

Abstract
The de novo design of molecular switching peptides is of increasing interest because it tests and extends our fundamental understanding of this process while laying the groundwork for the creation of new chemical and biological sensors. Here, an α-helical amphiphilic cell-lytic peptide, mastoparan X, was engineered to bind divalent cations. Binding of Zn(II) or Ni(II) to the designed peptide Mst-HH stabilizes the lytic amphiphilic structure and increases the activity of the peptide. Although both Zn(II) and Ni(II) activate Mst-HH for membrane lysis, they appear to do so via different mechanisms. Additionally, a series of metal binding-site mutants were synthesized to assess the relationship of charge and helical propensity to the toxicity and switchability. Additionally, by changing the characteristics of the metal-binding ligands, we can vary the selectivity of the site.
Introduction
A myriad of molecular switches is responsible for the tight regulation and proper function of cellular processes. The de novo design of peptides capable of selective conformational switching tests our understanding of the basic structural and chemical requirements for this phenomenon. Addressing this problem through protein design requires conditional stabilization of the target fold with concomitant destabilization of alternative folds. In this work, an amphiphilic cell-lytic peptide scaffold was manipulated to induce efficient membrane lysis only upon binding metal ligands.
In the study of metalloprotein folding and function, de novo peptide design has been successfully used to create simplified model systems that mimic more complicated native systems. Thus far, several groups have introduced metal ligands to stabilize the structure of helical peptides.1,2 In particular, Ghadiri et al. examined the effect of transition metal ion-binding to short partially stable model polyalanine peptides. By placing histidine residues at i and i+4 positions of the helix, they created a metal-binding site selective to divalent cations such as Cd(II), Cu(II), Ni(II), and Zn(II). Zn(II) bound the tightest to the peptide but showed only marginal increase in the stability of the α-helix. Although Ni(II) bound less tightly than Zn(II), it exhibited the greatest increase in helical content upon metal binding.
Metal-ion binding can also stabilize protein tertiary and quaternary structures.3–7 Studies by Ghadiri and McLendon have exploited the metal-binding properties of attached bipyridyl moieties to direct helix formation and trimerization of partially stable helices.5,8 The dueferri series of designed metal-binding proteins from the DeGrado laboratory exhibit metal-specific stabilization of a four-helix structure at pH values favorable for metal-coordination.9 The Pecoraro laboratory has detailed the metal-binding specific thermodynamic stabilization and folding kinetics of a their TRI series of peptides, three stranded coiled coils with a soft-metal binding site engineered into the hydrophobic core.3,6 By introducing His residues into the core of a coiled-coil, Suzuki et al. designed a triple stranded structure that undergoes metal-induced self-assembly,10 and Kohn used γ-carboxyglutamic acid residues as metal chelating side chains to link folding of a coiled coil to binding of hard metal ions.11 Furthermore, Ambroggio et al. computationally designed peptides that switch between a coiled-coil and zinc finger motif in a metal ion-dependent manner.12
The binding of metal ions can also be used to induce specific functions in natural systems. For example, the natural protein Bindin plays an important role in fertilization, triggered at the appropriate time by binding of Zn(II) ions.13 A fragment of this protein, named B18, has histidine residues placed at i and i+4 positions of the helix. Binding of this fragment to Zn(II) induces an increase in helical structure and activates the peptide for aggregation on membranes, lipid mixing, and vesicle fusion.
Here, we introduce metal-binding sites into an amphiphilic membrane-active peptide, providing a switch to induce lipid-binding and subsequent membrane lysis. Amphiphilic helical peptides capable of inducing membrane lysis are important components of host defense and innate immunity, as well as antifungal and antimicrobial interactions.14 Antimicrobial peptides are specific toward disrupting prokaryotic membranes in contrast with toxins, which lyse eukaryotic and prokaryotic membranes indiscriminately. There are a number of different classes of antimicrobial peptides with distinct secondary and tertiary structures, including β-hairpins, α-helices, and disulfide-rich folds. Despite this structural diversity they tend to have one common feature—an amphiphilic conformation in which hydrophobic and positively charged residues form extended surfaces on opposite sides of the peptide. While both toxins and antimicrobial peptides tend to be highly positively charged, toxins generally have a greater hydrophobicity, leading to indiscriminate interactions with membranes.15–19
Membrane-interactive peptides such as the magainins, melittin I and II, and the mastoparans form amphiphilic α-helices. These peptides are typically unstructured in the absence of lipid and then fold into α-helices when exposed to a membrane environment.14,20 The amphiphilic helical structure enhances the ability to bind to the anisotropic environment of the water-membrane interface. Binding to a biological membrane is the first step in the disruption process, occurring on the subsecond to minute time scale.21,22
The mastoparans are a family of 10–15 residue lytic peptides from wasp venom. They create inflammation due to mast cell degranulation and the concomitant release of histamine.23 Most of the mastoparans share an architecture containing lysine residues in the 4, 11, and 12 positions. Here, we introduce a metal-binding site into mastoparan X. Binding of metal ions to this peptide could promote membrane lysis by selectively stabilizing the helical conformation that facilitates membrane binding and disruption (Figure 1). Mastoparan X is too short to form a stable α-helix in water,24 but the introduction of helix-enhancing metal-binding sites should increase the fraction of helical states populated by the peptide—i.e., binding of metal ions will decrease the energy gap between the unfolded and helical states of the peptide. Thus, if the binding of metal ions is thermodynamically coupled to helix formation, and helix formation is thermodynamically coupled to membrane binding/disruption, then the potency of the peptides will be enhanced in the presence of metal ions, assuming that they do not interfere with peptide/membrane interactions. A secondary effect of binding of a divalent cation to the polar side of a lytic peptide, is that it will increase the local net positive charge (at pH > pKa of the ligating side chains), which might increase the affinity of the peptide for acidic bilayers and promote lysis, particularly of bacterial membranes.25,26
Figure 1.
