

Abstract
Excessive alcohol consumption is a leading cause of chronic liver disease in the Western world. Alcohol-induced hepatotoxicity and oxidative stress are important mechanisms contributing to the pathogenesis of alcoholic liver disease. However, emerging evidence suggests that activation of innate immunity involving TLR4 and complement also plays an important role in initiating alcoholic steatohepatitis and fibrosis, but the role of adaptive immunity in the pathogenesis of alcoholic liver disease remains obscure. Activation of a TLR4-mediated MyD88-independent (TRIF/IRF-3) signaling pathway in Kupffer cells contributes to alcoholic steatohepatitis, whereas activation of TLR4 signaling in hepatic stellate cells promotes liver fibrosis. Alcohol consumption activates the complement system in the liver by yet unidentified mechanisms, leading to alcoholic steatohepatitis. In contrast to activation of TLR4 and complement, alcohol consumption can inhibit natural killer cells, another important innate immunity component, contributing to alcohol-mediated acceleration of viral infection and liver fibrosis in patients with chronic viral hepatitis. Understanding of the role of innate immunity in the pathogenesis of alcoholic liver disease may help us identify novel therapeutic targets to treat this disease.
Keywords: alcoholic liver injury, complement, NK cells, TLR4
excessive ingestion of alcohol is the major cause of chronic liver disease in Western countries. It is well documented that chronic alcohol consumption leads to fatty liver; however, only up to 30% of heavy drinkers may develop more severe forms of chronic liver injury such as alcoholic hepatitis, fibrosis, cirrhosis, and hepatocellular carcinoma (83). Despite extensive research on alcoholic liver disease (ALD), how alcohol consumption damages the liver is still not completely understood. It is generally accepted that oxidative stress and ethanol metabolites (e.g., acetaldehyde, acetate) produced during alcohol metabolism are the major factors contributing to alcoholic liver injury. Emerging evidence suggests that activation or dysregulation of innate immunity also play important roles in the pathogenesis of ALD, whereas the role of adaptive immunity in ALD has not been given much attention and remains obscure.
Innate immunity, which is constitutively present, represents the first line of host defense against invading organisms without fine recognition of foreign antigens. It consists of anatomic barriers (e.g., skin, epidermis, dermis, and mucous membranes), physiological barriers (e.g., temperature, low pH, oxygen), humoral factors (e.g., pepsin, lysozyme, antimicrobial substances, interferons, complement), phagocytic cells (e.g., neutrophils and macrophages), and lymphocyte subsets [e.g., natural killer (NK) and NKT cells]. Many of these barriers and factors can prevent or destroy the invading pathogens nonspecifically; however, emerging evidence suggests that innate immunity can also specifically detect invading pathogens through pattern-recognition receptors (PRRs) expressed by host cells, which recognize common microbial patterns known as pathogen-associated molecular patterns (PAMPs) (54). Many PAMPs have been identified, including bacterial carbohydrates [e.g., lipopolysaccharide (LPS), mannose], bacterial peptides (flagellin), peptidoglycans and lipoteichoic acids (from gram-positive bacteria), N-formylmethionine, lipoproteins and fungal glucans, and nucleic acids (e.g., bacterial or viral DNA or RNA). These PAMPs can be recognized by secreted, membrane-bound, or phagocytic PRRs. Secreted PRRs include complements, pentraxins, and peptidoglycan-recognition proteins. Membrane-bound or intracellular PRRs include Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors, and retinoic acid-induced gene I-like helicases. Phagocytic (or endocytic) PRRs include scavenger receptors, macrophage mannose receptors, and β-glucan receptors.
Evidence suggests that the liver plays a key role in the innate immune response (34). First, hepatocytes are responsible for biosynthesis of 80–90% of innate immune proteins including complement components and many secreted PRRs. Second, the liver contains a large number of Kupffer cells (KCs), which account for 80–90% of the total population of fixed tissue macrophages in the body. KCs, in combination with liver sinusoidal cells, are responsible for the elimination of molecular wastes from the body. Third, liver lymphocytes are rich in innate immune cells including NK, NKT, and T cell receptor γδ T cells. Fourth, liver nonparenchymal cells also express high levels of membrane-bound PRRs, such as TLRs (104, 112). Moreover, there is evidence that innate immunity in the liver not only plays a key role in host defense against microbial infection and tumor formation but also contributes to the pathogenesis of acute and chronic liver diseases, including ALD. Here we summarize the role of several components of innate immunity in the pathogenesis of ALD.
LPS/TLR4 Signaling Pathway in Alcoholic Liver Disease
LPS/TLR4 signaling pathway.
LPS is a component of Gram-negative bacteria and, biochemically, consists of an O-antigen, a core polysaccharide, and a lipid-A component (71). The lipid-A of LPS represents one of the PAMPs recognized by the innate immune system as a pathogen-derived danger signal to initiate immune activation. PRRs, including TLRs, are expressed on innate immune cells and play a central role in sensing pathogens (71, 87). TLR4 can specifically recognize LPS as a danger signal and induce activation of inflammation-associated genes (71, 87). Although TLR4 cannot directly bind LPS, the TLR4 adapter molecule, MD-2, and its coreceptor, CD14, do have the capacity to bind LPS and bring it to the receptor complex for recognition by TLR4. Since MD-2 and CD14 have no intracellular domains, it is the TLR4 that triggers downstream signaling via its intracellular TIR domain upon LPS-induced activation (71, 87). Activation of TLR4 induces two downstream signaling pathways: First, the MyD88-dependent pathway is initiated by recruitment of MyD88 to the TLR4 complex, resulting in downstream activation of IRAK1/4 and TRAF6, followed by activation of NF-κB and subsequent induction of the expression of NF-κB-controlled genes including proinflammatory cytokine and chemokine genes. Second, the MyD88-independent pathway is initiated after recruitment of the TRIF adapter to the TLR4 complex and results in activation of IKK/TAK1 kinase and IRF-3 phosphorylation as well as late activation of NF-κB (71, 87). Phosphorylated IRF-3 forms a complex and translocates to the nucleus to subsequently activate the transcription of IFN-α and β, as well as other interferon-induced genes (71, 87).
LPS/TLR4-MyD88-independent signaling pathway in alcoholic steatohepatitis.
Among the various mechanisms contributing to the pathogenesis of alcoholic liver disease, gut-derived microbial products appear to play a significant role. This notion is supported by previous studies in which sterilization of the gut attenuated ALD in rodents (1). The bacterial endotoxin, LPS, has received particular attention since its levels are elevated in the plasma of both human alcoholics and in animal models of ALD (32, 81, 100).
The critical role of LPS in alcohol-induced steatohepatitis is believed to be mediated via targeting TLR4 on KCs (29, 118). In the liver, KCs are the primary cells that respond to LPS through TLR4 (104, 112). In the noninflamed liver, KCs secrete the anti-inflammatory cytokines IL-10 (63) and TGF-β (12); however, in response to LPS, they produce large amounts of proinflammatory cytokines including TNF-α, IL-1, IL-6, and IL-8, which contribute to liver inflammation (104, 112). Hepatocytes also express TLR4, but at low levels with a minimal response to LPS (51, 106). Excessive alcohol intake induces hepatocyte injury and lipid accumulation, which elevate TLR4 expression and potentially alters the sensitivity of TLR4 signaling on hepatocytes (41, 72). Recent studies by Machida et al. (72) have shown that elevated TLR4 expression on hepatocytes in hepatitis C virus (HCV) NS5A transgenic mice increases the sensitivity to alcohol and LPS, thereby exacerbating liver injury and tumorigenesis. In addition, TLR4 also recognizes endogenous ligands such as hyaluronan and high-mobility group box 1 (HMGB1) (58, 117). In particular, HMGB1 has been shown to be released from damaged hepatocytes and contribute to liver injury (16). Thus HMGB1 may also serve as an endogenous ligand for TLR4 to promote the progression of ALD.
