

Abstract
Emerging evidence suggests that renal endothelial function may be altered in ischemia-reperfusion injury. Acute kidney injury is sexually dimorphic, and estrogen protects renal tubular function after experimental ischemic injury. This study tested the hypothesis that during ischemia-reperfusion, estrogen alters glomerular endothelial function to prevent hyperpermeability. Glomerular endothelial cells were exposed to 8-h oxygen-glucose deprivation (OGD) followed by 4- and 8-h reoxygenation-glucose repletion. After 4-h reoxygenation-glucose repletion, transendothelial permeability to Ficoll-70 was reduced, and transendothelial resistance increased, by 17β-estradiol vs. vehicle treatment during OGD (OGD-vehicle: 91.0 ± 11.8%, OGD-estrogen: 102.6 ± 10.8%, P < 0.05). This effect was reversed by coadministration of G protein-coupled receptor 30 (GPR30) antagonist G15 with 17β-estradiol (OGD-estrogen-G15: 89.5 ± 6.9, P < 0.05 compared with 17β-estradiol). To provide preliminary confirmation of this result in vivo, Ficoll-70 was administered to mice 24 h after cardiac arrest and cardiopulmonary resuscitation (CA/CPR). Blood urea nitrogen (BUN) and serum creatinine (SCr) in these mice were elevated within 12 h following CA/CPR and reduced at 24 h by pretreatment with 17β-estradiol (BUN/SCr 17β-estradiol: 34 ± 19/0.2 ± 0.1 vehicle: 92 ± 49/0.5 ± 0.3, n = 8–12, P < 0.05). Glomerular sieving of Ficoll 70 was increased by CA/CPR within 2 h of injury and 17β-estradiol treatment (θ; 17β-estradiol: 0.74 ± 0.26 vs. vehicle: 1.05 ± 0.53, n = 14–15, P < 0.05). These results suggest that estrogen reduces postischemic glomerular endothelial hyperpermeability at least in part through GPR30 and that estrogen may regulate post CA/CPR glomerular permeability in a similar fashion in vivo.
Keywords: ischemia-reperfusion injury, renal ischemia, acute renal failure, acute kidney injury, oxygen-glucose deprivation, cardiac arrest, cardiopulmonary resuscitation, gender, sex, estrogen, estradiol
acute kidney injury (AKI) occurs in 10–30% of intensive care unit patients. There are few therapeutic options for this disease, which has extremely high mortality (up to 70%) and morbidity, with 10% of AKI patients progressing to dialysis-dependent end-stage renal disease (8, 23, 26, 45, 46). AKI is a sexually dimorphic disease, with both incidence and mortality being lower in women compared with men (15, 30, 49). Experimental models recapitulate the sexually dimorphic nature of AKI and implicate sex steroids in the sex differences observed, demonstrating that androgens increase and estrogens decrease kidney damage in animal models. Several studies have characterized the effects of sex steroids on AKI in animal models and strongly suggested a protective effect of estrogen, but a detailed understanding of the mechanism of this protection is lacking. Therefore, the current study aims at furthering our understanding of the beneficial effects of estrogen (18, 19, 31, 32, 39, 44).
Ischemic injury to the kidney results in injured renal epithelial tubular cells, causing functional renal failure in AKI. While tubular cell death is the hallmark of AKI, there is emerging evidence that renal ischemia alters endothelial cell function. Interestingly, the mechanism and cell type that mediate estrogen renoprotection remains unknown. There is evidence that estrogen acts directly on tubular epithelial cells via cytoskeletal and protective mechanisms (48). In addition, however, estrogen-mediated regulation of the endothelium is well described (10, 13, 34), and endothelial function is altered in renal ischemia (4, 5). For example, multiple investigators have shown that estrogen or female sex attenuates endothelin-1 [which reduces glomerular filtration rate and renal plasma flow (22) production after renal ischemia (28, 42, 44)]. We previously found that estrogen's renoprotective effect in vivo is not mediated by classic estrogen receptors (ER) α or β (17). We therefore hypothesized that during ischemia-reperfusion injury, estrogen alters the function of glomerular endothelium to prevent hyperpermeability and that in vitro this effect might be mediated through a third estrogen receptor, G protein-coupled receptor 30 (GPR30). To test this hypothesis, we developed and utilized a novel in vitro model employing cultured glomerular endothelial cells, and a novel in vivo model using macromolecular sieving to assess glomerular permeability following ischemia-reperfusion.
MATERIALS AND METHODS
Glomerular endothelial cell culture.
This conditionally immortalized cell line (1) was developed in the H-2Kb-tsA58 immortomouse by Dr. Michael Madaio, Temple University and was a kind gift of Dr. H. Abboud, University of Texas, San Antonio. Glomerular endothelial cells (gENCs) were cultured either under “standard” conditions or membrane integrity-enhancing conditions. Standard media consisted of DMEM with 10% fetal bovine serum and Ham's F12 (GIBCO, Invitrogen, Carlsbad, CA). Supplemented media was EGM2-MV (Lonza, Allendale, NJ). This proprietary media contains 5% fetal bovine serum and the supplements human fibroblast growth factor-2, vascular endothelial growth factor, R3-insulin-like growth factor-1, human endothelial growth factor, hydrocortisone, ascorbic acid, and gentamicin-amphotericin. After characterization and optimization of culture conditions for these experiments, gENCs were grown to confluence on fibronectin-coated (BD, San Jose, CA) membrane supports with 0.4-μm pores (Transwell, Corning, Corning NY) in supplemented media. Once confluent, media was changed daily, and transendothelial resistance (TEER) was measured daily. When the transendothelial resistance was >19, cells were considered ready for experimental use. All results reported represent repeated experiments.
RT-PCR.
gENCs were grown to 80% confluence. Media was removed, and cells were rinsed twice with sterile phosphate-buffered saline, then removed from culture dishes using RNA lysis buffer (Lysis Solution, Ambion, Austin, TX) and manual scraping. RNA was then isolated using the RNAqueous®-Micro Kit (Ambion) and further treated with DNase I, per the manufacturer's instructions. First-strand cDNA was reverse transcribed from 2 μg total RNA with a High Capacity cDNA archive Kit (Applied Biosystems, Foster City, CA), and PCR was then carried out using GoTaq DNA Polymerase (Promega, Madison WI). Primers were designed using National Institutes of Health Primer BLAST online software. Parameters for PCR were as follows: denaturing at 95°C for 2 min followed by 35 cycles of denaturation at 95°C for 30 s, annealing at 48°C for 1 min, and extension at 72°C for 1 min, with a final extension phase of 5 min at 72°C for 5 min. Twenty microliters of each sample were then loaded into a 1% agarose gel preloaded with ethidium bromide at 0.5 μg/ml, and 85 V were applied across the gel for 1 h. Gels were then imaged using a Typhoon 9400 scanner (GE Life Sciences, Piscataway, NJ). For each transcript, replicate PCR was performed on three separate samples collected from noncontemporaneous cultures. RNA extracted from female C57BL/6 mouse renal homogenate was used as a positive control, with the exception of ERβ, which produced a weak signal in the renal homogenate. Positive control for ERβ was RNA extracted from the uterine homogenate from C57BL/6 mice. The primers are described in Table 1.