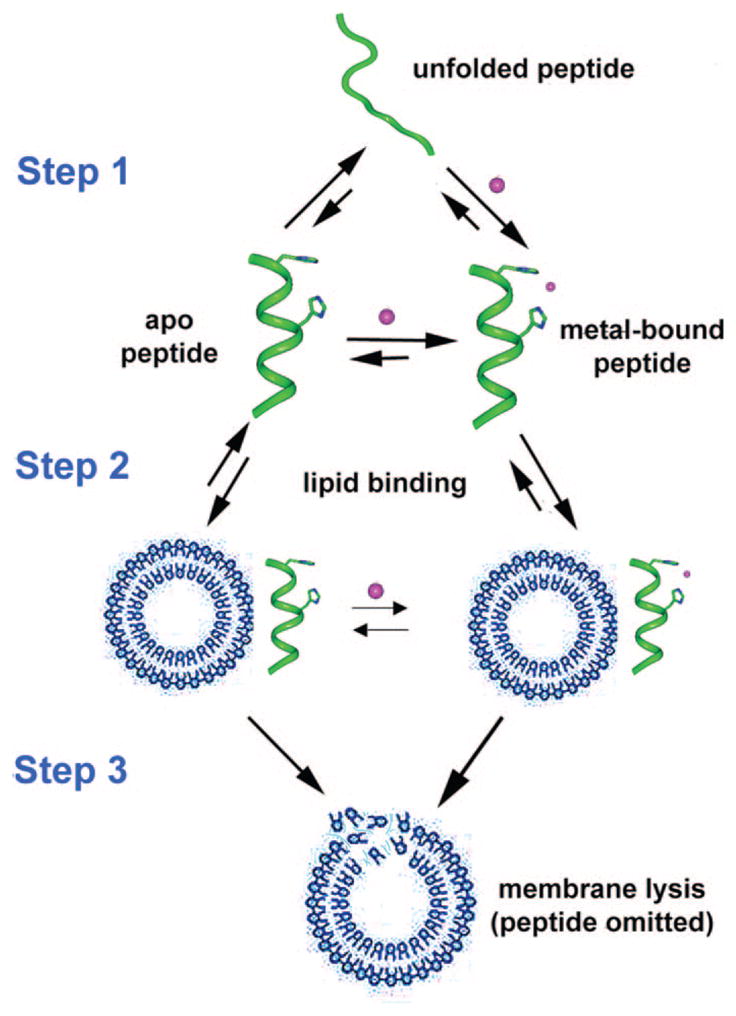
Schematic representing the possible mechanism for promotion of membrane lysis by metal ion-binding to a lytic helix. The lytic peptide is predominantly unfolded, but binding of metal ion enhances the stability of the helical conformation (Step 1). Binding of the peptide (Step 2) leads to lysis (Step 3).
Results
Design of the Switch
In order to incorporate a divalent metal binding site, two histidine residues or a glutamic acid and a histidine residue (shown in red below) were built into i, i+4 positions of the mastoparan X sequence. A model for each of the mastoparan mutants, viewed down the helix axis, is presented in Figure 2.
Figure 2.
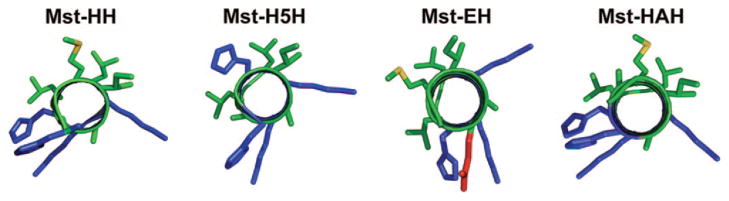
Model of the mastoparan X mutants viewed down the helix axis. Basic residues are colored blue. Acidic side chains are colored red. All other residues are colored according to atom type (carbon, green; sulfur, yellow).
The binding of metal ions to the Mst-HH peptide was expected to stabilize the amphiphilic α-helical conformation of the peptide, enhancing its lytic potential. To test the specificity of this effect, we also prepared the Mst-H5H mutant, that should be unable to bind metal ions in an α-helical conformation due to the i, i+5 spacing of the metal-binding histidine residues. The Mst-EH peptide retains an i, i+4 oriented metal-binding site, but should change the ion-binding specificity to favor harder metal ions than Mst-HH. Finally, Mst-HAH was designed to test further the relation between the helix-forming potential and lysis, by changing the helix-breaking glycine at the fifth position to alanine. Differences in net positive charge may play a secondary role in defining the lytic activity of these peptides. Hemolytic peptides such as melittin and mastoporans bear a high degree of positive charge; neutralization or elimination of the charged groups can reduce activity,27 although this is not as pronounced an effect as is seen in antimicrobial peptides. Thus, we would expect that the increase in positive charge associated with binding of metal ions to these peptides (particularly Mst-EH) may moderately enhance the lytic potential of the peptides, assuming that their hydrophobicities remain approximately constant.
Effect of Metal-Binding on the 2° Structure of Mst-HH
We used CD spectroscopy to investigate the secondary structure of the peptides. In dilute aqueous solution and in the absence of membranes the peptides gave spectra indicative of primarily random coil conformations, with a small fraction of α-helix, as expected from the known conformational properties of masto-paran.28 The differences in helicity as inferred from the ellipticity at 222 nm were consistent with the differences in their helical potential. For example, when one helix-destabilizing Gly residue in Mst-HH is replaced with a helix-stabilizing Ala residue in Mst-HAH, the magnitude of the ellipticity at 222 nm increased 3-fold (−2000 and −6000° cm2/dmol for Mst-HH and Mst-HAH, respectively). Similarly, a helix-destabilizing His in Mst-HH replaces a helix-stabilizing Glu residue in Mst-EH, and this substitution increases the magnitude of the ellipticity 2.75-fold (−2000 and −6000° cm2/dmol for Mst-HH and Mst-HAH, respectively).