Although the critical role of TLR4 and its coreceptor CD14 in alcohol-induced liver injury has been well documented (118, 119, 124), their downstream signaling pathways that contribute to the pathogenesis of ALD has just been revealed recently. Disruption of the TLR4 downstream signaling molecule MyD88 in mice failed to prevent alcohol-induced steatohepatitis, reactive oxygen species (ROS) production, and inflammatory cytokines in the liver (48), whereas disruption of the MyD88-independent signaling molecule TRIF in mice abolished alcohol-induced steatohepatitis (128), suggesting that the MyD88-independent pathway contributes to TLR4-mediated alcoholic liver injury. Further studies suggest that TRIF/IRF-3 plays a critical role in alcohol-induced transactivation of the TNF-α gene in Kupffer cells/macrophages in vitro and in vivo, thereby initiating alcoholic liver injury (128). Finally, the critical role of TLR4 in the development of alcoholic and nonalcoholic fatty liver has been well documented in animal models (48, 102, 109, 118); however, how TLR4 activation affects lipid metabolism in hepatocytes is not fully understood. It is generally believed that the increased intestinal translocation of bacterial LPS during alcohol consumption leads to TLR4-dependent activation of Kupffer cells and subsequent induction of inducible nitric oxide synthase, formation of ROS, and induction of TNF-α and prostaglandin E2 that affect lipid metabolism in hepatocytes (30, 94). For example, TNF-α increases mRNA expression of hepatic SREBP-1c, a master transcription factor, to promote lipid synthesis in mice and stimulate the maturation of SREBP-1 protein in human hepatocytes (68). Hepatocytes also express TLR4, and LPS can directly regulate glucose metabolism in hepatocytes by targeting TLR4 (97); however, it is not clear whether activation of TLR4 by LPS can also directly affect lipid metabolism in hepatocytes.
LPS/TLR4 in alcoholic liver fibrosis.
Like other forms of hepatocellular damage, chronic alcoholic liver injury also leads to liver fibrosis, which is characterized by the accumulation of extracellular matrix proteins in the liver (10, 31). It is well known that chronic alcohol consumption induces elevation of portal endotoxin levels followed by activation of Kupffer cells via targeting TLR4 on these cells. Activated Kupffer cells produce ROS, lipid peroxidation products, inflammatory cytokines, and profibrogenic factors (such as TGF-β and PDGF), which subsequently induce hepatic stellate cell (HSC) activation and liver fibrosis (93, 108). In addition, the ethanol metabolite acetaldehyde can also act as a profibrogenic factor to cause HSC activation (17, 26, 39, 108).
Recent studies have shown that TLR4 signaling in HSCs and liver sinusoidal endothelial cells (LSECs) also plays an important role in the pathogenesis of liver fibrogenesis (53, 106). HSCs express TLR4 and respond to LPS stimulation by enhancing TGF-β signaling and producing cytokines/chemokines that contribute to liver fibrogenesis (86, 106). Quiescent HSCs are resistant to TGF-β-induced HSC activation due to expressing a high level of Bambi (bone morphogenetic protein and activin membrane-bound inhibitor) that inhibits TGF-β receptor signaling (106). Upon TLR4 activation, Bambi expression is promptly downregulated, leading to unrestricted activation of the signaling via the receptor for TGF-β that is secreted by Kupffer cells (106). In addition, activation of TLR4 signaling pathway in LSECs regulates angiogenesis through its MyD88 effector protein by regulating extracellular protease production and promoting liver fibrogenesis (53). Therefore, enhanced TLR4 activation on HSCs and LSECs by elevated portal LPS levels during alcohol consumption likely contributes to the pathogenesis of alcoholic liver fibrosis.
TLR4 signaling also strongly induces the production of CC chemokines including CCL2, CCL4 (MIP-1β), and CCL5 from HSCs (10, 106). These chemokines can interact with chemokine receptors (such as CCR1 and CCR5) on macrophages and HSCs and subsequently cause migration of these cells into the injured sites, playing an important role in inducing liver fibrosis (6, 105), whereas the interaction of CXCL9 and its receptor CXCR3 suppresses liver fibrogenesis by modulating the antifibrotic Th1-associated immune response (120). Although the recruitment of Kupffer cells and HSCs is considered to be crucial for the progression of liver fibrosis, the contribution of chemokine-chemokine receptor systems in ALD has not been evaluated. Microarray analyses show that expression of the CXC subfamily members IL-8, Gro-α, CXCL5, CXCL6, CXCL10, and the CC chemokine CCL2, but not CCL5, are significantly upregulated in the livers from patients with alcoholic hepatitis, and such upregulation correlates with worse prognosis, neutrophil infiltration, and the severity of portal hypertension (28), suggesting that the chemokine-chemokine receptor system plays an important role in the pathogenesis of ALD.
The strong association of the TLR4 signaling pathway and liver fibrosis has been recently confirmed in patients with chronic HCV infection by studying TLR4 single-nucleotide polymorphisms (SNPs). Huang et al. (50) have identified a series of SNPs that create a “Cirrhosis Risk Score” to predict the likelihood of fibrosis progression in patients with chronic HCV infection (50). Among the seven components of this score, a major CC allele of TLR4 (pT399I) was the second most predicative SNP to indicate a protective role in the progression of liver fibrosis (50). The functional linkage of these TLR4 SNPs to HSC responses was further examined in a model of in vitro cultured cells, demonstrating that both the D299G and T399I TLR4 SNPs reduce TLR4-mediated inflammatory and fibrogenic signaling and lower the apoptotic threshold of activated HSCs (40). Thus the protective effect of the TLR4 SNP [c.1196C>T (rs4986791, p.T399I)] on liver fibrosis is explained at least in part by its ability to increase apoptosis and decrease fibrogenic signaling in HSCs (40). These findings presage a new era in which genetic risks of disease conferred by specific SNPs will both regulate clinical management of patients with ALD and uncover novel pathways of fibrogenesis. Although the SNPs have been characterized in patients with HCV, future studies should address whether they also confer risk of progression in ALD, a likely outcome given the important role of LPS signaling in this disease as a result of increased gut permeability (100).
Complement and Alcoholic Liver Disease
In addition to LPS/TLR4 signaling pathway, a growing body of evidence in mouse models of ethanol exposure suggests that activation of the complement system also plays an important role in the pathogenesis of ALD. The complement system is an ancient part of the immune system that bridges innate and adaptive immunity and is comprised of more than 30 proteins, the majority of which are produced and secreted by the liver (36). Complement can be activated via three pathways: the classical, lectin, or alternative pathway. These three pathways converge on the third component of the complement system (C3), which is cleaved by convertases resulting in the formation of C3a and C3b (36). In addition to producing complement proteins, cells in the liver also express complement factor receptors, as well as intrinsic regulatory proteins. Under basal conditions, Kupffer cells and HSCs express the anaphylatoxin C3a and C5a receptors (95). C5a receptor expression can be induced in proliferating hepatocytes or in response to inflammatory cytokines (95). Recent evidence suggests that complement activation also contributes significantly to the pathogenesis of various forms of liver diseases, including ALD, via the production of proinflammatory cytokines (95). The role of complement in the pathogenesis of ALD is summarized in Fig. 1. Chronic ethanol feeding to mice for 4–6 wk increases activation of C3, as evidenced by increased C3a in the circulation (92), as well as increased accumulation of C3 or its proteolytic end product C3b/iC3b/C3c in liver (55, 103). Mice deficient in C3 and C5 are protected against ethanol-induced increases in hepatic triglycerides and circulating alanine aminotransferase, respectively (14, 92). Conversely, chronic ethanol-induced liver injury is exacerbated in mice lacking CD55/DAF, a complement regulatory protein, compared with wild-type controls (92). In rats, chronic ethanol exposure increases C3 activity and decreases expression of Crry, the rat homologue of CD55/DAF, and CD59 in the liver (55). Additionally, rats deficient in complement component 6 (C6), a protein that makes up part of the terminal membrane attack complex, have increased hepatic steatosis and inflammation compared with wild-type controls (15).
