 mRNA in the cells were determined by real-time PCR. Data shown are the means ± SE of 3 separate experiments. *P < 0.05 cicaprost (Cica)-treated cells compared with vehicle (Veh)-treated cells.
DISCUSSION
Bleomycin has been used by many investigators as a model to study both ALI pathogenesis and to test the efficacy of potential therapeutic targets for this condition. In our study, we attempted to modulate ALI by overexpression of PGIS in the airway epithelium, with resultant production of PGI2. We found that PGIS OE protected against bleomycin-induced weight loss and mortality, lung consolidation, apoptosis, and F2-isoprostane production. Additionally, we report for the first time that PGI2 modulates mRNA expression of Nqo 1, both in vitro and in vivo. Nqo 1 is a cytosolic flavoprotein that detoxifies quinones and derivatives, leading to the protection of cells against oxidative stress (9, 19). PGIS OE+ mice had greater Nqo 1 expression in the lung both before and after bleomycin administration, whereas the PGI2 analog cicaprost induced Nqo 1 expression in alveolar Type II cells in vitro that had been exposed to bleomycin.
Work from other groups suggested that PGI2 protects against bleomycin-induced lung injury. Lovgren and colleagues (11) reported that the inability to signal through the PGI2 receptor IP exacerbated both the development of bleomycin-induced fibrosis and the resulting changes in lung mechanics. In contrast, the inability to synthesize PGE2 and deficiency of signaling through the G(s)-coupled E-prostanoid receptor 2 (EP2) and EP4 did not impact bleomycin-induced disease. In their studies, Lovgren and colleagues (11) did not directly test the ability of PGI2 or PGI2 analogs to impact bleomycin-induced lung injury, but their findings suggested that this prostanoid might protect against the augmented bleomycin-induced pulmonary fibrosis that occurred in COX-2-deficient and IP-deficient mice. Murakami and colleagues (13) reported that a novel long-acting PGI2 agonist with TxB synthase inhibitory activity (ONO-1301) reduced bleomycin-induced lung injury in mice. They found that ONO-1301 significantly reduced the number of inflammatory cells and total protein level in BAL fluid, and this change was associated with significantly increased plasma cAMP levels after ONO-1301 administration (13). Although we also found that PGIS OE reduced lung inflammation as reported by parenchymal consolidation, we did not find a reduction in the total protein level in BAL fluid in the PGIS OE+ mice compared with the nontransgenic littermates. Similar to our results, Murakami also found that ONO-1301 protected against bleomycin-induced mortality (13).
Overexpression of PGIS in the lung decreased the number of apoptotic cells in the lung. In our study, bleomycin appeared to induce apoptosis in cells with morphologies of resident and recruited inflammatory cells and lung structural cells. Although we are the first to find that PGI2 modulates apoptosis in the bleomycin model of ALI, other groups have shown that PGI2 decreases apoptosis in smoking-related lung disease. Nana-Sinkham and colleagues (15) reported that PGI2 prevented pulmonary endothelial cell apoptosis induced by cigarette smoke. Chen and colleagues (2) found that a PGI2 analog, beraprost, protected against the development of cigarette smoke extract-induced emphysema by significantly attenuating alveolar enlargement and pulmonary parenchymal destruction (2). This beraprost-mediated inhibition of pulmonary apoptosis was associated with induction of matrix metalloproteinase (MMP)-2 and MMP-9 activity (2). Therefore, these smoking-related models revealed that PGI2 decreased both endothelial and parenchymal apoptosis, findings that were similar to our results. PGI2 has also been reported to inhibit apoptosis and enhance embryo viability (17), protect renal tubular cells from both apoptosis induced by either gentamicin (8) or adriamycin (1), prevent radiocontrast nephropathy (27), and block apoptosis of cultured retinal pericytes (26). Therefore, PGI2 protects against apoptosis in many cell types and may be useful in preventing the apoptotic cell death that occurs in ALI.
Our data also suggest that PGI2 decreases oxidant stress as witnessed by the inhibited generation of F2-isoprostanes seen in our study. Others have also reported that PGI2 modulates F2-isoprostane levels. Robbins and colleagues (18) measured F2-isoprostanes in patients with primary pulmonary hypertension, both before and after initiation of intravenous PGI2. During treatment with PGI2, there was a significant decrease in the levels of F2-isoprostanes that strongly correlated with reduction in pulmonary vascular resistance (18). These results suggested that oxidant stress was inhibited by PGI2 therapy and was associated with hemodynamic and clinical improvement. This is supported by work of Muzaffar and colleagues (14), who showed that iloprost, a stable PGI2 analog, inhibited the induction of oxidant stress by a TxB analog, LPS, and the cytokines IL-α and tumor necrosis factor (14).