Following Ghadiri and Ruan,1,2 Ni(II) and Zn(II)were titrated into the Mst-HH peptide and the CD spectrum was acquired. Figure 3 shows the effect of the addition of metal ions on the n-π* transition at 222 nm, which is sensitive to the random coil to helix transition.
Figure 3.

Mean residue ellipticity of Mst-HH at 222 nm vs divalent cation concentration. A simple binding equilibrium (eq 1) was fit to the Ni(II) data and the resulting curve is drawn through the data points.
As expected from previous studies, only Ni(II) addition results in a significant increase in helical content. Ni(II) prefers to bind in a square planar geometry, which is ideal to bind and stabilize side chains of amino acid residues at the i and i+4 positions of an α-helix. The Kd value for Ni(II) binding to Mst-HH calculated from a nonlinear least-squares fit to a simple binding isotherm (eq 1) is 300 μM. This increase in helicity and Kd value is consistent with Ghadiri’s study on polyalanine peptides.2 The previous study by Ghadiri et al. showed that although zinc binds most tightly to an HX3H site it doesn’t increase the helical content. Similarly, the CD spectrum of Mst-HH does not change significantly upon addition of Zn(II) (Figure 3). We therefore used NMR spectroscopy to verify that Zn(II) binds to the Mst-HH histidine residues. The C4 protons on the histidine imidazole ring have sharp proton resonances (Figure 4) in the aromatic region of the NMR spectrum, that are extremely sensitive to changes in local chemical environment.29 These residues shift upon titration of Zn(II), until a single equivalent of Zn(II) per peptide is added, after which no further change is observed. The C4 proton peaks shift without significant broadening, indicating that exchange with the Zn(II) ligand is rapid relative to the difference in the Zn(II)-bound and free-peptide proton frequencies. From this data, we conclude that Zn(II) binds rapidly and reversibly to the HX3H site in a 1:1 complex with a Kd ≪ 1 mM.
Figure 4.

Chemical shift vs [Zn(II)] for the two Mst-HH C4 histidine proton resonances (1 mM Mst-HH, 10 mM Tris pH = 6.0, 150 mM NaCl).
Hemolytic Activity of Mst-HH
The ability of metal ions to enhance the activity of Mst-HH was evaluated by measuring peptide-induced erythrocyte lysis in the presence and absence of Zn(II) and Ni(II) (Figure 5). The curves were analyzed to obtain HC50 values (concentration of toxin at which 50% of the cells are lysed) and Hill coefficients (Table 1). As expected from its decreased helical potential, Mst-HH is less lytic than native mastoparan X in the absence of metal ions. Also, the hemolytic activity of Mst-HH in the presence of 300 μM Ni(II) is greater than both the apo Mst-HH peptide and the mastoparan X. The addition of 300 μM Zn(II) also increased the hemolytic potency of Mst-HH, which was unexpected given that this metal ion failed to increase the helical content of Mst-HH in aqueous solution. Although the Ni(II)-bound and the Zn(II)-bound Mst-HH peptides have similar HC50 values (33 μM and 38 μM, respectively), the dose response curve in the presence of Zn(II) is more cooperative (explored in more detail below).
Figure 5.
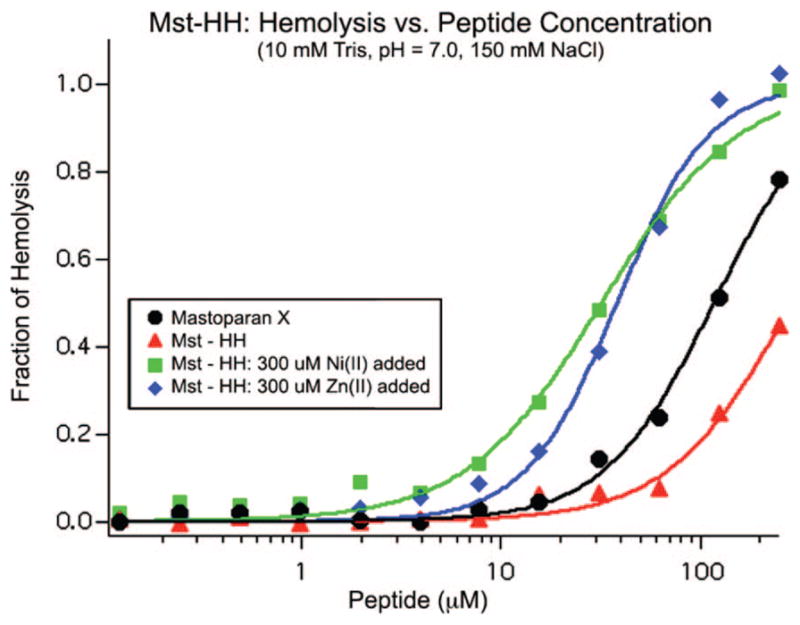
Fraction of hemolysis vs peptide concentration of Mst-HH in the presence and absence of Zn(II) and Ni(II) ions. The hemolytic activity of the native peptide, mastoparan X, is also shown. Results of fitting the Hill equation (eq 2) to the data are depicted as curves drawn through the data points.
Table 1.
HC50 Values Obtained for Mst-HH with and without Added Metal
| peptide/metal | HC50 (μM) | n |
|---|---|---|
| Mastoparan X, apo | 119 ± 4 | 1.6 ± 0.1 |
| Mst-HH, apo | 295 ± 17 | 1.4 ± 0.1 |
| Mst-HH, 300 μM Zn(II) added | 38 ± 2 | 1.9 ± 0.2 |
| Mst-HH, 300 μM Ni(II) added | 33 ± 2 | 1.3 ± 0.1 |
Hemolysis-Monitored Metal Titration of Mst-HH
The dependence of the hemolytic activity on the metal concentration was determined by varying the concentration of Zn(II) and Ni(II)
while maintaining the peptide concentration constant (Figure 6). Control studies in the absence of peptide showed that Ni(II) and Zn(II) did not cause lysis when the metal ions were added up to a concentration of 300 μM. However, at concentrations greater than 300 μM, Zn(II) actually decreased the total hemolytic activity of native mastoparan X, perhaps by interacting with the lipid head groups in the membrane, so we restricted the analysis to metal ion concentrations lower than this value. In the presence of Mst-HH, both metal ions showed a concentration-dependent increase in lysis; a plot of the metal ion concentration versus extent of lysis was well described by a single-site binding isotherm, yielding an apparent dissociation constant (Kdiss (app)) for binding of metal ions to the peptides in the presence of erythrocyte membranes.