Fig. 1.
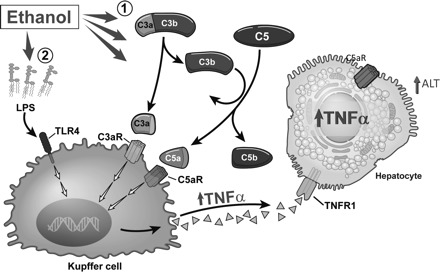
Activation of the complement system contributes the pathogenesis of alcoholic liver disease (ALD). 1: Alcohol consumption leads to an early activation of complement (C3 and C5). Activated C3a and C5a interact with their receptors on Kupffer cells, leading to TNF-α production that induces hepatocyte damage. 2: Long-term alcohol consumption activates both TLR4- and complement-dependent pathways that contribute to the pathogenesis of ALD. Adapted from Roychowdhury et al. (103) with permission.
Complement is activated early in the progression of ethanol-induced liver injury, prior to detectable increases in alanine aminotransferase/aspartate aminotransferase or accumulation of hepatic triglycerides (103). Early activation of complement contributes to increased inflammatory cytokine expression, mediated via the activation of the anaphylatoxin receptors, C3a and C5a, on Kupffer cells (103). The contribution of each pathway of complement activation in response to ethanol exposure is still unclear. It has also been suggested that ethanol-induced increases in LPS may contribute to activation of complement via the alternative pathway (55). It is also likely that activation of complement by any mechanism will initiate the alternative pathway-mediated feedback loop (36). Recent evidence shows that ethanol feeding activates the classical complement pathway via C1q binding to apoptotic cells in the liver, suggesting that the classical complement pathway also contributes to complement activation and the pathogenesis of ALD (21). Further studies are still needed to elucidate the specific roles of each pathway of complement activation in response to ethanol exposure. Although the critical role of complement in alcoholic liver injury has been well documented in rodent models, little is known about its role in human ALD. Recently, Rensen et al. (101) reported that the complement system is activated in human nonalcoholic fatty liver disease and its activation correlates with disease severity. Thus it will be very important to examine the role of complement in human ALD, which may provide novel therapeutic targets to treat this disease.
By using intercross studies in animal models of liver fibrosis, Hillebrandt et al. (45) demonstrated that C5 plays an important role in promoting liver fibrogenesis via targeting C5aR on activated HSCs and Kupffer cells in mice. Thus C5 activation during alcohol consumption, as discussed above, likely also contributes to the development of alcoholic liver fibrosis. In addition, Hillebrandt et al. also reported that two C5 htSNPs (rs 2300929 and rs17611) are associated with the high risk for developing advanced fibrosis in patients with chronic HCV infection. However, Halangk et al. (42) recently found no evidence for the association of these two C5 SNPs with advanced liver fibrosis in a large number of patients with chronic viral hepatitis, ALD, autoimmune hepatitis, and other liver diseases. Thus further studies are required to clarify the role of C5 in liver fibrogenesis in patients with chronic liver diseases, including ALD.
Innate Immune Cells and Alcoholic Liver Disease
Liver lymphocytes are enriched in innate immune cells including Kupffer cells, NK cells, NKT cells, and dendritic cells (DCs) (34). Recent studies show that chronic alcohol consumption dysregulates functions of these cells. Such dysregulation likely contributes to the pathogenesis of ALD and is discussed. In addition, alcoholic hepatitis is associated with infiltration of neutrophils, and alcohol consumption also alters the functions of neutrophils. The role of neutrophils in ALD is also briefly discussed.
Kupffer cells.
It is generally accepted that activation of Kupffer cells plays a key role in inducing alcoholic steatohepatitis. As mentioned above, an increased intestinal translocation of bacterial LPS during alcohol consumption is central to inducing Kupffer cell activation via targeting TLR4 on these cells (25, 116, 121). In addition, chronic alcohol intake also sensitizes Kupffer cell responses to LPS-mediated activation (115). Activated Kupffer cells produce inflammatory mediators (e.g., TNF-α and ROS) that contribute to hepatocyte necrosis and apoptosis and generation of extracellular matrix proteins leading to alcoholic liver injury and fibrosis (25, 82, 123). Many factors and their downstream signaling pathways attenuate Kupffer cell activation during alcohol consumption, such as adiponectin (74, 75, 90), IL-10 (75), STAT3 (47), Sirt1 (107), etc., and subsequently ameliorate alcoholic liver injury.
NK cells.
Liver lymphocytes are rich in NK cells, which play an important role in antiviral and antitumor defenses in the liver (35). NK cells are activated during hepatitis B virus (HBV) or HCV infection especially in the early stage of infection (3, 5, 38, 84, 127). Such activation not only plays a critical role in spontaneous recovery from HCV infection but may also contribute to the hepatocellular damage by killing hepatocytes (3, 127). In several animal models as well as in human nonalcoholic fatty liver disease, recent evidence suggests that NK cells may also contribute to liver injury by killing hepatocytes expressing elevated NK cell-activating ligands (18, 20, 60, 61). However, NK cell function is suppressed rather than activated in ALD (7, 13, 22, 24, 27, 65, 88, 91, 126). Thus it is very unlikely that NK cells contribute to ethanol-induced hepatocellular damage. In contrast, ethanol inhibition of NK cells may play an important role in accelerating hepatitis viral infection, liver fibrosis, and liver tumors in alcoholic patients with hepatitis viral infection.
The inhibitory effect of chronic alcohol consumption on NK cell functions has been observed for many years in alcoholic patients and rodents fed ethanol diets (7, 13, 22, 24, 27, 65, 88, 91, 126). Multiple mechanisms have been shown to contribute to alcohol inhibition of NK cells functions. First, alcohol consumption decreases expression of TRAIL, IFN-γ, and NK cell activating receptor NKG2D in liver NK cells (7, 56, 88). Second, alcohol consumption blocks NK cell release from the bone marrow and enhances splenic NK cell apoptosis (126). Third, alcohol consumption elevates serum levels of corticosterone, which inhibits NK cell functions (7). Lastly, alcohol consumption reduces central and peripheral levels of opioid peptide β-endorphin that can induce NK cell activation (13).
Clinical studies showed that individuals with a genetic predisposition to lower NK cell functions have greater propensity to develop chronic infection after acute HCV infection (62). Thus it is plausible to speculate that the high prevalence of chronic HCV infection in alcoholics could be partly attributed to ethanol's inhibition of NK cell functions in these patients. In addition to their antiviral function, NK cells also have antifibrotic effect by directly killing early-activated and senescent-activated HSCs that express elevated levels of NK cell activating ligands (37, 46, 64, 76, 79, 80, 96) and via producing IFN-γ that induces HSCs cycle arrest and apoptosis (57). Recent studies reveal that chronic ethanol consumption not only inhibits NK cells function (7, 13, 22, 24, 27, 65, 88, 91, 126), thereby reducing their cytotoxicity to HSCs, but also renders activated HSCs resistant to NK cell killing and to the inhibitory effect of IFN-γ (56). It was shown that HSCs isolated from ethanol-fed mice were less sensitive to NK cell killing than those from pair-fed mice. This reduced sensitivity is due to higher levels of TGF-β, a potent inhibitor for NK cells, produced by HSCs from ethanol-fed mice than those from pair-fed mice (56). HSCs from ethanol-fed mice were also resistant to IFN-γ-induced cell cycle arrest and apoptosis by expressing higher levels of SOCS1, an inhibitor of IFN-γ signaling, and by producing higher levels of oxidative stress that inhibits IFN-γ activation of STAT1 (56). In summary, disruption of the antifibrotic effects of NK/IFN-γ is an important mechanism contributing to ethanol-induced acceleration of liver fibrosis in alcoholics with viral hepatitis infection.
NKT cells.