Table 1.
Gene sequence identifiers and primers used for characterization of glomerular endothelial cells by RT-PCR
| Marker | RefSeq Gene ID | Forward Primer (5′→3′) | Reverse Primer (5′→3′) |
|---|---|---|---|
| CD102 | NM_010494 | CCAGGCTGAGCTGGATCTAC | GTGCTGGCCAAAGATAAAGC |
| CD31 | NM_008816.2 | TGCAGGAGTCCTTCTCCACT | ACGGTTTGATTCCACTTTGC |
| CD34 | NM_001111059.1 | GATGGCTGTTGGGAAGAAAA | ATGCCACTTTCCTGCATACC |
| EPCAM-1 | NM_008532.2 | TGACTCACAGCAAGTCTGGG | CCCTCCTCAGTTCAGCACTC |
| S100a | NM_011311.1 | CTTGGTCTGGTCTCAACGGT | ATGTGCGAAGAAGCCAGAGT |
| ERα | NM_029771.2 | ACGTTGTGCCCCTCTATGAC | CGCTTTGTCAACGACTTCAA |
| ERβ | NM_007956.4 | CTTTTGAAAGGCACTCTCGG | ACGTACTTTCCTCGGTCCCT |
| GPR30 | NM_010157.3 | CCCTTGACAGGCCACATAGT | GAGCTGGACAGTCAGGAAGG |
ER, estrogen receptor; GPR30, G protein-coupled receptor 30.
Preparation of fluorescent-conjugated Ficoll-70 solutions.
Ficoll-70 conjugated to tetramethyl rhodamine isothiocyanate (TRITC-Ficoll-70) or FITC-Ficoll-70 was purchased (TdB Consultancy, Uppsala, Sweden) and diluted in 0.9% sodium chloride solution which had been heated to 90°C. After cooling, the FITC- or TRITC-Ficoll-70 solution was dialyzed for 24 h in a 2,000-Dalton molecular mass cut-off dialysis cassette (Slide-a-lyzer, Pierce, Rockford, IL) to remove unbound fluorescent molecules. The solution was then filtered through a 0.22-μm filter (Millipore, Billerica, MA) to remove microbial and other contaminants. Concentration was then measured against a prederived standard curve on a plate reader (VICTOR 3, PerkinElmer, Waltham, MA) using excitation and emission wavelengths of 405 and 535 nM (for FITC) or 560 and 572 nM (for TRITC), respectively. Fluorescent tracers were protected from light during all experimental manipulations.
TEER measurements.
An endothelial microvolt-ohmmeter and concentric-ring electrode system (Evohm, Endohm, World Precision Instruments, Sarasota FL) were used to measure TEER. Measurements were performed to assess readiness for experimentation, immediately before oxygen-glucose deprivation (OGD) and after the specified reoxygenation-glucose repletion (RGR) period. After the cell support was placed in the measurement chamber, readings were allowed to stabilize for 10 s before recording. The resistance of a cell-free insert treated in parallel with cell-bearing inserts was subtracted from each measurement, and the resulting value was multiplied by the surface area of the membrane support to obtain TEER in Ω·cm2. This was then normalized to the baseline value for the same well and expressed as the percentage of baseline.
In vitro measurement of FITC-Ficoll flux.
At the specified time points, 50 μl of the media from the lower chamber of the membrane support system was aspirated and placed in one well of a 96-well cell culture plate. Fifty microliters of 0.5 M HEPES was then added to the sample to equalize pH between samples, as FITC fluorescence is pH dependent. FITC-Ficoll-70 concentration was then measured using a VICTOR3 plate reader (PerkinElmer) with excitation and emission wavelengths of 405 and 535 nM, respectively. A premeasured standard curve was used to determine concentration. Concentration was then multiplied by the area of the membrane support and divided by the time in hours to determine flux.
OGD.
Media above and below prepared monolayers on membrane supports was replaced with either growth media with 0.05% DMSO (normoxic control), serum- and glucose-free DMEM (Invitrogen) with 0.05% DMSO, serum- and glucose-free DMEM with 0.05% DMSO and 100 nM 17β-estradiol, or serum- and glucose-free DMEM with 0.05% DMSO, 1000 nM G15, and 100 nM 17β-estradiol. Cells were then placed in either of two environments for 8 h: normoxic control, 37°C incubator with 21% oxygen and 5% carbon dioxide; or OGD groups, 37°C anoxia chamber (COY Laboratory Products, Grass Lake, MI) filled with an anoxic gas mixture (5% CO2-5% H2-90% N2). Oxygen concentration was maintained at 0 parts per million (PPM) using a palladium catalyst. Anoxic conditions were monitored continuously with an oxygen monitor (COY Laboratory Products) placed inside the chamber. After 8 h, cells were removed from the chamber and media was changed to phenol red-free DMEM with 5% fetal bovine serum and 5.5 mM glucose. Four micrograms FITC-Ficoll-70 was added to the upper chamber of the membrane support. Cells were then housed in normoxic conditions until TEER and Ficoll concentrations were measured at 4 h of RGR and again at 8 h of RGR.
Animals and experimental groups.
This study was conducted in accordance with the National Institutes of Health guidelines for the care and use of animals in research, and all animal protocols were approved by the Oregon Health and Science University Institutional Animal Care and Use Committee. C57BL/6 mice (6- to 8-wk-old female, Charles River Laboratories, Boston, MA) were randomized to vehicle or 17-β-estradiol treatment groups and subsequently underwent experimental procedures.
Ovariectomy and 17β-estradiol treatment.
Under isoflurane anesthesia, female mice were ovariectomized 7 days before cardiac arrest/cardiopulmonary resuscitation (CA/CPR) to allow endogenous sex steroids to decrease. At the time of ovariectomy, all mice underwent subcutaneous placement of either sesame oil (vehicle)- or 17β-estradiol-containing (6.3 μg total dose) silastic implants. We previously demonstrated that this dose of 17β-estradiol reproducibly produces physiologic estradiol levels (3). All surgeons performing the ovariectomy in our laboratory have previously demonstrated proficiency measured by post-procedure serum estradiol levels.
In vivo whole-body ischemia-reperfusion (CA/CPR).
