

Abstract
Screening of the 50,000 ChemBridge compound library led to the identification of the oxadiazole-isopropylamide 1 (PI-1833) which inhibited CT-L activity (IC50 0.60 μM) with little effects on the other 2 major proteasome proteolytic activities, T-L and PGPH-L. LC/MS-MS and dialysis show that 1 is a non-covalent and rapidly reversible CT-L inhibitor. Focused library synthesis provided 11ad (PI-1840) with CT-L activity (IC50 27 nM). Detailed SAR studies indicate that the amide moiety and the 2 phenyl rings are sensitive toward modifications. Hydrophobic residues, such as propyl or butyl, in the para-position (not ortho or meta) of the A-ring and a meta-pyridyl group as B-ring significantly improve activity. Compound 11ad (IC50 0.37 μM) is more potent than 1 (IC50 3.5 μM) at inhibiting CT-L activity in intact MDA-MB-468 human breast cancer cells and inhibiting their survival. The activity of 11ad warrants further pre-clinical investigation of this class as non-covalent proteasome inhibitors.
INTRODUCTION
The ATP-dependent ubiquitin-proteasome pathway is responsible for the controlled degradation of proteins in eukaryotic cells.1-6 The 26S proteasome is a multifunctional complex consisting of a 19S regulatory particle (RP) and a 20S core particle (CP).7 The three main catalytic activities of the proteasome; peptidylglutamyl peptide hydrolyzing-like (PGPH-L), trypsin-like (T-L), and chymotrypsin-like (CT-L) are mediated by three distinct catalytic β-1, β-2, and β-5 subunits, respectively.8 For each of the catalytic β-subunits, the N-terminal Thr-1 serves as the nucleophile that initiate cleavage of the peptide bond.9,10,11 Development of inhibitors of CT-L activity has been the subject of considerable interest in the treatment of cancer due to its critical role in the degradation of apoptotic and tumor suppressor proteins.8,10,12 The proteasome inhibitors advanced to clinic or clinical trials are derived from 3 structural classes (Figure 1A): 1. Boronic acid containing compounds such as bortezomib, a dipeptide boronic acid (clinically approved by FDA),13, 14 MLN970815-17 and CEP-18770;18-21 (Figure 1A). 2. β-lactones such as salinosporamide A (NPI-0052)22,23 which is a marine microbial natural product (Figure 1A) and 3. Epoxyketone containing tetrapeptide carfilzomib24 (Figure 1A), clinically approved by FDA which is related to the natural product epoxomicin. Each inhibitor class reacts with the proteasome N-terminal threonine (Thr-1 at the active site) by a distinct mechanism. Peptide boronic acids form a covalent and slowly reversible tetrahedral adducts with the OH group of the catalytic Thr-1.11,25 For the β-lactone salinosporamide A, attack of the lactone ring by catalytic Thr-126 forms an ester bond (that undergoes intramolecular rearrangement) which makes this compound an irreversible inhibitor. The epoxyketone14 moiety of carfilzomib reacts with the OH and the α-amino group of Thr-1 to form 2 covalent bonds, also making the inhibition irreversible.
Figure 1.

A. Structures of clinically advanced covalent proteasome inhibitors B. Structures of non-covalent small molecule proteasome inhibitors; hydroxyurea pharmacophore38 and peptidic pharmacophore from Millennium.11,45
Covalent proteasome inhibitors are classified as slow reversible or irreversible inhibitors according to their chemical structure and mechanism of inhibition. Covalent irreversible or covalent slow reversible inhibitors as described above possess a chemically reactive group that bind to the proteasome covalently. In contrast, non-covalent inhibitors inhibit the proteasome through a network of interactions (hydrophobic, hydrogen bonds, electrostatic and/or van der Waals). Although some proteasome inhibitors have been suggested to bind non-covalently27-34 structural evidence to support this was provided for only three of these. The X-ray structures of TMC-95A,35-37 hydroxyl urea38 (Figure 1B) and the peptide11 from Millenium (Figure 1B) bound to proteasome demonstrate that the binding mode of these compounds are non-covalent.