NKT cells, which are abundant in the liver, are a heterogeneous group of T lymphocytes that recognize lipid antigens presented by the nonclassical MHC class I-like molecule CD1. Since they can rapidly produce a large amount of cytokines such as IFN-γ and IL-4 after stimulation with lipid antigens, NKT cells play an important role in regulating the innate and adaptive immunity. Although activation of NKT cells has been shown to induce hepatocellular damage in a variety of acute liver injury models, their role in chronic liver injury models seems more complex due to the existence of several subtypes of NKT cells that may have opposing functions. CD1d-dependent NKT cells can be divided into type I invariant NKT and type II NKT cells. Liver NKT cells consist of more than 90% type I NKT cells that promote liver injury induced by concanavalin A, α-Gal-cer, and ischemia-reperfusion but protect against liver injury induced by CCl4 and bile duct ligation (see review in Ref. 35). The detrimental effect of type I NKT cells on liver injury is mediated via killing of hepatocytes and production of various cytokines such as IFN-γ and IL-4 (see review in Ref. 35), whereas the hepatoprotective effect of type I NKT cells in CCl4 and bile duct ligation models is mediated via suppressing the neutrophil proinflammatory response (89, 122) and stimulating Kupffer cell-dependent IL-6 production that protects against liver injury (19). Type II NKT cells have been shown to protect against concanavalin A-induced liver injury via inhibiting the functions of type I NKT cells (43). Furthermore, it has been shown that, on one hand, NKT cells can kill activated HSCs and produce IFN-γ, which inhibits liver fibrosis (89), whereas on the other hand activation of NKT cells also promotes liver fibrosis via enhancing hepatocellular damage and promoting HSC activation (59, 111). Therefore, the final effect of NKT cells on liver fibrosis is determined by the balance between these inhibitory and stimulatory effects. However, the role of NKT cells in the pathogenesis of ALD remains unclear. In mice, NKT cell deficiency delayed alcohol-induced liver injury, and alcohol feeding enhanced NKT cell activator α-GalCer-induced liver injury (77), suggesting that activation of NKT cells may accelerate alcoholic liver injury.
Dendritic cells.
DCs are the most efficient antigen-presenting cells (APCs) of the immune system, playing a crucial role in innate and adaptive immune responses. Liver DCs are comprised of several subsets including myeloid CD8α−B220−, lymphoid CD8α+B220−, and plasmacytoid CD8α−B220− cells (49). Compared with peripheral DCs, liver DCs have a reduced ability to stimulate naive T cells but have enhanced ability to produce cytokines in response to TLR stimulation (49). In addition to acting as APCs, hepatic DCs also either aggravate or ameliorate hepatocellular damage via production of proinflammatory (23) or anti-inflammatory cytokines (9), respectively, in various liver injury models. Alcohol consumption can modulate the functions of DCs (66, 67) and subsequently impair the cellular response necessary for hepatitis viral clearance (4, 85, 113, 114), likely contributing to the synergistic effect of alcohol and viral hepatitis on liver injury. However, whether DCs directly contribute the pathogenesis of alcoholic liver injury via production of cytokines remains unknown.
Neutrophils.
Infiltration of a large number of neutrophils is a very prominent feature of alcoholic hepatitis (52, 98); however, the pathogenic role of neutrophils in ALD has not been investigated because most animal models of alcoholic liver injury do not develop neutrophil accumulation. Evidence from other models of liver injury suggests that neutrophils play an important role in inducing liver dysfunction and injury (99). These models include ischemia-reperfusion (8, 44) and certain drug toxicities (70, 125), etc. Ziol et al. (129) analyzed 35 patients with alcoholic hepatitis and found that the hepatocyte apoptotic index, but not the ballooning hepatocyte index, was strongly correlated with the neutrophil infiltration index. Collectively, neutrophils likely contribute to hepatocellular damage in patients with alcoholic hepatitis by producing ROS and proteases (52).
As is well known, almost all heavy drinkers develop fatty liver but only up to 35% develop alcoholic hepatitis with infiltration of neutrophils. However, how neutrophils are recruited into the fatty liver in heavy drinkers who develop alcoholic hepatitis remains obscure. It is believed that activated Kupffer cells produce a variety of cytokines and chemokines, including IL-8, RANTES, MCP-1, IL-17, etc., that subsequently recruit neutrophils into the liver (2, 11, 28, 69, 73). Targeting these chemokines and their receptors could be a potential therapeutic option to treat alcoholic hepatitis.
Future Studies
In summary, the liver is an organ with predominant innate immunity. Dysregulation of many components of innate immunity in the liver due to chronic alcohol consumption likely contributes additively or synergistically to alcohol-induced liver injury, acceleration of viral infection and tumor formation (Fig. 2). Further research is needed to further clarify and identify the interrelationships between innate immunity involved in ALD. Examples of research on the role of innate immunity in ALD need to include, but are not limited to, the following.
Fig. 2.
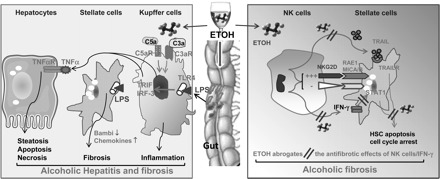
Ethanol (EtOH) dysregulation of innate immunity contributes to the pathogenesis of ALD. Chronic alcohol consumption results in activation of innate immunity components such as Kupffer cells/LPS/TLR4 and complements or inhibition of innate immunity components such as natural killer (NK) cells, contributing to the pathogenesis of ALD. First, alcohol consumption increases gut permeability and subsequently hepatic LPS levels via binding to TLR4; LPS then stimulates Kupffer cells to produce TNF-α in a TRIF/IRF-3-dependent manner. TNF-α induces hepatocellular damage. LPS can also directly target hepatic stellate cells (HSCs) and subsequently enhance TGF-β signaling and expression of chemokines, contributing to liver fibrogenesis. Second, alcohol consumption results in activation of complement components C3a and C5a, which then stimulate Kupffer cells to produce TNF-α, contributing to hepatocellular damage. Third, alcohol consumption inhibits the antifibrotic effect of NK cells and IFN-γ via multiple steps. Alcohol inhibits NK cell functions via blocking IFN-γ and TRAIL production, inhibits IFN-γ signaling in HSCs via induction of SOCS1, and renders HSC resistance to NK cell killing via production of TGF-β that inhibits NK cell function.
- 1) Further understanding is needed of the role of Kupffer cells/TLRs/NOD in ALD; for example:
- Identification of the correlation between TLR4 SNPs and the progression of ALD.
- Alcohol's effects on Kupffer cells to release cytokines, chemokines, eicosanoids, ROS, proteolytic enzymes, and NO.
- The effect, if any, of alcohol on Kupffer cell recruitment of NKT lymphocytes, neutrophils, and monocyte-derived macrophages via influencing adhesion molecules and processing and presenting antigens. Is alcohol-induced liver damage due to Kupffer cell inability to recognize and eliminate danger molecules, or excessive mobilization of cytotoxic molecules and failure to halt inflammation?
- Role of damage-associated molecular patterns (heat shock proteins, hyaluronan, ureate) and TIRAP/Myd88-dependent pathways in ALD progression.
- Further understanding of the role of TLR4 in ALD by neutralizing LPS or blocking LPS-signaling through the use of TLR4 antagonists (e.g., CyP, CRX-526, eritoran), or LPS signaling interfering molecules (e.g., TAK-242, besifloxacin, compound K, etc.).
- Improvement, if any, of ALD by probiotics such as VSL#3.
2) Recent evidence shows that IFN activation of NK cells plays an important role in inhibition of HCV replication in patients with chronic HCV infection (38, 110). It would be very important to determine the effect of alcohol consumption on IFN activation of NK cells, NK cell-mediated inhibition of HCV replication, and the expression and signaling of NK cell stimulatory and inhibitory receptors.
3) NKT cells seem to play a complex role in the pathogenesis of liver disease owing to existence of various subtypes of NKT cells and NKT cell tolerance induction. The role of NKT cells in the pathogenesis of ALD and the effects of ethanol on NKT cells remain largely unknown.