As we found that PGIS OE decreased F2-isoprostane levels, and therefore oxidant stress, we examined the effect of PGIS OE on genes that regulate oxidative stress. Whereas PGIS OE had no effect on lung mRNA expression of NRF2, there was a significant increase in lung mRNA expression of Nqo 1 gene at baseline and at days 3 and 11 after bleomycin administration. To our knowledge, there are no prior reports of PGI2 regulating Nqo 1 expression. We found, not only that PGIS OE increased Nqo 1 mRNA expression in the lung, but also that the PGI2 analog cicaprost increased Nqo 1 expression in alveolar Type II cells. This strongly suggests that PGI2 does indeed regulate Nqo 1 and may explain the effect of PGIS OE on the decreased F2-isoprostane levels. Nqo 1 is generally considered to be a detoxification enzyme, as it has the ability to reduce reactive quinones and quinine-imines to less reactive and less toxic hydroquinones (22). The two-electron reduction of quinones effected by Nqo 1 prevents the generation of reactive oxygen species such as O2−. Nqo 1 has the ability to deactivate many reactive species, including quinones, quinine-imines, and azo compounds, thus exhibiting its importance as a chemoprotective enzyme (22). Although Nqo 1 has these antioxidant effects, Nqo 1 may paradoxically also have some proinflammatory effects. Voynow and colleagues (24) published that Nqo 1 is essential for ozone-induced oxidative stress, as Nqo 1-deficient mice were resistant to ozone-induced airway responsiveness and actually had blunted F2-isoprostane generation. Therefore, the balance of Nqo 1 anti- and prooxidant activities regulates the redox state of the cell and thereby modulates the balance of downstream reactive oxidant species signaling molecules, such as the isoprostanes.
We did not find complete temporal concordance between the levels of the PGI2 metabolite in the urine of the PGIS OE mice and either the inflammatory or antioxidant effects witnessed in these mice. We found that the PGIS OE mice did not have significantly increased 2.3-dinor-6-keto-PGF1α at baseline or days 2-3, but a significant increase compared with the nontransgenic littermates was first seen at days 6-7. This is likely due to a low sensitivity of the urine levels of the PGI2 metabolite to reflect the higher PGI2 levels that are very likely present in the lungs of the PGIS OE+ mice. This possibility is supported by the greater magnitude in difference in the 2,3-dinor-6-keto-PGF1α levels seen in the lung 6 days after bleomycin administration compared with PGIS OE− mice or either groups of PBS-treated mice. The increase in lung 2,3-dinor-6-keto-PGF1α likely explains the changes in Nqo 1 mRNA expression between the PGIS OE+ mice and the PGIS OE− mice at baseline and other time points after bleomycin administration. Whereas there were increases in Nqo 1 mRNA expression in the PGIS OE+ mice at baseline and days 3 and 11, we saw that the PGIS OE+ mice had decreased F2-isoprostane levels at day 3 after bleomycin. Therefore, it is possible that the ongoing inflammatory changes caused by bleomycin may overwhelm the ability of PGIS OE and Nqo 1 to modulate F2-isoprostanes although it is also possible that the increased Nqo 1 lung mRNA expression may be coincidental and not related to the decreased F2-isoprostane levels. However, given the in vitro data showing that cicaprost regulates Nqo 1 expression in Type II cells in vitro, it seems likely that the increased mRNA expression of Nqo 1 in the PGIS OE+ mice compared with the PGIS OE− mice are truly related.