We performed normothermic CA/CPR as previously described (17, 18, 20). Briefly, after weighing, general anesthesia was induced with 4% isoflurane in a 2:1 air:oxygen mixture and then maintained with 1–2% isoflurane. Animals were then placed in a supine position on a warming pad, under a warming lamp. A rectal temperature probe was inserted. The pad and lamp were controlled by a proportional-integral-derivative algorithm-controlled temperature controller (Digi-Sense, Cole Parmer, Vernon Hills, IL) set to 37.0°C, and animal temperature was maintained between 36.5 and 37.5°C. After tracheal intubation with a 22-ga teflon catheter (InSyte-W, BD, Franklin, NJ), animals were mechanically ventilated and subcutaneous electrocardiography (EKG) electrodes were placed and secured. A PE-10 catheter was placed in the right jugular vein. After a 5-min equilibration period, cardiac arrest (CA) was induced with 40 μl of 0.5 M potassium chloride intravenously. CA was confirmed by isoelectric EKG signal and absence of visible cardiac contraction on the chest wall. Isoflurane and mechanical ventilation were discontinued, and the endotracheal tube was disconnected. At 7.5 min after the onset of CA, the endotracheal tube was reconnected and mechanical ventilation was resumed using 100% oxygen. At 8 min after the onset of CA, chest compressions were initiated at a rate of 300/min, measured by motion artifact on the EKG. Epinephrine, 5–16 μg in 0.9% sodium chloride solution (0.5–1.0 ml), was administered intravenously. Return of spontaneous circulation (ROSC) was confirmed by EKG and visible cardiac contractions on the chest wall. The jugular catheter was then removed, and hemostasis was obtained. The trachea was extubated when spontaneous respiratory rate was >60 breaths/min. Animals were then placed in a recovery cage, which was placed on a warming pad controlled at 37°C. Sham-treated animals underwent general anesthesia as described, with endotracheal intubation, mechanical ventilation, and incision over the jugular vein but no catheter placement.
In vivo measurement of transglomerular sieving of Ficoll-70.
At specified time points following CA/CPR, animals underwent induction of general anesthesia with isoflurane, followed by placement of a PE-10 catheter in the jugular vein. Then, 0.25 mg of bumetanide and 100 μg of TRITC-conjugated Ficoll-70 diluted in 1.5 ml 0.9% sodium chloride were administered via the jugular catheter. At 80 min after bumetanide/TRITC-Ficoll-70 administration, mice were euthanized by transcardial exsanguination and the bladder was completely aspirated under direct vision via a midline laparotomy. Blood samples were collected and serum-separated by centrifugation for serum TRITC fluorescence measurement. Urine volume was recorded, and TRITC fluorescence of both serum and urine was measured on a plate reader as described below.
Evaluation of functional renal outcomes.
Because of the limited blood volume of the mouse and the requirement for lithium heparin-preserved whole-blood samples, a separate cohort of animals was used for these measurements. Following acquisition of urine samples 24 h after CA/CPR, and immediately after euthanasia, a clamshell thoracotomy was performed and blood (100–500 μl) was drawn from the apex of the left ventricle. Blood samples were placed in lithium heparin tubes. A point-of-care enzyme-coupled analyzer (Abaxis Medical Diagnostics, Union City, CA) was used to measure blood urea nitrogen (BUN) and serum creatinine (SCr). This device was chosen because it employs a creatinine amidohydrolase catalyzed assay, which is not subject to artifactual elevation of creatinine caused by chromogens in mouse urine (21, 27).
Data analysis.
Analysis was performed with Prism 5.0 software (GraphPad Software, LaJolla CA). Treatment comparisons involving three or more groups were performed using one-way ANOVA. Posttest comparisons between individual groups within multiple group comparisons were performed using Bonferroni's algorithm. Two group comparisons for scalar values were performed using the unpaired t-test, with two-tailed P values. Two group comparisons for proportions (survival) were performed using Fisher's exact test. The Spearman correlation was performed to assess agreement between transendothelial resistance measurements and Ficoll flux. All data are shown as means ± SD. Statistical significance was inferred if the P value was <0.05.
RESULTS
gENCs express endothelial markers, ERα, ERβ, and GPR30.
We tested expression of endothelial-specific markers to verify that the gENC cell line retained endothelial specificity and expressed important molecules for estrogen signaling. Figure 1 illustrates that we observed mRNA for endothelial markers CD102, CD31, and CD34 in samples of cultured gENCs. Importantly, epithelial cell marker EPCAM-1 and fibroblast marker s100a were not expressed. ERα, ERβ, and GPR30 are expressed in this cell line.
Fig. 1.
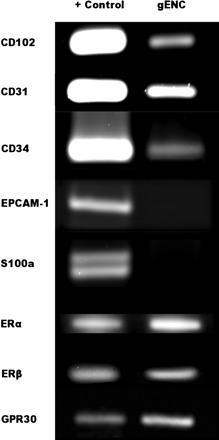
RT-PCR for cell markers on glomerular endothelial cells (gENCs). gENCs express endothelial cell markers CD102, CD31, and CD34. They do not express epithelial cell markers S100a and EPCAM -1. All 3 known estrogen receptors (ER), ERα, ERβ, and G protein-coupled receptor 30 (GPR30), are expressed on gENCs.
Supplemented media and fibronectin coating increase membrane integrity.
To determine ideal culture conditions for testing membrane integrity, gENCs were cultured using standard media with and without fibronectin surface treatment, or supplemented media with fibronectin surface treatment. We found that fibronectin substrate coating combined with supplemented media significantly enhanced endothelial membrane integrity as measured by TEER 4 days postconfluence (standard media alone 6.0 ± 0.53 Ω·cm2, standard media with fibronectin substrate 6.0 ± 0.0 Ω·cm2, supplemented media with fibronectin substrate 15.2 ± 2.17, P < 0.05, n = 8, 2, and 5, respectively).
Estrogen ameliorates OGD-induced endothelial monolayer injury in vitro.
OGD caused a significant decrease in gENC monolayer stability, as measured by TEER. gENC monolayers were exposed to 12-h OGD, and TEER was measured 4 h after RGR. OGD caused a significant decrease in resistance, decreasing TEER by 30% at 4 h. Glomerular endothelial monolayers treated with estrogen during OGD exposure were protected from OGD-induced decrease in TEER and were not significantly different from normoxic monolayers at the 4-h RGR time point. This effect was reversed by the coadministration of estrogen receptor GPR30 antagonist G15 with estrogen (Fig. 2). At 8 h of RGR, however, the effect of estrogen exposure during OGD was no longer evident, and estrogen-exposed monolayers were no longer significantly protected relative to vehicle (Fig. 3). Similarly, OGD caused increased gENC monolayer permeability to Ficoll flux. Figure 4 illustrates that 12-h OGD and 4-h RGR resulted in a significant increase in transendothelial Ficoll flux. Estrogen prevented OGD-induced hyperpermeability, reducing Ficoll flux to the level observed in normoxic control conditions. The protective effect of estrogen was reduced by the coadministration of G15, although the reversal of effect was nonsignificant. The estrogen effect was reversed at the 8-h time point, as seen in Fig. 5. Overall, there was significant correlation between endothelial permeability as measured by Ficoll flux and that measured by TEER (r = −0.3, P < 0.05, not shown).