4) Alcoholic hepatitis is associated with accumulation of neutrophils (69); however, the role of neutrophils in the pathogenesis of ALD remains obscure (52) and needs further study.
5) Hepatic DCs seem to determine the balance between liver tolerance and immunity. How alcohol disturbs this balance leading to ALD needs further investigation.
6) Since the liver is the major site for synthesis of complement components, alcohol may directly or indirectly affect the level of serum complements. Further work is needed to identify how alcohol derails the complement system, resulting in ALD.
7) Further investigation is needed into the role of epigenetic changes induced by alcohol on the innate immune system in ALD; e.g., both IFN-α and β have been shown to modulate expression of several microRNAs (miRNAs), and miRNA-146a/b is upregulated in response to TLR4 ligands in an NF-κB-dependent manner. Further insights into alcohol's effects on regulation of cytokine signaling by miRNAs, DNA methylation, and histone acetylation/deacetylation should help designing new approaches to modulate inflammation in ALD.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
ACKNOWLEDGMENTS
The authors greatly appreciate Dr. George Kunos for critical reading the manuscript.
The majority of the studies described in this review were presented in a symposium at Research Society of Alcoholism in 2009 in San Diego, CA.
REFERENCES
- 1. Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 108: 218–224, 1995 [DOI] [PubMed] [Google Scholar]
- 2. Afford SC, Fisher NC, Neil DA, Fear J, Brun P, Hubscher SG, Adams DH. Distinct patterns of chemokine expression are associated with leukocyte recruitment in alcoholic hepatitis and alcoholic cirrhosis. J Pathol 186: 82–89, 1998 [DOI] [PubMed] [Google Scholar]
- 3. Ahlenstiel G, Titerence RH, Koh C, Edlich B, Feld JJ, Rotman Y, Ghany MG, Hoofnagle JH, Liang TJ, Heller T, Rehermann B. Natural killer cells are polarized toward cytotoxicity in chronic hepatitis C in an interferon-alfa-dependent manner. Gastroenterology 138: 325–335.e1–2, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aloman C, Gehring S, Wintermeyer P, Kuzushita N, Wands JR. Chronic ethanol consumption impairs cellular immune responses against HCV NS5 protein due to dendritic cell dysfunction. Gastroenterology 132: 698–708, 2007 [DOI] [PubMed] [Google Scholar]
- 5. Amadei B, Urbani S, Cazaly A, Fisicaro P, Zerbini A, Ahmed P, Missale G, Ferrari C, Khakoo SI. Activation of natural killer cells during acute infection with hepatitis C virus. Gastroenterology 138: 1536–1545, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology 52: 1390–1400, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arjona A, Boyadjieva N, Sarkar DK. Circadian rhythms of granzyme B, perforin, IFN-gamma, and NK cell cytolytic activity in the spleen: effects of chronic ethanol. J Immunol 172: 2811–2817, 2004 [DOI] [PubMed] [Google Scholar]
- 8. Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology 51: 621–632, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bamboat ZM, Ocuin LM, Balachandran VP, Obaid H, Plitas G, DeMatteo RP. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J Clin Invest 120: 559–569, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 115: 209–218, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bautista AP. Neutrophilic infiltration in alcoholic hepatitis. Alcohol 27: 17–21, 2002 [DOI] [PubMed] [Google Scholar]
- 12. Bissell DM, Wang SS, Jarnagin WR, Roll FJ. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest 96: 447–455, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boyadjieva NI, Chaturvedi K, Poplawski MM, Sarkar DK. Opioid antagonist naltrexone disrupts feedback interaction between mu and delta opioid receptors in splenocytes to prevent alcohol inhibition of NK cell function. J Immunol 173: 42–49, 2004 [DOI] [PubMed] [Google Scholar]
- 14. Bykov I, Jauhiainen M, Olkkonen VM, Saarikoski ST, Ehnholm C, Junnikkala S, Vakeva A, Lindros KO, Meri S. Hepatic gene expression and lipid parameters in complement C3(-/-) mice that do not develop ethanol-induced steatosis. J Hepatol 46: 907–914, 2007 [DOI] [PubMed] [Google Scholar]
- 15. Bykov IL, Vakeva A, Jarvelainen HA, Meri S, Lindros KO. Protective function of complement against alcohol-induced rat liver damage. Int Immunopharmacol 4: 1445–1454, 2004 [DOI] [PubMed] [Google Scholar]
- 16. Cardinal J, Pan P, Dhupar R, Ross M, Nakao A, Lotze M, Billiar T, Geller D, Tsung A. Cisplatin prevents high mobility group box 1 release and is protective in a murine model of hepatic ischemia/reperfusion injury. Hepatology 50: 565–574, 2009 [DOI] [PubMed] [Google Scholar]
- 17. Casini A, Cunningham M, Rojkind M, Lieber CS. Acetaldehyde increases procollagen type I and fibronectin gene transcription in cultured rat fat-storing cells through a protein synthesis-dependent mechanism. Hepatology 13: 758–765, 1991 [PubMed] [Google Scholar]
- 18. Chen Y, Wei H, Sun R, Dong Z, Zhang J, Tian Z. Increased susceptibility to liver injury in hepatitis B virus transgenic mice involves NKG2D-ligand interaction and natural killer cells. Hepatology 46: 706–715, 2007 [DOI] [PubMed] [Google Scholar]
- 19. Cheng CW, Duwaerts C, Rooijen NV, Wintermeyer P, Mott S, Gregory SH. NK cells suppress experimental cholestatic liver injury by an interleukin-6-mediated, Kupffer cell-dependent mechanism. J Hepatol online doi:10.1016/j.jhep.2010.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng L, You Q, Yin H, Holt M, Franklin C, Ju C. Effect of polyI:C cotreatment on halothane-induced liver injury in mice. Hepatology 49: 215–226, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cohen JI, Roychowdhury S, McMullen MR, Stavitsky AB, Nagy LE. Complement and alcoholic liver disease: role of C1q in the pathogenesis of ethanol-induced liver injury in mice. Gastroenterology 139: 664–674, 674.e1, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Collier SD, Pruett SB. Mechanisms of suppression of poly I:C-induced activation of NK cells by ethanol. Alcohol 21: 87–95, 2000 [DOI] [PubMed] [Google Scholar]
- 23. Connolly MK, Bedrosian AS, Mallen-St Clair J, Mitchell AP, Ibrahim J, Stroud A, Pachter HL, Bar-Sagi D, Frey AB, Miller G. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. J Clin Invest 119: 3213–3225, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cook RT, Li F, Vandersteen D, Ballas ZK, Cook BL, LaBrecque DR. Ethanol and natural killer cells. I. Activity and immunophenotype in alcoholic humans. Alcohol Clin Exp Res 21: 974–980, 1997 [PubMed] [Google Scholar]
- 25. Cubero FJ, Nieto N. Kupffer cells and alcoholic liver disease. Rev Esp Enferm Dig 98: 460–472, 2006 [DOI] [PubMed] [Google Scholar]
- 26. Cubero FJ, Urtasun R, Nieto N. Alcohol and liver fibrosis. Semin Liver Dis 29: 211–221, 2009 [DOI] [PubMed] [Google Scholar]
- 27. Dokur M, Chen CP, Advis JP, Sarkar DK. Beta-endorphin modulation of interferon-gamma, perforin and granzyme B levels in splenic NK cells: effects of ethanol. J Neuroimmunol 166: 29–38, 2005 [DOI] [PubMed] [Google Scholar]
- 28. Dominguez M, Miquel R, Colmenero J, Moreno M, Garcia-Pagan JC, Bosch J, Arroyo V, Gines P, Caballeria J, Bataller R. Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology 136: 1639–1650, 2009 [DOI] [PubMed] [Google Scholar]