Figure 6.
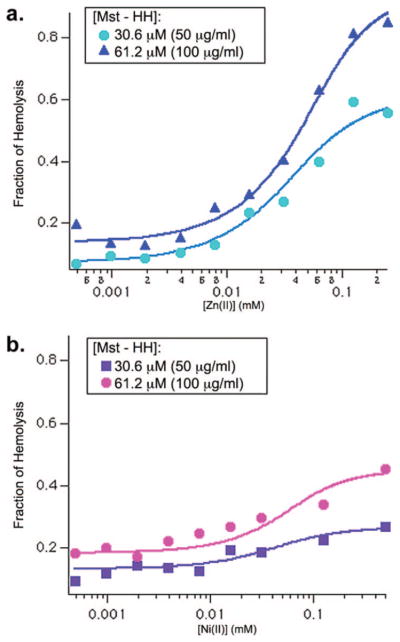
Hemolytic activity of Mst-HH vs (a) Zn(II) and (b) Ni(II) (10 mM Tris, pH = 7.0, 150 mM NaCl). The smooth curves represent the best fit to a single-site binding isotherm (eq 1).
The values of Kdiss (app) in the presence of erythrocytes are expected to be smaller (more favorable) than the actual Kdiss for metal binding to the peptide in aqueous buffer, due to additional favorable lipid-peptide interactions. The value of Kdiss (app) for Zn(II) and Ni(II) are similar, 20 μM and 26 μM, respectively. Interestingly, the maximal induction of hemolytic potency at saturating metal ion concentrations differs between the two peptides, as was further explored in the kinetic studies described below.
Hemolysis Induced by Variants of Mst-HH
Hemolysis experiments were also performed on the additional Mst variants (Figure 7, Table 2). The negative control, Mst-H5H, which contains an unproductive i, i+5 spacing of the ligating histidine residues, exhibited little hemolysis in the presence and absence of added metals. The Mst-EH peptide, which possesses a Glu-His metal-binding site, is weakly hemolytic in the absence of metal ions (apo HC50: 250 μM); importantly, 300 μM Ni(II) has no effect on its lytic activity. This result is expected from the soft character of the Ni(II), which is predicted to reduce its affinity for a Glu/His site relative to a His/His site. By contrast, Zn(II), which has intermediate chemical characteristics, increases the hemolytic potential 3-fold.
Figure 7.

Fraction hemolysis vs peptide concentration for Mst-HH peptide, Mst-H5H, Mst-EH, and Mst-HAH (10 mM Tris pH 7.0, 150 mM NaCl). The smooth curves through the data for the peptides, except the negative control Mst-H5H, were obtained from nonlinear least-squares fitting using the Hill equation.
Table 2.
HC50 and Hill Coefficients for All Mst Peptides with and without Added Metal
| peptide/metal | HC50 (μM) | n | fold increase in toxicity |
|---|---|---|---|
| Mastoparan X (apo) | 119 ± 4 | 1.6 ± 0.1 | |
| Mst-HH: | |||
| apo | 295 ± 17 | 1.4 ± 0.1 | |
| 300 μM Zn(II) added | 38 ± 2 | 1.9 ± 0.2 | 8 |
| 300 μM Ni(II) added | 33 ± 2 | 1.3 ± 0.1 | 9 |
| Mst-EH: | |||
| apo* | 250 ± 28 | 2.2 ± 0.6 | |
| 300 μM Zn(II) added | 81 ± 3 | 2.1 ± 0.1 | 3 |
| 300 μM Ni(II) added | 273 ± 6 | 2.2 ± 0.1 | |
| Mst-HAH: | |||
| apo | 69 ± 5 | 1.9 ± 0.2 | |
| 300 μM Zn(II) added | 119 ± 10 | 1.0 ± 0.1 | |
| 300 μM Ni(II) added | 6.7 ± 0.7 | 1.8 ± 0.7 | 10 |
Mst-HAH, maintains the same metal-binding site as the original Mst-HH peptide, but has the helix-breaking residue Gly converted to helix-promoting Ala. This peptide was synthesized following the unexpected finding that the hemolytic potency of Mst-HH was enhanced in the presence of Zn(II), despite the fact that the binding of this metal ion failed to increase the helical content of Mst-HH in aqueous solution. As expected from its enhanced helix-forming potential, the Mst-HAH has greater hemolytic activity than the original Mst-HH peptide in the absence of metal ions. Ni(II) further activates Mst-HAH membrane binding and lysis by 10-fold. On the other hand, Zn(II) failed to increase the hemolytic potency of this peptide, which is in line with the original expectation that Zn(II) would not increase hemolysis because it does not increase the helical content of this peptide.
The Ni(II) activation of the Mst-HAH peptide allowed further analysis by hemolysis-monitored metal titrations. Figure 8 shows the effect of Ni(II) additions on the toxicity of Mst-HAH. A Kdiss (app) value of 12 μM was obtained from analysis of the hemolysis-monitored metal titrations. Thus, Mst-HAH exhibits the lowest Kdiss (app) for binding to Ni(II), as well as the greatest increase in toxicity upon metal-binding.
Figure 8.

Hemolytic activity of Mst-HAH vs [Ni(II)]. The fit of a curve through the data was accomplished using a simple binding equilibrium (eq 1).