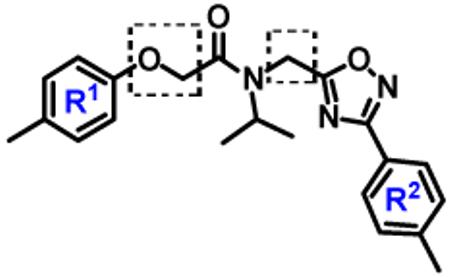
Since non-covalent inhibitors do not have a reactive electrophilic moiety or ‘warhead’, which is often associated with metabolic instability, poor specificity, and excessive reactivity, they have the advantage of exerting fewer side effects over the covalent ones. It has been shown that the proteasome activity recovers at the same rate with covalent irreversible inhibitors as with covalent slow reversible inhibitors, presumably via de novo proteasome synthesis.39 The clinical advantages/benefits of non-covalent proteasome inhibitors in cancer treatment are not well understood. Figure 1B shows the structures of small molecules that have been identified as non-covalent proteasome inhibitors.11,38 We have been actively engaged in the discovery of novel proteasome inhibitors.40,41 We reported the discovery of the compound 1 as a proteasome inhibitor in a poster at the 2011 American Association for Cancer Research (AACR) meeting.42 Villoutreix et al also have reported oxadiazole-isopropylamide containing compounds as proteasome modulators.43,44 Although Villoutreix et al and our group have independently identified similar scaffolds, each group focused on different modifications of the hits that led to important findings that are complementary but not overlapping. In our study, we have extensively explored SAR (Figure 2) on the oxadiazole-isopropylamide containing compounds as proteasome inhibitors by systematically synthesizing focused libraries around key features of the pharmacophore. We present compound 1 and its most potent analogs as non-peptidic, non-covalent and reversible proteasome inhibitors that have the potential to become clinical candidates.
Figure 2.
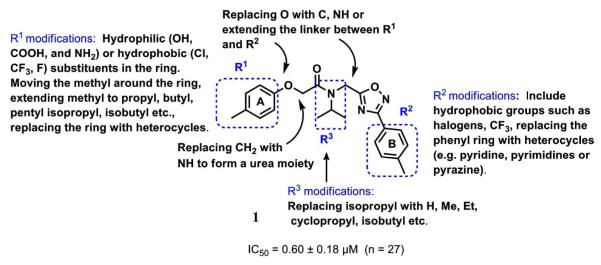
Modifications and library synthesis around 1 for design of new proteasome inhibitors and SAR studies.
CHEMISTRY
The screening hit 1 was identified as a CT-L proteasome inhibitor with an IC50 value of 0.60 ± 0.18 μM (in-vitro). Validation of the synthetic route to 1 was undertaken to confirm both the structure and the in vitro CT-L inhibitory activity. Synthesis of 1 was achieved using the route shown in Scheme 1. The substituted acetyl chloride building block library 5 (Scheme 1) was synthesized from readily available phenol derivatives via the ester 3 and acid 4 using reported protocols.46-50 The oxadiazole portion of the compound 1 was synthesized from readily available nitrile building blocks 6. The nitrile building blocks were reacted with hydroxylamine hydrochloride and sodium carbonate at 70 °C in water to yield the hydroxyamidines51 7 (Scheme 1, condition g). The 5-substituted pyrimidinehydroxyamidine 7h (see supporting information) was synthesized starting from the commercially available methyl pyrimidine-5-carboxylate via amide 24 and nitrile 25.52 The intermediate hydroxyamidine library 7 was reacted with chloroacetyl chloride (Scheme 1, condition h) to provide the library 8,53 