Fig. 2.

Transendothelial resistance after 8-h oxygen-glucose deprivation (OGD) and 4-h reoxygenation-glucose repletion (RGR). Cells were treated with vehicle (VEH), 100 nM 17β-estradiol (EST), or 100 nM 17β-estradiol (EST) and 1,000 nM G15 (a selective GPR30 antagonist). Control cells were exposed to normoxic conditions in vehicle (Normox). Drug treatments were removed following OGD. Estrogen-treated cell monolayers exhibited significantly more electrical resistance than those treated with vehicle and were not significantly different from non-OGD exposed cells. This protective effect was reversed by the addition of G15. NS, not significant. Values are means ± SD.
Fig. 3.

Transendothelial resistance after 8-h OGD and 8-h RGR. Cells were treated with vehicle, 100 nM 17β-estradiol, or 100 nM 17β-estradiol and 1,000 nM G15 (a selective GPR30 antagonist). Control cells were exposed to normoxic conditions in vehicle (Normox). Drug treatments were removed following OGD. At the 8-h time point, the protective effect of estrogen was lost. Drug-treated cells were not different from vehicle-treated cells, and all OGD-exposed cell monolayers exhibited reduced electrical resistance compared with control (Normox) cell monolayers. Values are means ± SD.
Fig. 4.
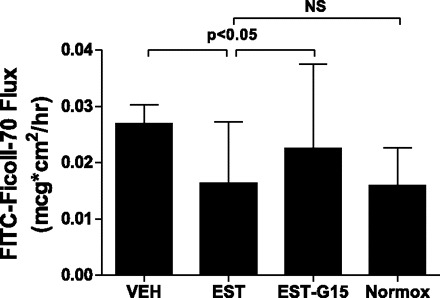
Transendothelial flux of FITC-labeled Ficoll-70 after 8-h OGD and 4-h RGR. Cells were treated with vehicle, 100 nM 17β-estradiol, or 100 nM 17β-estradiol and 1,000 nM G15 (a selective GPR30 antagonist). Control cells were exposed to normoxic conditions in vehicle. Drug treatments were removed following OGD. Estrogen-treated cell monolayers transited significantly less FITC-Ficoll-70 than those treated with vehicle and were not significantly different from non-OGD exposed cells. This protective effect was reversed by the addition of G15. Values are means ± SD.
Fig. 5.
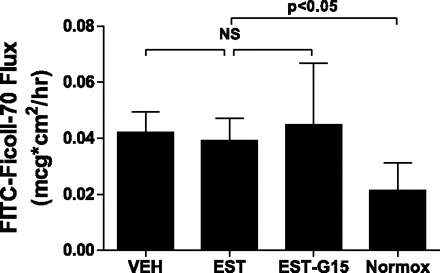
Transendothelial flux of FITC-Ficoll-70 after 8-h OGD and 8-h RGR. Cells were treated with vehicle, 100 nM 17β-estradiol, or 100 nM 17β-estradiol and 1,000 nM G15 (a selective GPR30 antagonist). Control cells were exposed to normoxic conditions in vehicle. Drug treatments were removed following OGD. At the 8-h time point, the protective effect of estrogen was lost. Drug-treated cells were not different from vehicle-treated cells, and all OGD-exposed cell monolayers transited more FITC-Ficoll-70 compared with control (Normox) cell monolayers. Values are means ± SD.
CA/CPR induces renal functional injury.
To determine the time course of renal functional injury after CA/CPR, we measured serum blood urea nitrogen and creatinine in sham-exposed mice and mice subjected to CA/CPR and 2, 6, 12, 18, 24, or 48 h of recovery. Figure 6 illustrates that peak serum markers (blood urea nitrogen 91 ± 68 mg/dl, creatinine 0.5 ± 0.4 mg/dl, n = 5–7/group, P < 0.05 with respect to sham) occurred at 12 h after CA/CPR, with a plateau of both blood urea nitrogen for all time points measured thereafter (18, 24, and 48 h not significantly different from 12 h, n = 7/group).
Fig. 6.

Serum creatinine and blood urea nitrogen (BUN) following sham or cardiac arrest/cardiopulmonary resuscitation (CA/CPR) surgery. After sham surgery (time 0) or 2, 6, 12, 18, 24, or 48 h after CA/CPR, separate cohorts of mice were assessed for serum content of creatinine and BUN. Values peaked and were significantly elevated relative to sham (P < 0.05, n = 5–7/group) at 12 h post-CA/CPR.
Glomerular sieving of macromolecules is increased 2 h after CA/CPR.
To determine the effect of CA/CPR and that of ablation of physiological estrogen on glomerular sieving of the macromolecule Ficoll-70, we measured the glomerular sieving coefficient (the urine/serum ratio of TRITC-Ficoll-70 concentration; θFicoll-70) in CA/CPR naive, gonadally intact or ovariectomized female mice. Figure 7 illustrates that θFicoll-70 was not affected by ovariectomy (0.47 ± 0.16 in CA/CPR-naive, gonadally intact mice, 0.53 ± 0.15 in CA/CPR-naive, ovariectomized mice, n = 8/group, P > 0.05). Following this, we measured θFicoll-70 in ovariectomized mice at 2, 6, 12, and 24 h after CA/CPR. Two hours following CA/CPR, θFicoll-70 was significantly elevated relative to baseline (0.71 ± 0.24, n = 10, P < 0.05). By 6 h following CA/CPR, θFicoll-70 returned to baseline (0.48 ± 0.11, n = 10, P > 0.05) and remained similar to baseline thereafter (12 h: 0.50 ± 0.15, n = 7, 24 h: 0.48 ± 0.21, n = 6 respectively, P > 0.05).
Fig. 7.

Glomerular sieving coefficient for Ficoll-70 (θFicoll-70) in gonadally intact CA/CPR-naive mice (Naive-INT), ovariectomized CA/CPR-naive mice (Naive-OVX), and CA/CPR-treated mice at 2, 6, 12, and 24 h following arrest and resuscitation. At the specified time point, mice were reanesthetized and administered fluorescent-labeled Ficoll-70, bumetanide, and intravenous 0.9% sodium chloride solution. Eighty minutes later, the mice were euthanized and blood and serum fluorescence were evaluated to determine Ficoll-70 concentration. Removal of physiological estrogen by ovariectomy did not alter θFicoll-70. Twenty-four hours following CA/CPR, θFicoll-70 was significantly elevated, but then rapidly declined to values similar to those found in CA/CPR-naive animals. Numbers in bars are n.
Estrogen is renoprotective and alters glomerular sieving of macromolecules after CA/CPR in vivo.