Analysis of Binding by Mst-HH to Phospholipid Vesicles in the Presence of Zn(II)
Zn(II) is hypothesized to enhance the lytic ability of Mst-HH by increasing the acidity with which it binds to membranes. Therefore, we evaluated the binding of Mst-HH to phospholipid vesicles in the presence and absence of Zn(II) by monitoring the intrinsic fluorescence of its Trp residue. To help simplify the interpretation of the results and to minimize electrostatic effects, the titrations were conducted using vesicles composed of the zwitterionic phospholipid 5 μM 1-palmitoyl-2-oleoyl-3-phosphocholine (POPC). Mst-HH contains a tryptophan residue at position 3. Titration of Mst-HH with phospholipid vesicles gives rise to a blue-shift in the emission spectrum, which is indicative of transfer of the Trp to a more rigid and/or hydrophobic environment (Figure 9). A plot of the emission maximum versus [vesicles] is well described by a Langmuir isotherm (eq 3), allowing computation of the number of lipids/site as well as the dissociation constant. From the fitting procedure as well as the raw data, it is apparent that Mst-HH binds tighter and with its tryptophan residue in a more hydrophobic or rigid environment in the presence of Zn(II). The increase in affinity is ~5-fold which compares reasonably well to the 8-fold enhancement in HC50 observed with Zn(II). Therefore, Zn(II)-binding facilitates the binding and penetration of Mst-HH into membrane bilayers
Figure 9.

Mst-HH wavelength of maximum tryptophan emission vs [POPC]. Results from fitting the data to a simple dissociation equilibrium (eq 3) with a lipid site size of 80 are depicted as curves through the data points.
Kinetics of Vesicle Lysis by Mst-HH
To gain additional insight into the mechanism by which metal-bound Mst-HH disrupts bilayers, we examined the kinetics of leakage of the aqueous contents of zwitterionic POPC large unilamellar vesicles (LUV). LUVs were loaded with the water-soluble fluorescent dye ANTS (8-aminonaphthalene-1,3,6-trisulfonic acid, disodium salt) and its quencher DPX (p-xylene-bis-pyridinium bromide). Leakage of the aqueous contents of the LUV results in dilution of ANTS and DPX, resulting in an increase in fluorescence at 530 nm. Figure 10 illustrates the time course of dye leakage induced by Mst-HH at several different concentrations of Zn(II) and Ni(II). The biphasic curves have a fast initial phase followed by a slow continued release of dye (Figure 10), as has been seen previously in hemolysis and vesicle assays.20,30–32 There is less total dye leakage with the Ni(II) additions as compared with the Zn(II) leakage studies. This is consistent with the previous hemolysis-monitored metal titrations of Mst-HH using both Zn(II) and Ni(II) (Figure 6), which showed that, under titration conditions, Ni(II) was less effective at inducing hemolysis.
Figure 10.
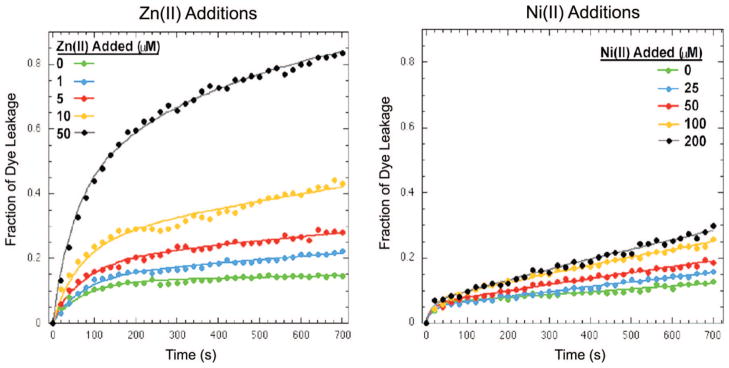
Fraction of leakage from POPC LUV’s by Mst-HH vs time for several concentrations of added Zn(II) (left) and Ni (right). A simple biphasic leakage model (eq 4) was fit to the data and depicted as a solid line through the data points for clarity.
The leakage profiles in Figure 10 were analyzed with a minimal biphasic leakage scheme (eq 4) to highlight the differences in leakage upon metal binding as well as the differences in activation by Zn(II) and Ni(II). The data suggests different mechanisms of activation of Mst-HH by Zn(II) and Ni(II). With increasing additions of Zn(II) there is a significant increase in the initial fast leakage process while Ni(II) appears to activate vesicle lysis through enhancement of the second slower leakage process.
Discussion
Overall, the results of these studies are in agreement with the expectation that enhancing the helix-forming potential by amino acid substitutions or introduction of helix-stabilizing metal-ion binding sites increased their potency, because helix formation and membrane-binding are thermodynamically linked. Potency was decreased by replacing helix-favoring Lys4 and Ala8 of mastoparan X with His, which has a low helix-forming potential. Furthermore, the helical propensity was increased as was the potency by replacing Gly5 with Ala, or by stabilizing the helix via binding Ni(II) to Mst-HH and Mst-HAH. One puzzling discovery, however, was that the addition of Zn(II) increased the potency of Mst-HH, although it failed to increase the helical content of the monomeric peptide in dilute aqueous solution. One possible reason for the increased activity was that the binding of metal ions might increase the net positive charge, increasing the affinity for anionic lipids. However, this does not seem to be the dominant effect, because Zn(II) increased the affinity of the peptide for zwitterionic phospholipid vesicles.