which was cyclized in refluxing toluene to provide the oxadiazole portion of the pharmacophore 9. The library 9 was subsequently reacted with appropriate alkyl amines (isopropyl-, isobutyl-, methyl-. ethyl-, cyclopropyl- and tert-butyl amines) to obtain the amine building block library 1054 (Scheme 1, condition j). For the synthesis of 10n (R3 = H), compound 9a (R2 = para-tolyl, see supporting information compound 26) was first reacted with phthalimide in the presence of potassium carbonate in refluxing acetonitrile, followed by reaction with hydrazine to obtain the compound 10n in high yield.55 We were able to generate library 10 with a variety of substituted aryl and hetero-aryl R2 moieties and library 5 with substituted/unsubstituted aromatic R1 moieties (Scheme 1). Modifications around 1 for library synthesis are shown in Figure 2, and initially we focused our synthetic efforts to modify R1, R2 and R3 moieties. The two key building block libraries 10 and 5 were then reacted in the presence of triethylamine to provide the compound 1, library 11 and 12 (Scheme 1, Tables 1 and 3) in good yields. The route described in Scheme 1 was efficient and convenient for rapid synthesis and optimization of the substituted phenyl and amide moieties. The final libraries 11, 12 and compound 1 were characterized using NMR, LC-MS, HRMS and the purity was > 95% as determined by HPLC. The final compound library 11 and 12 (including compound 1) showed formation of approximately 3:1 atropisomers (hindered rotation about the C-N amide bond) by 1H NMR and 13C NMR spectroscopy (see the experimental section and supporting information). Analysis of 1H NMR spectra of compound 1 at variable temperatures (20 °C to 50 °C) showed that the peaks from the minor rotamer coalesced with the major rotamer as the temperature increased.
Scheme 1.

Synthetic route to compound 1, libraries 11 and 12. Reagents and conditions: a. Ethyl bromoacetate, K2CO3, Acetone, reflux, 14 h. b. tert-Butyl bromoacetate, DMF, 80 °C, 14 h. c. Ethyl bromoacetate, K2CO3, DMF, r.t., 14 h. d. NaOH, THF, reflux, 2 h. (R= Ethyl). e. CF3COOH, DCM, r.t., 2 h (R = tBu). f. SOCl2, benzene, reflux, 3 h. g. NH2OH.HCl, Na2CO3, water, 70 °C, 14 h. h. Chloroacetyl chloride, acetone, r.t., 30 min. i. toluene, reflux, 2 h. j. Alkylamine, K2CO3, CH3CN, reflux, 30 min. k. Et3N, THF, r.t., 15 min.
Table 1.
Synthetic analogs of Library 12 and SAR.
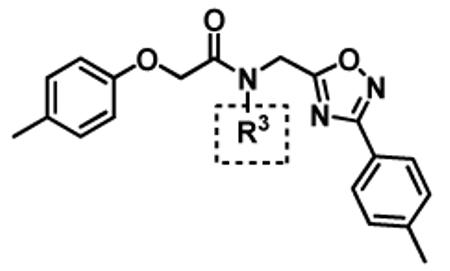
| Entry | Compound ID |
R3 | IC50 (μM) CT-La |
|---|---|---|---|
| 1 | 1 | isopropyl | 0.60 ± 0.18 (n=27) |
| 2 | 12a | isobutyl | 2.37 ± 0.40 (n=2) |
| 3 | 12b | ethyl | 6.04 ± 1.34 (n=3) |
| 4 | 12c | methyl | 29.90 ± 3.9 (n=2) |
| 5 | 12d | H | No inhibition @ 10 μM |
| 6 | 12e | tert-butyl | No inhibition @ 10 μM |
| 7 | 12f | cyclopropyl | No inhibition @ 10 μM |
IC50 values are given as the average of 2 or more determinations; n = number of determinations.
Table 3.
Compound 1, synthetic analogs of library 11 and SAR.