- 29. Enomoto N, Ikejima K, Bradford BU, Rivera CA, Kono H, Goto M, Yamashina S, Schemmer P, Kitamura T, Oide H, Takei Y, Hirose M, Shimizu H, Miyazaki A, Brenner DA, Sato N, Thurman RG. Role of Kupffer cells and gut-derived endotoxins in alcoholic liver injury. J Gastroenterol Hepatol 15 Suppl: D20–D25, 2000 [DOI] [PubMed] [Google Scholar]
- 30. Enomoto N, Ikejima K, Yamashina S, Enomoto A, Nishiura T, Nishimura T, Brenner DA, Schemmer P, Bradford BU, Rivera CA, Zhong Z, Thurman RG. Kupffer cell-derived prostaglandin E2 is involved in alcohol-induced fat accumulation in rat liver. Am J Physiol Gastrointest Liver Physiol 279: G100–G106, 2000 [DOI] [PubMed] [Google Scholar]
- 31. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology 134: 1655–1669, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fujimoto M, Uemura M, Nakatani Y, Tsujita S, Hoppo K, Tamagawa T, Kitano H, Kikukawa M, Ann T, Ishii Y, Kojima H, Sakurai S, Tanaka R, Namisaki T, Noguchi R, Higashino T, Kikuchi E, Nishimura K, Takaya A, Fukui H. Plasma endotoxin and serum cytokine levels in patients with alcoholic hepatitis: relation to severity of liver disturbance. Alcohol Clin Exp Res 24: 48S–54S, 2000 [PubMed] [Google Scholar]
- 33. Gao B. Innate immunity and steatohepatitis: a critical role of another toll (TLR-9). Gastroenterology 139: 27–30, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gao B, Jeong WI, Tian Z. Liver: an organ with predominant innate immunity. Hepatology 47: 729–736, 2008 [DOI] [PubMed] [Google Scholar]
- 35. Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol 86: 513–528, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gasque P. Complement: a unique innate immune sensor for danger signals. Mol Immunol 41: 1089–1098, 2004 [DOI] [PubMed] [Google Scholar]
- 37. Glassner AEM, Kramer B, Koerner C, Sauerbruch T, Spengler U, Nattermann J. NK cells from HCV-infected patients are highly efficient in inducing apoptosis of activated primary human hepatic stellate cells (Abstract). Hepatology 52: 747A, 2010. [DOI] [PubMed] [Google Scholar]
- 38. Golden-Mason L, Cox AL, Randall JA, Cheng L, Rosen HR. Increased natural killer cell cytotoxicity and NKp30 expression protects against hepatitis C virus infection in high-risk individuals and inhibits replication in vitro. Hepatology 52: 1581–1589, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Greenwel P, Dominguez-Rosales JA, Mavi G, Rivas-Estilla AM, Rojkind M. Hydrogen peroxide: a link between acetaldehyde-elicited alpha1(I) collagen gene up-regulation and oxidative stress in mouse hepatic stellate cells. Hepatology 31: 109–116, 2000 [DOI] [PubMed] [Google Scholar]
- 40. Guo J, Loke J, Zheng F, Hong F, Yea S, Fukata M, Tarocchi M, Abar OT, Huang H, Sninsky JJ, Friedman SL. Functional linkage of cirrhosis-predictive single nucleotide polymorphisms of Toll-like receptor 4 to hepatic stellate cell responses. Hepatology 49: 960–968, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gustot T, Lemmers A, Moreno C, Nagy N, Quertinmont E, Nicaise C, Franchimont D, Louis H, Deviere J, Le Moine O. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology 43: 989–1000, 2006 [DOI] [PubMed] [Google Scholar]
- 42. Halangk J, Sarrazin C, Neumann K, Puhl G, Mueller T, Teuber G, Klinker H, Hinrichsen H, Buggisch P, Landt O, Weich V, Bergk A, Wiedenmann B, Neuhaus P, Berg T, Witt H. Evaluation of complement factor 5 variants as genetic risk factors for the development of advanced fibrosis in chronic hepatitis C infection. J Hepatol 49: 339–345, 2008 [DOI] [PubMed] [Google Scholar]
- 43. Halder RC, Aguilera C, Maricic I, Kumar V. Type II NKT cell-mediated anergy induction in type I NKT cells prevents inflammatory liver disease. J Clin Invest 117: 2302–2312, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hamada T, Fondevila C, Busuttil RW, Coito AJ. Metalloproteinase-9 deficiency protects against hepatic ischemia/reperfusion injury. Hepatology 47: 186–198, 2008 [DOI] [PubMed] [Google Scholar]
- 45. Hillebrandt S, Wasmuth HE, Weiskirchen R, Hellerbrand C, Keppeler H, Werth A, Schirin-Sokhan R, Wilkens G, Geier A, Lorenzen J, Kohl J, Gressner AM, Matern S, Lammert F. Complement factor 5 is a quantitative trait gene that modifies liver fibrogenesis in mice and humans. Nat Genet 37: 835–843, 2005 [DOI] [PubMed] [Google Scholar]
- 46. Hintermann E, Bayer M, Pfeilschifter JM, Luster AD, Christen U. CXCL10 promotes liver fibrosis by prevention of NK cell mediated hepatic stellate cell inactivation. J Autoimmun 35: 424–435, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Horiguchi N, Wang L, Mukhopadhyay P, Park O, Jeong WI, Lafdil F, Osei-Hyiaman D, Moh A, Fu XY, Pacher P, Kunos G, Gao B. Cell type-dependent pro- and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology 134: 1148–1158, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, Kurt-Jones E, Szabo G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 48: 1224–1231, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hsu W, Shu SA, Gershwin E, Lian ZX. The current immune function of hepatic dendritic cells. Cell Mol Immunol 4: 321–328, 2007 [PubMed] [Google Scholar]
- 50. Huang H, Shiffman ML, Friedman S, Venkatesh R, Bzowej N, Abar OT, Rowland CM, Catanese JJ, Leong DU, Sninsky JJ, Layden TJ, Wright TL, White T, Cheung RC. A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology 46: 297–306, 2007 [DOI] [PubMed] [Google Scholar]
- 51. Isogawa M, Robek MD, Furuichi Y, Chisari FV. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol 79: 7269–7272, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jaeschke H. Neutrophil-mediated tissue injury in alcoholic hepatitis. Alcohol 27: 23–27, 2002 [DOI] [PubMed] [Google Scholar]
- 53. Jagavelu K, Routray C, Shergill U, O'Hara SP, Faubion W, Shah VH. Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology 52: 590–601, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol 20: 197–216, 2002 [DOI] [PubMed] [Google Scholar]
- 55. Jarvelainen HA, Vakeva A, Lindros KO, Meri S. Activation of complement components and reduced regulator expression in alcohol-induced liver injury in the rat. Clin Immunol 105: 57–63, 2002 [DOI] [PubMed] [Google Scholar]
- 56. Jeong WI, Park O, Gao B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology 134: 248–258, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jeong WI, Park O, Radaeva S, Gao B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology 44: 1441–1451, 2006 [DOI] [PubMed] [Google Scholar]
- 58. Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 11: 1173–1179, 2005 [DOI] [PubMed] [Google Scholar]
- 59. Jin ZSR, Wei H, Gao X, Chen Y, Tian Z. Accelerated liver fibrosis in HBV transgenic mice: involvement of NKT cells. Hepatology 53: 219–229, 2011. [DOI] [PubMed] [Google Scholar]
- 60. Kahraman A, Barreyro FJ, Bronk SF, Werneburg NW, Mott JL, Akazawa Y, Masuoka HC, Howe CL, Gores GJ. TRAIL mediates liver injury by the innate immune system in the bile duct-ligated mouse. Hepatology 47: 1317–1330, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kahraman ASM, Kocabayoglu P, Yildiz-Meziletoglu Schlensak M, Fingas C, Wedemeyer I, Marquitan G, Gieseler R, Baba H, Gerken G, Canbay A. Major histocompatibility complex class I-related chains A and B (MICA/B): a novel role in non-alcoholic steatohepatitis. Hepatology 51: 92–102, 2010 [DOI] [PubMed] [Google Scholar]