This anomaly is interesting when one considers the stability of metalloproteins with more complex architectures. For example, DF2 proteins and zinc fingers, each of which have two metal-binding ligands projecting from a single α-helix, are actually stabilized in their native conformation by the binding of Zn(II).33,34 Examination of their structures suggests that the binding of Zn(II) causes small deviations of the intervening ϕ − ψ angles from the most preferred α-helical region of the Ramachandran plot. Evidently, these deviations can be better accommodated in a globular protein domain or a surface-bound helix than in a monomeric helix, where a more canonical helical conformation may be essential for propagation of this marginally stable structure. Furthermore, we reasoned that if deviations from an ideal α-helical conformation were required to allow Zn(II) to bind, then these excursions might be encouraged by the intrinsic flexibility of Gly5 of Mst-HH (which lies between the two ligating His residues). To test this concept, we synthesized the HAH peptide (which has an Ala residue in this position) expecting that this strongly helix-favoring residue will accommodate fewer excursions from ideal helical conformations. This peptide was indeed found to have an enhancement in its α-helical content in aqueous solution, and binding Ni(II) but not Zn(II) enhanced its potency. These observations are consistent with—but do not prove—the concept of thermodynamic linkage between metal ion-binding, helicity, and lytic potency. However, we note that there is no simple relationship between helix-forming potential and lysis,35 and it is possible that Zn(II) induction of lysis also includes other less well-defined effects. Indeed, the differences in lysis kinetics induced by Ni(II) versus Zn(II) indicate that their mechanisms of activation are not identical.
From a more practical perspective, this work demonstrates a successful strategy for selectively inducing membrane interactions, as the binding of transition metal ions amplified the lytic capability of mastoparan X in a switchable manner. Furthermore, it is possible to modulate the metal ion selectivity by altering the hard/soft characteristics of the side-chain ligands. Mst-EH is highly selective for Zn(II), while Mst-HH binds both Zn(II) and Ni(II). It should also be possible to enhance the affinity and specificity of the switches by introducing unnatural amino acids into lytic peptides.
Materials and Methods
Molecular Modeling
Models of the designed lytic peptides were constructed in Insight by constraining the backbone to helical ϕ and ψ angles and decorating with the lowest energy amino acid rotamers in the appropriate sequence order. Approximate ϕ and ψ angles for the metal-coordinating histidine were obtained from the structure of hemerythrin (1A7D). Once the model helices were constructed they were then rendered in Pymol (DeLano Scientific).
Peptide Synthesis
All peptides were synthesized using standard Fmoc (9-fluorenylmethoxycarbonyl) amino acid chemistry on an Applied Biosystems model 433A solid phase peptide synthesizer or by hand. Peptides were synthesized on a 0.25 mM scale on PAL resin (Applied Biosystems), which yields a C-terminal carboxyamide. After the final deprotection, the peptides were acetylated at the N-terminus. Peptides were cleaved from the resin for 3 h with TFA:thioanisol:anisol:EDT (90:5:3:2 v/v) and precipitated with cold ether. All peptides were purified using reversed-phase HPLC on a preparative C4 column (Vydac) with the appropriate acidified (0.1% TFA) water/acetonitrile gradient. Sample purity was verified at 98% with analytical reversed-phase HPLC and MALDI-TOF mass spectrometry. All peptides contain a tryptophan at the position 3, which was used to spectroscopically determine peptide concentration by the absorbance at 280 nm (ε280 = 5690 M−1 cm−1).
CD Spectroscopy
CD spectra were acquired on a Jasco J-810 CD spectrometer using 1 cm path length quartz cuvettes. Standard CD samples contained 20 μM Mst peptides made up in 10 mM MOPS, pH = 7.0, with no added salt. CD titration studies were carried out with 1500 μL total sample volume in a 1 cm path length cuvette. Each wavelength scan was taken from 200 to 260 nm as an average of 3 scans, with a 1 nm step and an averaging time of 4 s per point. All scans were taken at 25 °C. Mean residue ellipticity at 222 nm was monitored to observe the change in helical content vs environmental conditions. Control experiments were performed to confirm that the hemoly-sis behavior of these peptides is identical in MOPS or Tris buffer (not shown).
The CD-monitored and hemolysis-monitored metal-titrations were globally analyzed using a simple binding equilibrium:
Pfree = unbound peptide
Pbound = M2+ − peptide complex = C
K = equilibrium dissociation constant
PT = total concentration of peptide
MT = total concentration of M2+
We assume that once we form the peptide-metal complex, it is free to interact with the membrane and cause increased lysis:
F = fractional value measured at a specific [M2+]
H0 = initial value (no metal added)
HMax = maximum value with added metal
PT and MT are known experimental values. We can calculate the fraction of the metal-peptide complex, as opposed to total metal concentration, using the following relationships:
| (1) |
The final value for C is calculated from the solution of the following:
solve for Pfree:
The value of C calculated is used in eq 1 and fit to the hemolysis-monitored and CD-monitored metal titrations. There are two adjustable parameters, Kd and HMax.
Hemolysis Assays
Human red blood cells, without platelets, were obtained from the Bennet Laboratory (UPENN). Erythrocytes were separated from plasma by gentle centrifugation at 4000 rpm for 30 min. Every hemolysis sample contained 0.25% red blood cells in hemolysis buffer (10 mM Tris pH = 7.0, 150 mM NaCl). The total reaction volume for each sample was 100 μL. Samples containing the appropriate peptide and metal concentrations were added to the red blood cells in hemolysis buffer. Samples were prepared in 200 μL thin walled PCR tubes and incubated at 37 °C with gentle shaking for 1 h. After incubation, samples were centrifuged at 4000 rpm for 4 min to separate cellular materials from the supernatant. Twenty μL of the sample supernatant was removed from each sample and diluted with 80 μL of deionized water in a single well of flat bottom polystyrene 96-well plate (Costar). The total absorbance of each sample at 414 nm, corresponding to the absorbance of released hemoglobin, was measured with a Thermolab Systems Multiskan UV–vis plate reader. 100% lysis was obtained by incubating 0.25% red blood cells with 200 μg/mL of melittin. 0% lysis was obtained by incubating 0.25% red blood cells in hemolysis buffer. Fraction of cells lysed was calculated from the following equation: . No increase of hemolysis was observed by addition of divalent metal ions in hemolysis buffer (Zn, Ni, Cd) under reaction conditions. Each point in a hemolysis assay represents a point in the kinetic trace of the hemolysis reaction and a 10% error is assumed.