| Entry | Compound ID |
R1 | R2 | IC50 (μM) CT-L a |
|---|---|---|---|---|
| 13 | 1 |
|
|
0.60 ± 0.18 (n=27) |
| 14 | 11a |
|
|
1.08 ± 0.33 (n=4) |
| 15 | 11b |
|
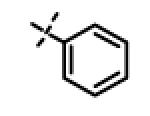
|
0.43 ± 0.12 (n=4) |
| 16 | 11c |
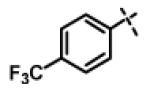
|
|
2.53 ± 0.95 (n=5) |
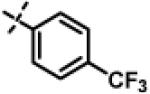
| 17 | 11d |
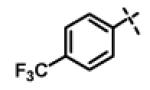
|
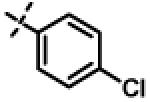
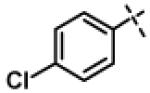
|
0.94 ± 0.26 (n=8) |
| 18 | 11e |
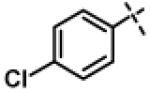
|
|
1.12 ± 0.33 (n=5) |
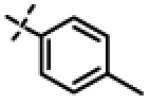
| 19 | 11f |
|
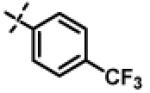
|
1.41 ± 0.19 (n=2) |
| 20 | 11g |
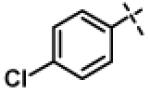
|
|
1.07 ± 0.05 (n=4) |
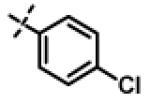
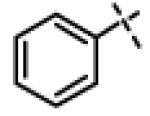
| 21 | 11h |
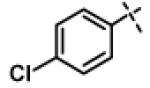
|
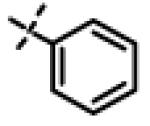
|
0.51 ± 0.16 (n=6) |
| 22 | 11i |
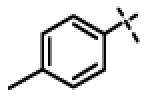
|
|
0.45 ± 0.15 (n=4) |
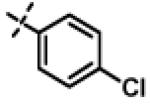
| 23 | 11j |
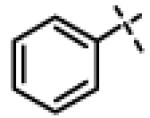
|
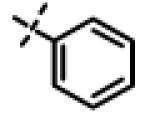
|
6.20 ± 1.1 (n=2) |
| 24 | 11k |
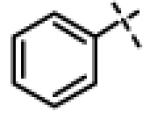
|
|
8.50 ± 0.60 (n=2) |
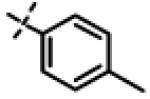
| 25 | 11l |
|
|
11.20 ± 2.1 (n=3) |
| 26 | 11m |
|
|
0.31 ± 0.08 (n=4) |
| 27 | 11n |
|
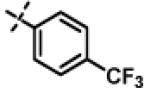
|
0.97 ± 0.15 (n=2) |
| 28 | 11o |
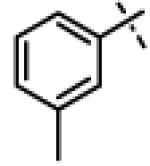
|
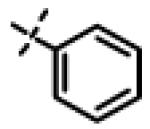
|
No inhibition @ 10 μM |
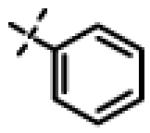
| 29 | 11p |
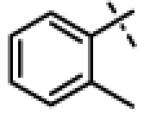
|
|
No inhibition @ 10 μM |
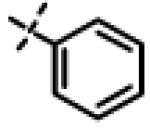
| 30 | 11q |
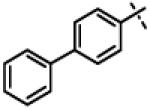
|
|
No inhibition @ 10 μM |
| 31 | 11r |
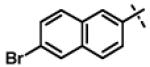
|
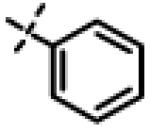
|
No inhibition @ 10 μM |
| 32 | 11s |
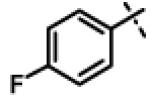
|
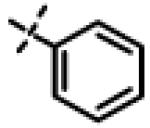
|
10.09 ± 1.63(n=3) |
| 33 | 11t |
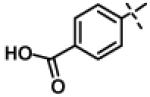
|
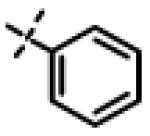
|
No inhibition @ 10 μM |
| 34 | 11u |