To confirm the renoprotective effect of estrogen in the intact animal, we performed CA/CPR in female mice following ovariectomy and estrogen replacement (compared with ovariectomy without estrogen replacement). Intravenous injection of potassium chloride led to immediate asystolic cardiac arrest in all mice. Total ischemia time, epinephrine dose, epinephrine dose indexed to weight (not shown), and survival rate were not different between ovariectomized and ovariectomized+17β-estradiol-treated groups (Table 2). In a single cohort, we measured serum functional markers at 24 h after CA/CPR. CA/CPR resulted in significant renal injury 24 h following insult, indicated by significant increase in blood urea nitrogen and serum creatinine. Figure 8, A and B, illustrates that serum from 17β-estradiol-replaced mice contained significantly less urea nitrogen and creatinine compared with ovariectomized, vehicle-treated mice, consistent with our previous observation of estrogen protection of renal function following ischemia (P < 0.05, n = 8–12/group) (16, 18, 20). TRITC-labeled Ficoll was administered to a separate cohort of mice at 2 or 24 h following CA/CPR to assess the effect of ischemia on transglomerular permeability. Consistent with our in vitro data, Fig. 9, A and B, illustrates that estrogen significantly reduced θFicoll-70 in the early postinjury phase at 2 h (vehicle 1.1 ± 0.5, n = 14, 17β-estradiol 0.7 ± 0.3, n = 15, P < 0.05), but the effect was no longer significant at 24 h post-insult (vehicle 1.1 ± 0.9, n = 16, 17β-estradiol 0.8 ± 0.4, n = 17, P > 0.05).
Table 2.
Baseline weight, metabolic, and resuscitation data for animals subjected to CA/CPR
| Naive/NT (n = 8) | Naive/OVX (n = 8) | Vehicle, 2 h (n = 14) | Estrogen, 2 h (n = 15) | P | Vehicle, 24 h (n = 16) | Estrogen, 24 h (n = 17) | P | |
|---|---|---|---|---|---|---|---|---|
| Prearrest weight, g | N/A | N/A | 21.6 ± 1.2 | 21.7 ± 1.6 | 0.94 | 22.0 ± 0.9 | 22.1 ± 1.0 | 0.88 |
| Time to ROSC, s | N/A | N/A | 83 ± 31 | 83 ± 30 | 0.95 | 113 ± 37 | 89 ± 33 | 0.06 |
| Epinephrine dose, μg | N/A | N/A | 8 ± 8 | 8 ± 2 | 0.75 | 10 ± 3 | 10 ± 2 | 0.60 |
| Survival, % | 100 | 100 | 88 | 100 | 0.49 | 81 | 90 | 0.66 |
| Urine output, ml | 0.16 ± 0.12 | 0.26 ± 0.11 | 0.19 ± 0.12 | 0.30 ± 0.16 | >0.05 | 0.32 ± 0.14 | 0.47 ± 0.18 | <0.05 |
Values are means ± SD. CA/CPR, cardiac arrest/cardiopulmonary resuscitation; Naïve-NT, gonadally intact CA/CPR-naive mice; Naïve-OVX, ovariectomized CA/CPR-naive mice; ROSC, return of spontaneous circulation. Resuscitation parameters were not different between groups. Urine output was greater in estrogen-treated animals at 24 h post-CA/CPR.
Fig. 8.
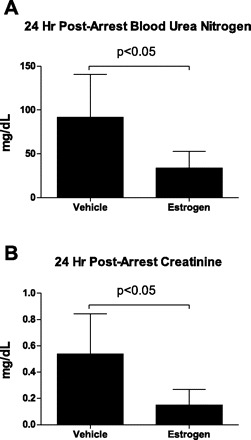
Serum BUN (A) and creatinine (B) were measured 24 h following CA/CPR and were significantly lower in estrogen-treated animals than in controls (P < 0.05, n = 8–12/group).
Fig. 9.
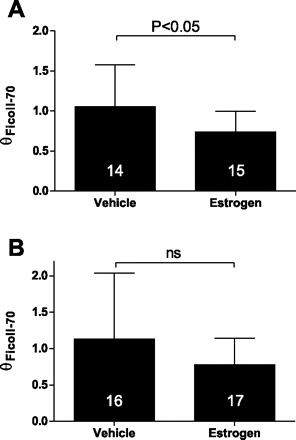
θFicoll-70 in vehicle- and estrogen-treated animals 2 h (A) and 24 h (B) following CA/CPR. At the specified time point, mice were reanesthetized and administered fluorescent-labeled Ficoll-70, bumetanide, and intravenous 0.9% sodium chloride solution. Eighty minutes later, the mice were euthanized and blood and serum fluorescence were evaluated to determine Ficoll-70 concentration. Administration of 17β-estradiol (estrogen) significantly reduced glomerular sieving of Ficoll-70 at the 2-h, but not the 24-h post-CA/CPR time point. Numbers in bars are n.
DISCUSSION
This study provides novel evidence that estrogen reduces glomerular endothelial permeability following ischemia-reperfusion injury. In vitro, macromolecular flux was attenuated, and TEER was augmented by transient estrogen exposure at physiological concentration. This effect was reversed in vitro by coadministration of the GPR30 antagonist g15 and estrogen. In vivo, administration of estrogen to mice reduced glomerular sieving of the macromolecule Ficoll-70. Our in vivo findings that estrogen protects renal tubular function as measured by blood urea nitrogen, serum creatinine, and urine output are in line with prior studies (18, 19, 31, 32, 39, 44). In addition, we describe barrier-optimizing culture conditions and report for the first time the expression of ERα, ERβ, and GPR30 mRNA in cultured glomerular endothelial cells. The novel pairing of in vitro and in vivo glomerular functional modeling offers a potentially powerful set of tools for further investigation. Overall, our findings suggest that estrogen reduces the permeability of ischemia-reperfusion-injured glomerular endothelium and that this effect may occur via the novel estrogen receptor GPR30.
The failure to translate putative renoprotective agents to the clinical setting suggests that alternative mechanisms may need to be explored (38). While it is clear that estrogen exerts a renoprotective effect, the mechanism of this effect remains unclear. Estrogen is a pleiotropic molecule with effects which might be undesirable in a therapeutic context (for example, increased incidence of thromboembolism) (47), particularly in the critically ill patient. As selective ER modulators are available, understanding which receptors are involved in our observed effect is important. Previously, we found that in vivo, ERα and ERβ were not involved in the renoprotective effect after CA/CPR (18). We therefore hypothesized that GPR30, a G protein-coupled ER, might mediate estrogen's effect. Available data suggest it is possible that estrogen mediates endothelial function through GPR30 (14, 24). To elucidate the role of GPR30, we confirmed that it was expressed in glomerular endothelial cells, and then coadministered estrogen with the GPR30-specific antagonist G15 in our cell culture model. G15 reversed the protective effect of estrogen in vitro. This finding suggests that estrogen reduces the permeability of OGD-exposed glomerular endothelial cells, at least in part through GPR30. The current study did not assess the effect of other ERs; therefore, it remains possible that GPR30 is not the only contributor to estrogen-mediated protection of renal endothelial cells. We did not assess the effect of other receptor antagonists. It is possible that nonspecific binding or a mixed agonist/antagonist effect of G15 confounds this finding. However, G15 is a highly specific antagonist with no apparent agonist activity in vivo (11). Nonetheless, our data suggest that specific GPR30 agonists may provide a new therapeutic strategy to prevent ischemia-induced endothelial hyperpermeability and improve renal function.