- 62. Khakoo SI, Thio CL, Martin MP, Brooks CR, Gao X, Astemborski J, Cheng J, Goedert JJ, Vlahov D, Hilgartner M, Cox S, Little AM, Alexander GJ, Cramp ME, O'Brien SJ, Rosenberg WM, Thomas DL, Carrington M. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 305: 872–874, 2004 [DOI] [PubMed] [Google Scholar]
- 63. Knolle PA, Loser E, Protzer U, Duchmann R, Schmitt E, zum Buschenfelde KH, Rose-John S, Gerken G. Regulation of endotoxin-induced IL-6 production in liver sinusoidal endothelial cells and Kupffer cells by IL-10. Clin Exp Immunol 107: 555–561, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell 134: 657–667, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Laso FJ, Madruga JI, Giron JA, Lopez A, Ciudad J, SanMiguel JF, Alvarez-Mon M, Orfao A. Decreased natural killer cytotoxic activity in chronic alcoholism is associated with alcohol liver disease but not active ethanol consumption. Hepatology 25: 1096–1100, 1997 [DOI] [PubMed] [Google Scholar]
- 66. Laso FJ, Vaquero JM, Almeida J, Marcos M, Orfao A. Chronic alcohol consumption is associated with changes in the distribution, immunophenotype, and the inflammatory cytokine secretion profile of circulating dendritic cells. Alcohol Clin Exp Res 31: 846–854, 2007 [DOI] [PubMed] [Google Scholar]
- 67. Lau AH, Abe M, Thomson AW. Ethanol affects the generation, cosignaling molecule expression, and function of plasmacytoid and myeloid dendritic cell subsets in vitro and in vivo. J Leukoc Biol 79: 941–953, 2006 [DOI] [PubMed] [Google Scholar]
- 68. Lawler JF, Jr, Yin M, Diehl AM, Roberts E, Chatterjee S. Tumor necrosis factor-alpha stimulates the maturation of sterol regulatory element binding protein-1 in human hepatocytes through the action of neutral sphingomyelinase. J Biol Chem 273: 5053–5059, 1998 [DOI] [PubMed] [Google Scholar]
- 69. Lemmers A, Moreno C, Gustot T, Marechal R, Degre D, Demetter P, de Nadai P, Geerts A, Quertinmont E, Vercruysse V, Le Moine O, Deviere J. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology 49: 646–657, 2009 [DOI] [PubMed] [Google Scholar]
- 70. Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology 43: 1220–1230, 2006 [DOI] [PubMed] [Google Scholar]
- 71. Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine 42: 145–151, 2008 [DOI] [PubMed] [Google Scholar]
- 72. Machida K, Tsukamoto H, Mkrtchyan H, Duan L, Dynnyk A, Liu HM, Asahina K, Govindarajan S, Ray R, Ou JH, Seki E, Deshaies R, Miyake K, Lai MM. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci USA 106: 1548–1553, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Maltby J, Wright S, Bird G, Sheron N. Chemokine levels in human liver homogenates: associations between GRO alpha and histopathological evidence of alcoholic hepatitis. Hepatology 24: 1156–1160, 1996 [DOI] [PubMed] [Google Scholar]
- 74. Mandal P, Park PH, McMullen MR, Pratt BT, Nagy LE. The anti-inflammatory effects of adiponectin are mediated via a heme oxygenase-1-dependent pathway in rat Kupffer cells. Hepatology 51: 1420–1429, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mandal P, Pritchard MT, Nagy LE. Anti-inflammatory pathways and alcoholic liver disease: role of an adiponectin/interleukin-10/heme oxygenase-1 pathway. World J Gastroenterol 16: 1330–1336, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Melhem A, Muhanna N, Bishara A, Alvarez CE, Ilan Y, Bishara T, Horani A, Nassar M, Friedman SL, Safadi R. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol 45: 60–71, 2006 [DOI] [PubMed] [Google Scholar]
- 77. Minagawa M, Deng Q, Liu ZX, Tsukamoto H, Dennert G. Activated natural killer T cells induce liver injury by Fas and tumor necrosis factor-alpha during alcohol consumption. Gastroenterology 126: 1387–1399, 2004 [DOI] [PubMed] [Google Scholar]
- 78. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 139: 323–334.e7, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Muhanna N, Abu Tair L, Doron S, Amer J, Azzeh M, Mahamid M, Friedman S, Safadi R. Amelioration of hepatic fibrosis by NK cell activation. Gut 60: 90–98, 2011 [DOI] [PubMed] [Google Scholar]
- 80. Muhanna N, Doron S, Wald O, Horani A, Eid A, Pappo O, Friedman SL, Safadi R. Activation of hepatic stellate cells after phagocytosis of lymphocytes: a novel pathway of fibrogenesis. Hepatology 48: 963–977, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nanji AA, Khettry U, Sadrzadeh SM, Yamanaka T. Severity of liver injury in experimental alcoholic liver disease. Correlation with plasma endotoxin, prostaglandin E2, leukotriene B4, and thromboxane B2. Am J Pathol 142: 367–373, 1993 [PMC free article] [PubMed] [Google Scholar]
- 82. Nath B, Szabo G. Alcohol-induced modulation of signaling pathways in liver parenchymal and nonparenchymal cells: implications for immunity. Semin Liver Dis 29: 166–177, 2009 [DOI] [PubMed] [Google Scholar]
- 83. O'Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Hepatology 51: 307–328, 2010 [DOI] [PubMed] [Google Scholar]
- 84. Oliviero B, Varchetta S, Paudice E, Michelone G, Zaramella M, Mavilio D, De Filippi F, Bruno S, Mondelli MU. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology 137: 1151–1160, 2009 [DOI] [PubMed] [Google Scholar]
- 85. Osna NA. Hepatitis C virus and ethanol alter antigen presentation in liver cells. World J Gastroenterol 15: 1201–1208, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 37: 1043–1055, 2003 [DOI] [PubMed] [Google Scholar]
- 87. Palsson-McDermott EM, O'Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 113: 153–162, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pan HN, Sun R, Jaruga B, Hong F, Kim WH, Gao B. Chronic ethanol consumption inhibits hepatic natural killer cell activity and accelerates murine cytomegalovirus-induced hepatitis. Alcohol Clin Exp Res 30: 1615–1623, 2006 [DOI] [PubMed] [Google Scholar]
- 89. Park O, Jeong WI, Wang L, Wang H, Lian ZX, Gershwin ME, Gao B. Diverse roles of invariant natural killer T cells in liver injury and fibrosis induced by carbon tetrachloride. Hepatology 49: 1683–1694, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Park PH, Thakur V, Pritchard MT, McMullen MR, Nagy LE. Regulation of Kupffer cell activity during chronic ethanol exposure: role of adiponectin. J Gastroenterol Hepatol 21, Suppl 3: S30–S33, 2006 [DOI] [PubMed] [Google Scholar]
- 91. Perney P, Portales P, Corbeau P, Roques V, Blanc F, Clot J. Specific alteration of peripheral cytotoxic cell perforin expression in alcoholic patients: a possible role in alcohol-related diseases. Alcohol Clin Exp Res 27: 1825–1830, 2003 [DOI] [PubMed] [Google Scholar]
- 92. Pritchard MT, McMullen MR, Stavitsky AB, Cohen JI, Lin F, Medof ME, Nagy LE. Differential contributions of C3, C5, and decay-accelerating factor to ethanol-induced fatty liver in mice. Gastroenterology 132: 1117–1126, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Purohit V, Brenner DA. Mechanisms of alcohol-induced hepatic fibrosis: a summary of the Ron Thurman Symposium. Hepatology 43: 872–878, 2006 [DOI] [PubMed] [Google Scholar]