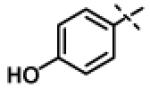
|
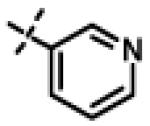
|
0.98 ± 0.50 (n=3) |
| 35 | 11v |
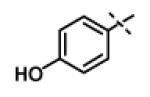
|
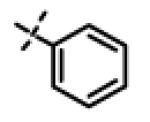
|
10.12 ± 4.5 (n=3) |
| 36 | 11w |
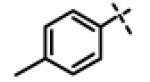
|
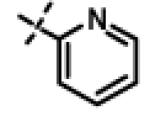
|
3.75 ± 1.47 (n=6) |
| 37 | 11x |
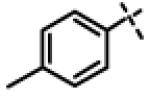
|
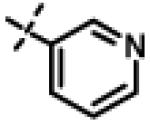
|
0.22 ± 0.084 (n=6) |
| 38 | 11y |
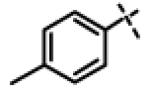
|
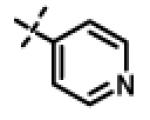
|
0.37 ± 0.05 (n=4) |
| 39 | 11z |
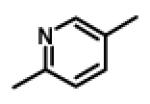
|
|
4.0 ± 0.90 (n=3) |
| 40 | 11aa |
|
|
0.26 ± 0.05 (n=6) |
| 41 | 11ab |
|
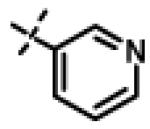
|
0.099 ± 0.032 (n=14) |
| 42 | 11ac |
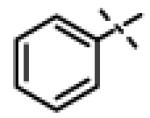
|
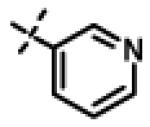
|
2.71 ± 0.56 (n=5) |
| 43 | 11ad |
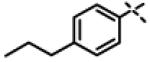
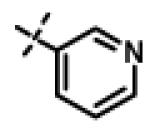
|
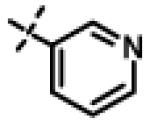
|
0.027 ± 0.014 (n=20) |
| 44 | 11ae |
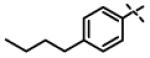
|
|
0.039 ± 0.01 (n=7) |
| 45 | 11af |
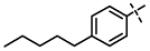
|
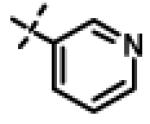
|
0.120 ± 0.04 (n=3) |
| 46 | 11ag |
|
|
0.43 ± 0.04 (n=3) |
| 47 | 11ah |
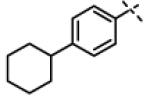
|
|
1.36 ± 0.18 (n=3) |
| 48 | 11ai |
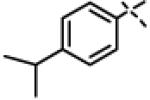
|
|
0.44 ± 0.07 (n=3) |
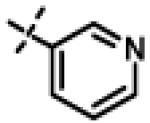
| 49 | 11aj |
|
|
46.49 ± 1.23(n=3) |
| 50 | 11ak |
|
|
0.140 ± 0.05 (n=3) |
| 51 | 11al |
|
|
0.032 ± 0.003 (n=3) |
| 52 | 11am |
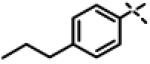
|
|
0.105 ± 0.031 (n=3) |
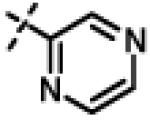
| 53 | 11an |
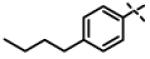
|
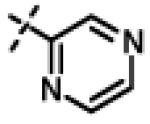
|
0.107 ± 0.013 (n=3) |
| 54 | 11ao |
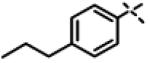
|
|
1.27 ± 0.22 (n=3) |
| 55 | 11ap |
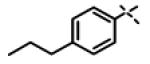
|
|
0.273 ± 0.10 (n=3) |
| 56 | 11aq |
|
|
No inhibition @ 10 μM |
| 57 | 11ar |
|
|
1.93 ± 0.61 (n=4) |
| 58 | 11as |
|
|
0.14 ± 0.064 (n=4) |
IC50 values are given as the average of 2 or more determinations; n = number of determinations.