Investigators have focused on the tubular epithelium as it is these cells which die and directly impair renal function after ischemic injury. However, evidence supports the idea that glomerular barrier injury (of which the endothelium is an important part) might precede or induce tubular dysfunction (reviewed in Ref. 35). Sutton et al. (43) demonstrated endothelial injury, cytoskeletal alteration, impaired cell-cell adhesion, and leakage of dextran from the renal microvasculature into the interstitium after bilateral renal pedicle occlusion (43). Using a rat renal pedicle occlusion model, Rippe et al. (37) reported that renal permeability to Ficoll was increased by 60 min of unilateral renal ischemia, primarily large (>55 Å) Ficoll molecules. However, Andersson et al. (2) found damage to both the size and charge barriers after mild (15 min) ischemia in isolated, perfused rat kidneys without morphological change as assessed by electron microscopy, suggesting that the glycocalyx (an endothelium-derived structure not imaged by electron microscopy) might be the source of barrier damage from ischemia. Because the glomerular endothelium is both sero- and uroluminally upstream from the tubular epithelium, it is uniquely positioned to affect tubular function once it is compromised by ischemia-reperfusion. Together with the emerging data on renal endothelium in ischemia, this observation prompted our hypothesis that estrogen protected renal function following ischemia via improved glomerular endothelial function. To test this hypothesis, we used paired in vitro and in vivo models and complementary techniques to measure permeability: TEER (40) and transendothelial flux of the macromolecule Ficoll (29, 40). Transient estrogen exposure reduced the permeability of glomerular endothelial monolayers after 8-h OGD and 4-h RGR. OGD is a limited model of ischemia-reperfusion. Cells in culture may be phenotypically different from the same cells in vivo, and the challenge of OGD (unlike ischemia) is that cells in the culture system are under limited, if any, physiological demand. For example, in preliminary experiments, we found that it took 24-h OGD and 24-h RGR to produce 75% cell death in the glomerular endothelial cell culture model, yet endothelial apoptosis is evident in the systemic circulation within 3 h of cardiac arrest in humans (12). Because of this disparity, we also used the CA/CPR model to evaluate estrogen's effect on glomerular function.
The loss of a protective effect at the 8-h RGR time point could suggest that estrogen's effect is transient. It is also possible, however, that cell proliferation after OGD in all treatments obscured the differences that were visible at the 4-h RGR time point. Although increased TEER in all treatments at 8 h suggests this interpretation, Ficoll flux also increased across treatments at 8-h compared with 4-h RGR. Understanding these effects would be important to translational investigation, and we are focusing further experiments on this question.
CA/CPR is an established model of renal ischemia-reperfusion injury (7, 17, 18, 20). Although the most extensive in vivo findings about renal ischemia have been derived from focal renal pedicle occlusion models, most human AKI occurs after whole body ischemia-reperfusion (25, 46). CA/CPR is a titratable and reliable inducer of renal injury which functionally and histologically resembles human prerenal AKI, although it is limited in that in the murine model it is not possible to exclude the effects of ischemic distant organs on renal injury. Our finding that estrogen protects tubular function is in line with previous work in both pedicle occlusion and CA/CPR (18, 28, 32, 44). The most significant finding from the in vivo model in the present study is that glomerular sieving of Ficoll-70 increases rapidly after CA/CPR, resolves quickly, and is reduced 2 h after CA/CPR by estrogen administration. We believe this finding is concordant with our in vitro findings and suggests that estrogen acts to protect the barrier function of the glomerular endothelium after ischemia-reperfusion injury. This conclusion must be drawn with some caution. First, Ficoll-70 is a polydisperse compound with an average Stokes-Einstein radius of 50 Å. We did not evaluate the size fraction of excreted Ficoll and cannot be sure that the apparent reduced permeability in estrogen-treated animals includes larger Ficoll particles. Since albumin has a Stokes radius of 36 Å, it is possible that if the change in permeability was only to small Ficoll molecules, there would be less physiological relevance to the observed result. Second, we quantified θFicoll-70 by measuring serum and urine TRITC fluorescence. The urine was obtained from the bladder after administration of intravenous fluid and a diuretic, necessary to produce adequate urine output in mice after CA/CPR. The urine output was slightly higher in estrogen-treated animals than in vehicle-treated animals. It is therefore possible that some, but not all, of the effect observed on glomerular sieving is explained by concentration of the urine in the vehicle-treated animals. We do not believe this contributes to the observed result because the difference at 2 h was neither large nor statistically significant between treatments or between CA/CPR-exposed mice and CA/CPR-naive mice. Although we describe a defect in glomerular integrity induced by CA/CPR, the methods we (36, 41) have employed in vivo do not allow us to localize the effect precisely to the mouse glomerular endothelium, although the in vitro results of this study suggest that is a potential site of barrier disruption. Finally, we measured permeability to Ficoll-70 because it is uncharged and neither secreted nor reabsorbed. We speculate that the observed effect extends to albumin and other similarly sized physiological mediators, as has been observed in other models (36, 41).
In summary, the present study demonstrates that estrogen reduces OGD-induced hyperpermeability of glomerular endothelium at least in part through the receptor GPR30 and that estrogen mediates in vivo post-CA/CPR glomerular permeability in a similar fashion. These findings support the hypothesis that glomerular endothelial function is critically altered by renal ischemia-reperfusion injury. Further study based on this premise will focus on the critical link between glomerular endothelial dysfunction and tubular functional failure, a link with likely implications for prevention and therapy of AKI.
GRANTS
This work was supported in part by NIDDK Grant DK090754-01 (to M. P. Hutchens).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: M.P.H., R.K., P.S.H., and S.A. provided conception and design of research; M.P.H. and T.F. performed experiments; M.P.H. and T.F. analyzed data; M.P.H., T.F., R.K., P.S.H., and S.A. interpreted results of experiments; M.P.H. prepared figures; M.P.H. drafted manuscript; M.P.H., R.K., P.S.H., and S.A. edited and revised manuscript; M.P.H., T.F., R.K., P.S.H., and S.A. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors express their gratitude to Simon Satchell, MRCP, PhD, for helpful discussions.