In summary, we found that PGIS OE and PGI2 significantly protected against bleomycin-induced ALI. This survival benefit in PGI2-overexpressing mice was linked to decreased lung inflammation and apoptosis. The PGI2-mediated decrease in oxidant stress was associated with increased Nqo 1 mRNA expression and suggests that PGI2 may be a potential therapeutic strategy in the treatment of ALI, for which there are currently few effective treatments.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
REFERENCES
- 1. Chen CH, Lin H, Hsu YH, Sue YM, Cheng TH, Chan P, Chen TH. The protective effect of prostacyclin on adriamycin-induced apoptosis in rat renal tubular cells. Eur J Pharmacol 529: 8–15, 2006 [DOI] [PubMed] [Google Scholar]
- 2. Chen Y, Hanaoka M, Chen P, Droma Y, Voelkel NF, Kubo K. Protective effect of beraprost sodium, a stable prostacyclin analog, in the development of cigarette smoke extract-induced emphysema. Am J Physiol Lung Cell Mol Physiol 296: L648–L656, 2009 [DOI] [PubMed] [Google Scholar]
- 3. Daniel VC, Minton TA, Brown NJ, Nadeau JH, Morrow JD. Simplified assay for the quantification of 2,3-dinor-6-keto- prostaglandin F1 alpha by gas chromatography-mass spectrometry. J Chromatogr B Biomed Appl 653: 117–122, 1994 [DOI] [PubMed] [Google Scholar]
- 4. Dworski R, Murray JJ, Roberts LJ, Oates JA, Morrow JD, Fisher L, Sheller JR. Allergen-induced synthesis of F(2)-isoprostanes in atopic asthmatics. Evidence for oxidant stress. Am J Respir Crit Care Med 160: 1947–1951, 1999 [DOI] [PubMed] [Google Scholar]
- 5. Fam SS, Morrow JD. The isoprostanes: unique products of arachidonic acid oxidation-a review. Curr Med Chem 10: 1723–1740, 2003 [DOI] [PubMed] [Google Scholar]
- 6. Geraci MW, Gao B, Shepherd DC, Moore MD, Westcott JY, Fagan KA, Alger LA, Tuder RM, Voelkel NF. Pulmonary prostacyclin synthase overexpression in transgenic mice protects against development of hypoxic pulmonary hypertension. J Clin Invest 103: 1509–1515, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Howell DC, Johns RH, Lasky JA, Shan B, Scotton CJ, Laurent GJ, Chambers RC. Absence of proteinase-activated receptor-1 signaling affords protection from bleomycin-induced lung inflammation and fibrosis. Am J Pathol 166: 1353–1365, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hsu YH, Chen CH, Hou CC, Sue YM, Cheng CY, Cheng TH, Lin H, Tsai WL, Chan P, Chen TH. Prostacyclin protects renal tubular cells from gentamicin-induced apoptosis via a PPARalpha-dependent pathway. Kidney Int 73: 578–587, 2008 [DOI] [PubMed] [Google Scholar]
- 9. Iskander K, Jaiswal AK. Quinone oxidoreductases in protection against myelogenous hyperplasia and benzene toxicity. Chem Biol Interact 153–154: 147–157, 2005 [DOI] [PubMed] [Google Scholar]
- 10. Jenkins RG, Su X, Su G, Scotton CJ, Camerer E, Laurent GJ, Davis GE, Chambers RC, Matthay MA, Sheppard D. Ligation of protease-activated receptor 1 enhances alpha(v)beta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J Clin Invest 116: 1606–1614, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lovgren AK, Jania LA, Hartney JM, Parsons KK, Audoly LP, Fitzgerald GA, Tilley SL, Koller BH. COX-2-derived prostacyclin protects against bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 291: L144–L156, 2006 [DOI] [PubMed] [Google Scholar]
- 12. Mercer PF, Johns RH, Scotton CJ, Krupiczojc MA, Konigshoff M, Howell DC, McAnulty RJ, Das A, Thorley AJ, Tetley TD, Eickelberg O, Chambers RC. Pulmonary epithelium is a prominent source of proteinase-activated receptor-1-inducible CCL2 in pulmonary fibrosis. Am J Respir Crit Care Med 179: 414–425, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Murakami S, Nagaya N, Itoh T, Kataoka M, Iwase T, Horio T, Miyahara Y, Sakai Y, Kangawa K, Kimura H. Prostacyclin agonist with thromboxane synthase inhibitory activity (ONO-1301) attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol 290: L59–L65, 2006 [DOI] [PubMed] [Google Scholar]