Next, we focused our efforts to modify the chemical space between the amide moiety, A and B rings (Figure 2) in compound 1. First, we introduced a urea moiety to assess the SAR. To install the urea moiety, the intermediate 10d was reacted with commercially available isocyanate 13 in the presence of triethylamine in refluxing benzene, and under these conditions urea 14 was obtained in good yield (Scheme 2, condition q). Further modifications included replacement of the ether moiety (H-bond acceptor) in 1 (Figure 2) with an NH (H-bond acceptor/donor) group. The amine 10d was first reacted with chloroacetyl chloride in the presence of triethylamine in THF at room temperature to obtain intermediate 15 (Scheme 2) followed by coupling 15 with para-methylaniline using sodium acetate in refluxing ethanol to obtain the final compound 16 (Scheme 2, conditions r and s respectively) also in good yield. The ether moiety in 1 (Figure 2) was also replaced by a methylene unit using 3-(4-(trifluoromethyl)phenyl)propanoic acid building block (17a). The acid starting material 17a (Scheme 2) was converted to the corresponding acid chloride 18a and coupled with 10d to provide the oxadiazole 19a (Scheme 2). The final compound 19b with bulky R-groups was synthesized following the route in Scheme 2 starting from benzofuran-2-carboxylic acid (17b) via the formation of acid chloride 18b and subsequent coupling with 10f. The intermediate 10d was chosen for synthesis of compounds 14, 16, and 19a since our early SAR indicated unsubstituted B ring is desirable to retain in vitro CT-L potency together with para-CH3 group on the A ring and the isopropyl moiety in the amide group. Extension of the linker between the oxadiazole moiety and amide group in 1 (Figure 2) by one carbon atom was achieved by first reacting the intermediate 7d with 3-chloropropanoyl chloride to generate intermediate 20 which provided 21 upon refluxing in toluene. The intermediate 21 was next reacted with isopropylamine to obtain 22, which was further reacted with intermediate 5a (see supporting information) to obtain the final compound 23 in good yield. The intermediates and final compounds were characterized by 1H NMR, 13C NMR, HRMS, LC-MS; the purity of final compounds was determined using HPLC.
Scheme 2.

Reagents and conditions: q. Et3N, benzene, reflux, 14 h, 78%. r. Chloroacetyl chloride, Et3N, THF, r.t., 15 min., 80%. s. para-Methylaniline, NaOAc, ethanol, reflux, 15 h, 78%. f. SOCl2, benzene, reflux, 3 h, 94%. i. toluene, reflux, 2 h, 81%. j. Isopropylamine, K2CO3, CH3CN, reflux, 30 min., 86%. k. Et3N, THF, r.t., 15 min., 88% (19a), 82% (19b), 87%, (23). l. DCM, r.t., 14 h, 76%.
RESULTS AND DISCUSSIONS
Unlike lactacystin, 1 is a non-covalent and reversible CT-L inhibitor
To determine whether our hit compound 1 is a non-covalent CT-L inhibitor, we used two approaches, Liquid Chromatography Tandem Mass Spectrometry (LC/MS-MS) and dialysis. With both approaches we used lactacystin, a known covalent and irreversible CT-L inhibitor as a control.56,57 For LC/MS-MS, 1 or lactacystin were incubated with the 20S proteasome, digested with trypsin and the resulting peptides purified by HPLC and analyzed by LC/MS-MS as described under Methods. Figure 3A shows that peptides purified from the vehicle-treated samples contained unmodified TTTLAFK peptide (observed as a protonated molecule at m/z 781.4401) corresponding to the N-terminal tryptic peptide of rabbit proteasome subunit β type-5. Figure 3B shows that peptides purified from the compound 1 treated samples also contained the unmodified Thr-1 containing TTTLAFK peptide (Figure 3D). Both intact mass spectrum and tandem mass spectrum indicate unmodified Thr-1 containing peptide. Figure 3C shows that lactacystin-treated samples contained a lactacystin-modified Threonine adduct corresponding to the doubly charged modified peptide at m/z 497.7795 (structure shown in Figure 3E). The searches matching experimental data to peptides, from the database of rabbit proteasome subunits produced only one modified peptide, which indicated that Thr-1 on β-5 was modified by lactacystin. No other modifications to β-5 subunit from samples treated with DMSO, compound 1 or lactacystin were observed. In addition, no modifications were detected for other proteasome subunits, such as β-1 and β-2 subunits included in the database (n =21). These results suggest that, unlike lactacystin, 1 does not bind covalently to the proteasome.
Figure 3.