REFERENCES
- 1. Akis N, Madaio MP. Isolation, culture, and characterization of endothelial cells from mouse glomeruli. Kidney Int 65: 2223–2227, 2004 [DOI] [PubMed] [Google Scholar]
- 2. Andersson M, Nilsson U, Hjalmarsson C, Haraldsson B, Nystrom JS. Mild renal ischemia-reperfusion reduces charge and size selectivity of the glomerular barrier. Am J Physiol Renal Physiol 292: F1802–F1809, 2007 [DOI] [PubMed] [Google Scholar]
- 3. Ardelt AA, McCullough LD, Korach KS, Wang MM, Munzenmaier DH, Hurn PD. Estradiol regulates angiopoietin-1 mRNA expression through estrogen receptor-alpha in a rodent experimental stroke model. Stroke 36: 337–341, 2005 [DOI] [PubMed] [Google Scholar]
- 4. Basile DP. Rarefaction of peritubular capillaries following ischemic acute renal failure: a potential factor predisposing to progressive nephropathy. Curr Opin Nephrol Hypertens 13: 1–7, 2004 [DOI] [PubMed] [Google Scholar]
- 5. Basile DP, Friedrich JL, Spahic J, Knipe N, Mang H, Leonard EC, Changizi-Ashtiyani S, Bacallao RL, Molitoris BA, Sutton TA. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol 300: F721–F733, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Benton J, Powers A, Eiselein L, Fitch R, Wilson D, Villablanca AC, Rutledge JC. Hyperglycemia and loss of ovarian hormones mediate atheroma formation through endothelial layer disruption and increased permeability. Am J Physiol Regul Integr Comp Physiol 292: R723–R730, 2007 [DOI] [PubMed] [Google Scholar]
- 7. Burne-Taney MJ, Kofler J, Yokota N, Weisfeldt M, Traystman RJ, Rabb H. Acute renal failure after whole body ischemia is characterized by inflammation and T cell-mediated injury. Am J Physiol Renal Physiol 285: F87–F94, 2003 [DOI] [PubMed] [Google Scholar]
- 8. Chertow GM, Levy EM, Hammermeister KE, Grover F, Daley J. Independent association between acute renal failure and mortality following cardiac surgery. Am J Med 104: 343–348, 1998 [DOI] [PubMed] [Google Scholar]
- 9. Cho MM, Ziats NP, Pal D, Utian WH, Gorodeski GI. Estrogen modulates paracellular permeability of human endothelial cells by eNOS- and iNOS-related mechanisms. Am J Physiol Cell Physiol 276: C337–C349, 1999 [DOI] [PubMed] [Google Scholar]
- 10. Delarue F, Daunes S, Elhage R, Garcia A, Bayard F, Faye J. Estrogens modulate bovine vascular endothelial cell permeability and HSP 25 expression concomitantly. Am J Physiol Heart Circ Physiol 275: H1011–H1015, 1998 [DOI] [PubMed] [Google Scholar]
- 11. Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, Bologa CG, Leitao A, Brailoiu E, Deliu E, Dun NJ, Sklar LA, Hathaway HJ, Arterburn JB, Oprea TI, Prossnitz ER. In vivo effects of a GPR30 antagonist. Nat Chem Biol 5: 421–427, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fink K, Schwarz M, Feldbrugge L, Sunkomat JN, Schwab T, Bourgeois N, Olschewski M, von Zur Muhlen C, Bode C, Busch J. Severe endothelial injury and subsequent repair in patients after successful cardiopulmonary resuscitation. Crit Care 14: R104, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Groten T, Pierce AA, Huen AC, Schnaper HW. 17 Beta-estradiol transiently disrupts adherens junctions in endothelial cells. FASEB J 19: 1368–1370, 2005 [DOI] [PubMed] [Google Scholar]
- 14. Haas E, Meyer MR, Schurr U, Bhattacharya I, Minotti R, Nguyen HH, Heigl A, Lachat M, Genoni M, Barton M. Differential effects of 17beta-estradiol on function and expression of estrogen receptor alpha, estrogen receptor beta, and GPR30 in arteries and veins of patients with atherosclerosis. Hypertension 49: 1358–1363, 2007 [DOI] [PubMed] [Google Scholar]
- 15. Hamel MB, Phillips RS, Davis RB, Desbiens N, Connors AF, Jr, Teno JM, Wenger N, Lynn J, Wu AW, Fulkerson W, Tsevat J. Outcomes and cost-effectiveness of initiating dialysis and continuing aggressive care in seriously ill hospitalized adults. SUPPORT Investigators Study to Understand Prognoses and Preferences for Outcomes and Risks of Treatments. Ann Intern Med 127: 195–202, 1997 [DOI] [PubMed] [Google Scholar]
- 16. Hutchens MP, Dunlap J, Nakano T, Traystman RJ, Hurn PD. Cardiac arrest-induced acute kidney injury is sexually dimorphic and mediated by estrogen (Abstract). Anesthesiology 107: A1857, 2007 [Google Scholar]
- 17. Hutchens MP, Nakano T, Dunlap J, Traystman RJ, Hurn PD, Alkayed NJ. Soluble epoxide hydrolase gene deletion reduces survival after cardiac arrest and cardiopulmonary resuscitation. Resuscitation 76: 89–94, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hutchens MP, Nakano T, Kosaka Y, Dunlap J, Zhang W, Herson PS, Murphy SJ, Anderson S, Hurn PD. Estrogen is renoprotective via a non-receptor dependent mechanism after cardiac arrest in vivo. Anesthesiology 112: 395–405, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hutchens MP, Dunlap J, Hurn PD, Jarnberg PO. Renal ischemia: does sex matter? Anesth Analg 107: 239–249, 2008 [DOI] [PubMed] [Google Scholar]
- 20. Hutchens MP, Traystman RJ, Fujiyoshi T, Nakayama S, Herson PS. Normothermic cardiac arrest and cardiopulmonary resuscitation: a mouse model of ischemia-reperfusion injury. J Vis Exp. [Epub before print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Keppler A, Gretz N, Schmidt R, Kloetzer HM, Groene HJ, Lelongt B, Meyer M, Sadick M, Pill J. Plasma creatinine determination in mice and rats: an enzymatic method compares favorably with a high-performance liquid chromatography assay. Kidney Int 71: 74–78, 2007 [DOI] [PubMed] [Google Scholar]
- 22. Kon V, Yoshioka T, Fogo A, Ichikawa I. Glomerular actions of endothelin in vivo. J Clin Invest 83: 1762–1767, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lassnigg A, Schmidlin D, Mouhieddine M, Bachmann LM, Druml W, Bauer P, Hiesmayr M. Minimal changes of serum creatinine predict prognosis in patients after cardiothoracic surgery: a prospective cohort study. J Am Soc Nephrol 15: 1597–1605, 2004 [DOI] [PubMed] [Google Scholar]