- 14. Muzaffar S, Shukla N, Lobo C, Angelini GD, Jeremy JY. Iloprost inhibits superoxide formation and gp91phox expression induced by the thromboxane A2 analog U46619, 8-isoprostane F2alpha, prostaglandin F2alpha, cytokines and endotoxin in the pig pulmonary artery. Br J Pharmacol 141: 488–496, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nana-Sinkam SP, Lee JD, Sotto-Santiago S, Stearman RS, Keith RL, Choudhury Q, Cool C, Parr J, Moore MD, Bull TM, Voelkel NF, Geraci MW. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. Am J Respir Crit Care Med 175: 676–685, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oury TD, Thakker K, Menache M, Chang LY, Crapo JD, Day BJ. Attenuation of bleomycin-induced pulmonary fibrosis by a catalytic antioxidant metalloporphyrin. Am J Respir Cell Mol Biol 25: 164–169, 2001 [DOI] [PubMed] [Google Scholar]
- 17. Pakrasi PL, Jain AK. Cyclooxygenase-2-derived endogenous prostacyclin reduces apoptosis and enhances embryo viability in mouse. Prostaglandins Leukot Essent Fatty Acids 79: 27–33, 2008 [DOI] [PubMed] [Google Scholar]
- 18. Robbins IM, Morrow JD, Christman BW. Oxidant stress but not thromboxane decreases with epoprostenol therapy. Free Radic Biol Med 38: 568–574, 2005 [DOI] [PubMed] [Google Scholar]
- 19. Ross D, Siegel D. NAD(P)H:quinone oxidoreductase 1 (Nqo 1, DT-diaphorase), functions and pharmacogenetics. Methods Enzymol 382: 115–144, 2004 [DOI] [PubMed] [Google Scholar]
- 20. Rubenfeld GD, Herridge MS. Epidemiology and outcomes of acute lung injury. Chest 131: 554–562, 2007 [DOI] [PubMed] [Google Scholar]
- 21. Scotton CJ, Krupiczojc MA, Konigshoff M, Mercer PF, Lee YC, Kaminski N, Morser J, Post JM, Maher TM, Nicholson AG, Moffatt JD, Laurent GJ, Derian CK, Eickelberg O, Chambers RC. Increased local expression of coagulation factor X contributes to the fibrotic response in human and murine lung injury. J Clin Invest 119: 2550–2563, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Siegel D, Gustafson DL, Dehn DL, Han JY, Boonchoong P, Berliner LJ, Ross D. NAD(P)H:quinone oxidoreductase 1: role as a superoxide scavenger. Mol Pharmacol 65: 1238–1247, 2004 [DOI] [PubMed] [Google Scholar]
- 23. Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, Sherrill TP, Plieth D, Neilson EG, Blackwell TS, Lawson WE. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med 180: 657–665, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Voynow JA, Fischer BM, Zheng S, Potts EN, Grover AR, Jaiswal AK, Ghio AJ, Foster WM. NAD(P)H quinone oxidoreductase 1 is essential for ozone-induced oxidative stress in mice and humans. Am J Respir Cell Mol Biol 41: 107–113, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 25. Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med 27: 337–349, 2006 [DOI] [PubMed] [Google Scholar]
- 26. Yamagishi S, Amano S, Inagaki Y, Okamoto T, Takeuchi M, Makita Z. Beraprost sodium, a prostaglandin I2 analog, protects against advanced gycation end products-induced injury in cultured retinal pericytes. Mol Med 8: 546–550, 2002 [PMC free article] [PubMed] [Google Scholar]
- 27. Yano T, Itoh Y, Kubota T, Sendo T, Koyama T, Fujita T, Saeki K, Yuo A, Oishi R. A prostacyclin analog prevents radiocontrast nephropathy via phosphorylation of cyclic AMP response element binding protein. Am J Pathol 166: 1333–1342, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou W, Blackwell TS, Goleniewska K, O'Neal JF, Fitzgerald GA, Lucitt M, Breyer RM, Peebles RS., Jr Prostaglandin I2 analogs inhibit Th1 and Th2 effector cytokine production by CD4 T cells. J Leukoc Biol 81: 809–817, 2007 [DOI] [PubMed] [Google Scholar]
- 29. Zhou W, Hashimoto K, Goleniewska K, O'Neal JF, Ji S, Blackwell TS, Fitzgerald GA, Egan KM, Geraci MW, Peebles RS., Jr Prostaglandin I2 analogs inhibit proinflammatory cytokine production and T cell stimulatory function of dendritic cells. J Immunol 178: 702–710, 2007 [DOI] [PubMed] [Google Scholar]