To obtain HC50 values (concentration of toxin at which 50% of the cells are lysed), the following standard equation was fit to the peptide vs hemolysis data.
| (2) |
POPC Vesicle Preparation
A 10 mg/mL solution of POPC (1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phosphocholine) in chloroform was obtained from Avanti polar lipids. For all vesicle preparations, the appropriate amount of POPC solution was dried down under nitrogen gas and lyophilized for at least 1 h. For small unilamellar vesicles, buffer was added to the dried lipid suspension then sonicated until the solution became clear. For large unilamellar dye-loaded vesicles, a solution of 12.5 mM ANTS (8-aminonaphthalene-1,3,6-trisulfonic acid, disodium salt) and 45 mM DPX (p-xylene-bis-pyridinium bromide) in 10 mM Tris pH = 7.0 was added to the dried lipid. The lipid suspension was vortexed for 10 min until homogeneous. The lipid-dye mixture was freeze thawed 10× and then extruded (Avestin Liposofast-Basic) at least 25× with a 200 nm pore polycarbonate membrane filter (Avestin). After extrusion, the LUV’s were isolated by gel filtration through a G-25 superfine sephadex column equilibrated with 10 mM Tris pH = 7.0, 150 mM NaCl as the mobile phase. Before the drying step, the POPC solution was doped with C-14 labeled lipid. Exact vesicle concentration was obtained through analysis of total C-14 radioactivity after extrusion compared to that after gel filtration.
NMR Spectroscopy
1-D proton NMR spectra were collected on a Bruker 500 MHz spectrometer. All samples were prepared in 100% D2O to reduce the water protons’ resonance. Samples were prepared at 1 mM total peptide with 10 mM Tris pH = 7.0, and 150 mM NaCl at various Zn(II) concentrations. The final sample pH = 6.0. All samples were incubated for 12 h to allow exchange of the amide protons with deuterium. Each spectrum is an average of 256 acquired spectra.
Fluorescence SUV-Binding and LUV-Leakage Assays
All SUV-binding fluorescence experiments were performed at room temperature, 25 °C. SUV-binding experiments were completed on a Hitachi F-2500 FL Spectrophotometer. SUV-binding samples were prepared at a total volume of 2.0 mL with 0.5 μM peptide or 5 μM peptide (respectively, with and without 100 μM or 300 μM metal added) in 10 mM Tris, pH = 7.0, 150 mM NaCl. A 10 mM SUV stock was prepared in identical buffer conditions and added in 1–10 μL increments. All samples were excited at 280 nm and the emission spectrum was collected from 250–500 nm with a scan speed of 300 nm/min and a response time of 0.08 s. Excitation and emission slit widths were set at 2 nm. The PMT voltage was 400 V. The peak apex for the tryptophan emission was recorded for each spectrum.
LUV-leakage studies were performed on an Aviv model ATF-105 spectrometer. Vesicles were loaded with the ANTS/DPX cocktail (12.5 mM ANTS/45 mM DPX). The excitation wavelength was set at 353 nm. The emission at 530 nm was monitored for 800 s with data points taken at 20 s intervals with an averaging time of 2 s. Excitation and emission bandwidths were set at 2 nm and the signal PMT setpoint was 850. The first 100 s of the experiments were monitored before the addition of peptide to confirm the presence of a stable baseline. A 0% dye release was determined by this initial fluorescence, The peptide was added to the sample and the fluorescent dye leakage was monitored for the remaining 700 s. The value for 100% dye release was determined from adding 0.1% Triton X-100 to the sample, mixing well, and averaging the resultant fluorescence signal for an additional 100 s. All leakage data was normalized to with the values for 0% and 100% release, Fraction of dye leakage = (Fsample – F0%release)/(F100%release – F0%release).
Curve Fitting the SUV-Binding Data
A simple dissociation equilibrium was fit to tryptophan fluorescence monitored SUV titrations:
| (3) |
The wavelength of maximum emission, λMax, was measured vs the concentration of phospholipid to determine Kd and m (the number of lipids required to form a peptide-binding site). In order to determine an accurate value for these parameters, two SUV titrations were performed at 0.5 μM and 5 μM Mst-HH with and without the addition of excess Zn(II). Equation 3 was used to globally fit curves for the apo and metal-bound 5 μM Mst-HH data, which yielded a value of 80 for m. This value was fixed when fitting Kd to the low-concentration data, because m was less well defined when the peptide concentration was 0.5 μM.
Curve Fitting LUV Leakage Data
To further analyze metal-activated LUV leakage data, a minimal biphasic leakage scheme was fit to the Mst-HH LUV-leakage time courses:
With rearrangement and integration the final fitting equation becomes:
| (4) |
For both Zn(II) and Ni(II) leakage data, the value of k did not systematically change and was a fixed parameter. Equation 4 was fit to all of the leakage time courses.
Table 3.
Mst-HH SUV-Binding Fitted Parameters
| fitted parameters | Mst-HH, no metal added | Mst-HH, 100 μM Zn(II) |
|---|---|---|
| λmax-saturation (nm) | 330 | 328 |
| Kdiss (per site) | 33.0 μM | 6.76 μM |
| no. of lipids bound | 80 | 80 |
Acknowledgments
We thank Dr. James Lear for many helpful conversations, Dr. Soni Basra for help with the NMR spectroscopy, and NIH Grant No. R0156423 for financial support. We also received support from the NSF MRSEC grant to the LRSM of the University of Pennsylvania.