- 24. Lindsey SH, Carver KA, Prossnitz ER, Chappell MC. Vasodilation in response to the GPR30 agonist G-1 is not different from estradiol in the mRen2.Lewis female rat. J Cardiovasc Pharmacol 57: 598–603, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mehta RL, Pascual MT, Soroko S, Savage BR, Himmelfarb J, Ikizler TA, Paganini EP, Chertow GM, Program to Improve Care in Acute Renal Disease Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int 66: 1613–1621, 2004 [DOI] [PubMed] [Google Scholar]
- 26. Metnitz PG, Krenn CG, Steltzer H, Lang T, Ploder J, Lenz K, Le Gall JR, Druml W. Effect of acute renal failure requiring renal replacement therapy on outcome in critically ill patients. Crit Care Med 30: 2051–2058, 2002 [DOI] [PubMed] [Google Scholar]
- 27. Meyer MH, Meyer RA, Jr, Gray RW, Irwin RL. Picric acid methods greatly overestimate serum creatinine in mice: more accurate results with high-performance liquid chromatography. Anal Biochem 144: 285–290, 1985 [DOI] [PubMed] [Google Scholar]
- 28. Muller V, Losonczy G, Heemann U, Vannay A, Fekete A, Reusz G, Tulassay T, Szabo AJ. Sexual dimorphism in renal ischemia-reperfusion injury in rats: possible role of endothelin. Kidney Int 62: 1364–1371, 2002 [DOI] [PubMed] [Google Scholar]
- 29. Oliver JD, 3rd, Anderson S, Troy JL, Brenner BM, Deen WH. Determination of glomerular size-selectivity in the normal rat with Ficoll. J Am Soc Nephrol 3: 214–228, 1992 [DOI] [PubMed] [Google Scholar]
- 30. Paganini EP, Halstenberg WK, Goormastic M. Risk modeling in acute renal failure requiring dialysis: the introduction of a new model. Clin Nephrol 46: 206–211, 1996 [PubMed] [Google Scholar]
- 31. Park KM, Cho HJ, Bonventre JV. Orchiectomy reduces susceptibility to renal ischemic injury: a role for heat shock proteins. Biochem Biophys Res Commun 328: 312–317, 2005 [DOI] [PubMed] [Google Scholar]
- 32. Park KM, Kim JI, Ahn Y, Bonventre AJ, Bonventre JV. Testosterone is responsible for enhanced susceptibility of males to ischemic renal injury. J Biol Chem 279: 52282–52292, 2004 [DOI] [PubMed] [Google Scholar]
- 34. Patschan D, Patschan S, Wessels JT, Becker JU, David S, Henze E, Goligorsky MS, Muller GA. Epac-1 activator 8-O-cAMP augments renoprotective effects of allogeneic murine EPCs in acute ischemic kidney injury. Am J Physiol Renal Physiol 298: F78–F85, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Remuzzi G. Abnormal protein traffic through the glomerular barrier induces proximal tubular cell dysfunction and causes renal injury. Curr Opin Nephrol Hypertens 4: 339–342, 1995 [DOI] [PubMed] [Google Scholar]
- 36. Rippe C, Asgeirsson D, Venturoli D, Rippe A, Rippe B. Effects of glomerular filtration rate on Ficoll sieving coefficients (theta) in rats. Kidney Int 69: 1326–1332, 2006 [DOI] [PubMed] [Google Scholar]
- 37. Rippe C, Rippe A, Larsson A, Asgeirsson D, Rippe B. Nature of glomerular capillary permeability changes following acute renal ischemia-reperfusion injury in rats. Am J Physiol Renal Physiol 291: F1362–F1368, 2006 [DOI] [PubMed] [Google Scholar]
- 38. Rosen S, Heyman SN. Difficulties in understanding human “acute tubular necrosis”: limited data and flawed animal models. Kidney Int 60: 1220–1224, 2001 [DOI] [PubMed] [Google Scholar]
- 39. Satake A, Takaoka M, Nishikawa M, Yuba M, Shibata Y, Okumura K, Kitano K, Tsutsui H, Fujii K, Kobuchi S, Ohkita M, Matsumura Y. Protective effect of 17beta-estradiol on ischemic acute renal failure through the PI3K/Akt/eNOS pathway. Kidney Int 73: 308–317, 2008 [DOI] [PubMed] [Google Scholar]
- 40. Satchell SC, Anderson KL, Mathieson PW. Angiopoietin 1 and vascular endothelial growth factor modulate human glomerular endothelial cell barrier properties. J Am Soc Nephrol 15: 566–574, 2004 [DOI] [PubMed] [Google Scholar]
- 41. Sharma M, Zhou Z, Miura H, Papapetropoulos A, McCarthy ET, Sharma R, Savin VJ, Lianos EA. ADMA injures the glomerular filtration barrier: role of nitric oxide and superoxide. Am J Physiol Renal Physiol 296: F1386–F1395, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shibata Y, Takaoka M, Maekawa D, Kuwahara C, Matsumura Y. Involvement of nitric oxide in the suppressive effect of 17beta-estradiol on endothelin-1 overproduction in ischemic acute renal failure. J Cardiovasc Pharmacol 44, Suppl 1: S459–S461, 2004 [DOI] [PubMed] [Google Scholar]
- 43. Sutton TA, Mang HE, Campos SB, Sandoval RM, Yoder MC, Molitoris BA. Injury of the renal microvascular endothelium alters barrier function after ischemia. Am J Physiol Renal Physiol 285: F191–F198, 2003 [DOI] [PubMed] [Google Scholar]
- 44. Takaoka M, Yuba M, Fujii T, Ohkita M, Matsumura Y. Oestrogen protects against ischaemic acute renal failure in rats by suppressing renal endothelin-1 overproduction. Clin Sci (Lond) 103, Suppl 48: 434S–437S, 2002 [DOI] [PubMed] [Google Scholar]
- 45. Triverio PA, Martin PY, Romand J, Pugin J, Perneger T, Saudan P. Long-term prognosis after acute kidney injury requiring renal replacement therapy. Nephrol Dial Transplant 24: 2186–2189, 2009 [DOI] [PubMed] [Google Scholar]
- 46. Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Ronco C, and Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA 294: 813–818, 2005 [DOI] [PubMed] [Google Scholar]
- 47. Vessey MP. Oral contraceptives and thromboembolic disease. Am Heart J 77: 153–157, 1969 [DOI] [PubMed] [Google Scholar]
- 48. Voss MR, Stallone JN, Li M, Cornelussen RN, Knuefermann P, Knowlton AA. Gender differences in the expression of heat shock proteins: the effect of estrogen. Am J Physiol Heart Circ Physiol 285: H687–H692, 2003 [DOI] [PubMed] [Google Scholar]
- 49. Xue JL, Daniels F, Star RA, Kimmel PL, Eggers PW, Molitoris BA, Himmelfarb J, Collins AJ. Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J Am Soc Nephrol 17: 1135–1142, 2006 [DOI] [PubMed] [Google Scholar]