- 94. Purohit V, Gao B, Song BJ. Molecular mechanisms of alcoholic fatty liver. Alcohol Clin Exp Res 33: 191–205, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Qin XB, Gao B. The complement system in liver diseases. Cell Mol Immunol 3: 333–340, 2006 [PubMed] [Google Scholar]
- 96. Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 130: 435–452, 2006 [DOI] [PubMed] [Google Scholar]
- 97. Raetzsch CF, Brooks NL, Alderman JM, Moore KS, Hosick PA, Klebanov S, Akira S, Bear JE, Baldwin AS, Mackman N, Combs TP. Lipopolysaccharide inhibition of glucose production through the Toll-like receptor-4, myeloid differentiation factor 88, and nuclear factor kappa b pathway. Hepatology 50: 592–600, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ramaiah SK, Jaeschke H. Hepatic neutrophil infiltration in the pathogenesis of alcohol-induced liver injury. Toxicol Mech Methods 17: 431–440, 2007 [DOI] [PubMed] [Google Scholar]
- 99. Ramaiah SK, Jaeschke H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol 35: 757–766, 2007 [DOI] [PubMed] [Google Scholar]
- 100. Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 50: 638–644, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rensen SS, Slaats Y, Driessen A, Peutz-Kootstra CJ, Nijhuis J, Steffensen R, Greve JW, Buurman WA. Activation of the complement system in human nonalcoholic fatty liver disease. Hepatology 50: 1809–1817, 2009 [DOI] [PubMed] [Google Scholar]
- 102. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol 47: 571–579, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Roychowdhury S, McMullen MR, Pritchard MT, Hise AG, van Rooijen N, Medof ME, Stavitsky AB, Nagy LE. An early complement-dependent and TLR-4-independent phase in the pathogenesis of ethanol-induced liver injury in mice. Hepatology 49: 1326–1334, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 48: 322–335, 2008 [DOI] [PubMed] [Google Scholar]
- 105. Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, Llovet JM, Brenner DA, Schwabe RF. CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest 119: 1858–1870, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 13: 1324–1332, 2007 [DOI] [PubMed] [Google Scholar]
- 107. Shen Z, Ajmo JM, Rogers CQ, Liang X, Le L, Murr MM, Peng Y, You M. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-alpha production in cultured macrophage cell lines. Am J Physiol Gastrointest Liver Physiol 296: G1047–G1053, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Siegmund SV, Dooley S, Brenner DA. Molecular mechanisms of alcohol-induced hepatic fibrosis. Dig Dis 23: 264–274, 2005 [DOI] [PubMed] [Google Scholar]
- 109. Spruss AKG, Wagnerberger S, Haub S, Bischoff S, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 50: 1094–1104, 2009 [DOI] [PubMed] [Google Scholar]
- 110. Stegmann KA, Bjorkstrom NK, Veber H, Ciesek S, Riese P, Wiegand J, Hadem J, Suneetha PV, Jaroszewicz J, Wang C, Schlaphoff V, Fytili P, Cornberg M, Manns MP, Geffers R, Pietschmann T, Guzman CA, Ljunggren HG, Wedemeyer H. Interferon-alpha-induced TRAIL on natural killer cells is associated with control of hepatitis C virus infection. Gastroenterology 138: 1885–1897, 2010 [DOI] [PubMed] [Google Scholar]
- 111. Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, Witek RP, Choi SS, Guy CD, Fearing CM, Teaberry V, Pereira FE, Adams DH, Diehl AM. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 51: 1998–2007, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Szabo G, Dolganiuc A, Mandrekar P. Pattern recognition receptors: a contemporary view on liver diseases. Hepatology 44: 287–298, 2006 [DOI] [PubMed] [Google Scholar]
- 113. Szabo G, Dolganiuc A, Mandrekar P, White B. Inhibition of antigen-presenting cell functions by alcohol: implications for hepatitis C virus infection. Alcohol 33: 241–249, 2004 [DOI] [PubMed] [Google Scholar]
- 114. Szabo G, Wands JR, Eken A, Osna NA, Weinman SA, Machida K, Joe Wang H. Alcohol and hepatitis C virus—interactions in immune dysfunctions and liver damage. Alcohol Clin Exp Res 34: 1675–1686, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Thakur V, McMullen MR, Pritchard MT, Nagy LE. Regulation of macrophage activation in alcoholic liver disease. J Gastroenterol Hepatol 22, Suppl 1: S53–S56, 2007 [DOI] [PubMed] [Google Scholar]
- 116. Thurman RG., 2nd Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol Gastrointest Liver Physiol 275: G605–G611, 1998 [DOI] [PubMed] [Google Scholar]
- 117. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med 201: 1135–1143, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 34: 101–108, 2001 [DOI] [PubMed] [Google Scholar]
- 119. Uesugi T, Froh M, Arteel GE, Bradford BU, Wheeler MD, Gabele E, Isayama F, Thurman RG. Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J Immunol 168: 2963–2969, 2002 [DOI] [PubMed] [Google Scholar]
- 120. Wasmuth HE, Lammert F, Zaldivar MM, Weiskirchen R, Hellerbrand C, Scholten D, Berres ML, Zimmermann H, Streetz KL, Tacke F, Hillebrandt S, Schmitz P, Keppeler H, Berg T, Dahl E, Gassler N, Friedman SL, Trautwein C. Antifibrotic effects of CXCL9 and its receptor CXCR3 in livers of mice and humans. Gastroenterology 137: 309–319, 319.e1–3, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wheeler MD, Kono H, Yin M, Nakagami M, Uesugi T, Arteel GE, Gabele E, Rusyn I, Yamashina S, Froh M, Adachi Y, Iimuro Y, Bradford BU, Smutney OM, Connor HD, Mason RP, Goyert SM, Peters JM, Gonzalez FJ, Samulski RJ, Thurman RG. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic Biol Med 31: 1544–1549, 2001 [DOI] [PubMed] [Google Scholar]
- 122. Wintermeyer P, Cheng CW, Gehring S, Hoffman BL, Holub M, Brossay L, Gregory SH. Invariant natural killer T cells suppress the neutrophil inflammatory response in a mouse model of cholestatic liver damage. Gastroenterology 136: 1048–1059, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wu D, Cederbaum AI. Oxidative stress and alcoholic liver disease. Semin Liver Dis 29: 141–154, 2009 [DOI] [PubMed] [Google Scholar]
- 124. Yin M, Bradford BU, Wheeler MD, Uesugi T, Froh M, Goyert SM, Thurman RG. Reduced early alcohol-induced liver injury in CD14-deficient mice. J Immunol 166: 4737–4742, 2001 [DOI] [PubMed] [Google Scholar]
- 125. You Q, Cheng L, Reilly TP, Wegmann D, Ju C. Role of neutrophils in a mouse model of halothane-induced liver injury. Hepatology 44: 1421–1431, 2006 [DOI] [PubMed] [Google Scholar]
- 126. Zhang H, Meadows GG. Exogenous IL-15 in combination with IL-15R alpha rescues natural killer cells from apoptosis induced by chronic alcohol consumption. Alcohol Clin Exp Res 33: 419–427, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zhang ZZS, Zou Z, Shi J, Zhao J, Fan R, Qin E, Li B, Li Z, Xu X, Fu J, Zhang J, Gao B, Tian Z, Wang FS. Hypercytolytic activity of hepatic natural killer cells correlates with liver injury in chronic hepatitis B patients. Hepatology 53: 73–85, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zhao XJ, Dong Q, Bindas J, Piganelli JD, Magill A, Reiser J, Kolls JK. TRIF and IRF-3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J Immunol 181: 3049–3056, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ziol M, Tepper M, Lohez M, Arcangeli G, Ganne N, Christidis C, Trinchet JC, Beaugrand M, Guillet JG, Guettier C. Clinical and biological relevance of hepatocyte apoptosis in alcoholic hepatitis. J Hepatol 34: 254–260, 2001 [DOI] [PubMed] [Google Scholar]