References
- 1.Ruan FQ, Chen YQ, Hopkins PB. J Am Chem Soc. 1990;112:9403–9404. [Google Scholar]
- 2.Ghadiri MR, Choi C. J Am Chem Soc. 1990;112:1630–1632. [Google Scholar]
- 3.Ghosh D, Pecoraro VL. Inorg Chem. 2004;43:7902–7915. doi: 10.1021/ic048939z. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh D, Pecoraro VL. Curr Opin Chem Biol. 2005;9:97–103. doi: 10.1016/j.cbpa.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Ghadiri MR, Soares C, Choi C. J Am Chem Soc. 1992;114:825–831. [Google Scholar]
- 6.Farrer BT, Pecoraro VL. Proc Natl Acad Sci U S A. 2003;100:3760–3765. doi: 10.1073/pnas.0336055100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiyokawa T, Kanaori K, Tajima K, Koike M, Mizuno T, Oku JI, Tanaka T. J Peptide Res. 2004;63:347–353. doi: 10.1111/j.1399-3011.2004.00109.x. [DOI] [PubMed] [Google Scholar]
- 8.Case MA, Ghadiri MR, Mutz MW, McLendon GL. Chirality. 1998;10:35–40. [PubMed] [Google Scholar]
- 9.Calhoun JR, Nastri F, Maglio O, Pavone V, Lombardi A, DeGrado WF. Biopolymers. 2005;80:264–278. doi: 10.1002/bip.20230. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki K, Hiroaki H, Kohda D, Nakamura H, Tanaka T. J Am Chem Soc. 1998;120:13008–13015. [Google Scholar]
- 11.Kohn WD, Kay CM, Sykes BD, Hodges RS. J Am Chem Soc. 1998;120:1124–1132. [Google Scholar]
- 12.Ambroggio XI, Kuhlman B. J Am Chem Soc. 2006;128:1154–1161. doi: 10.1021/ja054718w. [DOI] [PubMed] [Google Scholar]
- 13.Ulrich AS, Otter M, Glabe CG, Hoekstra D. J Biol Chem. 1998;273:16748–16755. doi: 10.1074/jbc.273.27.16748. [DOI] [PubMed] [Google Scholar]
- 14.Tossi A, Sandri L, Giangaspero A. Biopolymers. 2000;55:4–30. doi: 10.1002/1097-0282(2000)55:1<4::AID-BIP30>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y, Guarnieri MT, Vasil AI, Vasil ML, Mant CT, Hodges RS. Antimicrob Agents Chemother. 2007;51:1398–406. doi: 10.1128/AAC.00925-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Mant CT, Farmer SW, Hancock RE, Vasil ML, Hodges RS. J Biol Chem. 2005;280:12316–29. doi: 10.1074/jbc.M413406200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maloy WL, Kari UP. Biopolymers. 1995;37:105–22. doi: 10.1002/bip.360370206. [DOI] [PubMed] [Google Scholar]
- 18.Park Y, Hahm KS. J Biochem Mol Biol. 2005;38:507–16. doi: 10.5483/bmbrep.2005.38.5.507. [DOI] [PubMed] [Google Scholar]
- 19.Frecer V. Bioorg Med Chem. 2006;14:6065–74. doi: 10.1016/j.bmc.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Degrado WF, Musso GF, Lieber M, Kaiser ET, Kezdy FJ. Biophys J. 1982;37:329–338. doi: 10.1016/S0006-3495(82)84681-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwarz G, Reiter R. Biophys Chem. 2001;90:269–277. doi: 10.1016/s0301-4622(01)00149-1. [DOI] [PubMed] [Google Scholar]
- 22.Tang J, Signarvic RS, DeGrado WF, Gai F. Biochemistry. 2007;46:13856–63. doi: 10.1021/bi7018404. [DOI] [PubMed] [Google Scholar]
- 23.Mendes MA, de Souza BM, Marques MR, Palma MS. Toxicon. 2004;44:67–74. doi: 10.1016/j.toxicon.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 24.Scholtz JM, Barrick D, York EJ, Stewart JM, Baldwin RL. Proc Natl Acad Sci U S A. 1995;92:185–9. doi: 10.1073/pnas.92.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang Z, Vasil AI, Hale JD, Hancock RE, Vasil ML, Hodges RS. Biopolymers. 2008;90:369–83. doi: 10.1002/bip.20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abraham T, Marwaha S, Kobewka DM, Lewis RN, Prenner EJ, Hodges RS, McElhaney RN. Biochim Biophys Acta. 2007;1768:2089–98. doi: 10.1016/j.bbamem.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Degrado WF. Adv Protein Chem. 1988;39:51–124. doi: 10.1016/s0065-3233(08)60375-7. [DOI] [PubMed] [Google Scholar]
- 28.Higashijima T, Wakamatsu K, Takemitsu M, Fujino M, Nakajima T, Miyazawa T. FEBS Lett. 1983;152:227–30. doi: 10.1016/0014-5793(83)80385-8. [DOI] [PubMed] [Google Scholar]
- 29.Wèuthrich K. NMR of Proteins and Nucleic Acids. Wiley; New York: 1986. [Google Scholar]
- 30.van den Bogaart G, Guzman JV, Mika JT, Poolman B. J Biol Chem. 2008;283:33854–7. doi: 10.1074/jbc.M805171200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamba Y, Yamazaki M. Biochemistry. 2005;44:15823–33. doi: 10.1021/bi051684w. [DOI] [PubMed] [Google Scholar]
- 32.Epand RF, Zhang YL, Mirzabekov T, Kagan B, Silberstein A, Hubbell WL, Epand RM, Chakraborti S, Dimitrov DS, Anderson WF, Rozenberg-Adler Y. Exp Mol Pathol. 2008;84:9–17. doi: 10.1016/j.yexmp.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 33.Calhoun JR, Kono H, Lahr S, Wang W, DeGrado WF, Saven JG. J Mol Biol. 2003;334:1101–15. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 34.Berg JM. J Biol Chem. 1990;265:6513–6. [PubMed] [Google Scholar]
- 35.Schmitt MA, Weisblum B, Gellman SH. J Am Chem Soc. 2007;129:417–28. doi: 10.1021/ja0666553. [DOI] [PubMed] [Google Scholar]