A: LC/MS-MS analysis of tryptic digests from proteasome CT-L subunit after incubation with vehicle. LC/MS-MS analysis of Thr-1 containing peptide from the proteasome CT-L subunit β5 after tryptic digestion is shown. Singly charged unmodified peptide was observed at m/z 781.4401, which represents a mass error of 6.9 ppm. The tandem mass spectrum was matched to peptide TTTLAFK. The b ions (labeled in red), contain the N-terminus of the peptide; and y ions, (labeled in blue), contain the C-terminus of the peptide. The number associated with each ion indicates the number of amino acids in that fragment (for example, y4 contains LAFK from C-terminus of the peptide). B: LC/MS-MS analysis of tryptic digests from proteasome CT-L subunit after incubation with compound 1. The Thr-1 containing peptide didn’t show any modification. Both intact mass spectrum and tandem mass spectrum indicate unmodified Thr-1 containing peptide. C: LC/MS-MS analysis of tryptic digests from proteasome CT-L subunit after incubation with lactacystin shows clasto-lactacystin-modified Thr-1 containing peptide. Doubly charged lactacystin-Thr modified peptide was detected at m/z 497.7795, which represents a mass error 6.1 ppm. The tandem mass spectrum confirms the modification of the peptide by lactacystin. D: Structure of the unmodified TTTLAFK tryptic peptide. E: Structure of the clasto-lactacystin modified peptide.
To confirm these results, we incubated 1 and lactacystin with the 20S proteasome and determined the reversibility of binding by dialysis as described under Methods. The Figure 4 shows that more than 70% of the CT-L activity in the dialysis compartment from the sample that was treated with 1 was recovered within the first 20 min., and recovered fully by 2 hours of dialysis. In contrast, very little CT-L activity was recovered from the sample that was treated with lactacystin even after 18 hours of dialysis (Figure 4). Taken together these results confirm the well established fact that lactacystin is a covalent and irreversible CT-L inhibitor and indicate that 1 is a non-covalent and reversible CT-L inhibitor. This is consistent with the chemical structure of 1 that lacks any reactive groups, unlike clasto-lactacystin that contains the reactive beta-lactone moiety. The β-lactone moiety of the clasto-lactacystin covalently modifies the β-subunits of the proteasome and is responsible for inhibiting 20S proteasome in an irreversible mode of action57.
Figure 4.

Recovery of CT-L activity upon dialysis of the 20S proteasome-compound complexes after pre-incubation with lead 1 and lactacystin.
Structure Activity Relationship studies and chemistry leading to compound 11ad
Our screening efforts of the 50,000 in-house ChemBridge compound library led to the discovery of the hit 1, an inhibitor of the CT-L activity of the proteasome. After confirming the structure and in vitro CT-L activity of the in-house synthesized 1 (Scheme 1), we embarked on synthetic modifications to develop structure and activity relationship (SAR) data to identify novel, potent and selective CT-L proteasome inhibitors that block the action of the proteasome in a non-covalent manner. Proteasome CT-L activity was measured using a fluorogenic assay as previously described.41 Focused library synthesis was undertaken by independently varying the R1, R2 and R3 groups in compound 1 (Figure 2). Initially, we replaced the isopropyl R3 group in 1 with H, isobutyl, ethyl, methyl, tert-butyl and cyclopropyl moieties (Table 1). The loss of CT-L activities of these analogs indicated that the isopropyl group is essential and optimal for proteasome inhibitory activity (see Table 1). The isobutyl amide 12a and ethyl amide 12b (Table 1, Entries 2 and 3) showed 4- to 10-fold loss of activity with IC50 values of 2.37 and 6.02 μM respectively compared to 1. Methyl, H, tert-butyl and cyclopropyl as R3 groups were detrimental for CT-L activity (12c, 12d, 12e, 12f in Table 1 showed weak or no proteasome inhibition at 10 μM). The 1H and 13C NMR spectra of the compound 1 with isopropyl amide group indicated the presence of a mixture of 3:1 atropisomers (isomers that exist due to the hindered rotation about the carbon-nitrogen bond). We observed a similar ratio of atropisomers with 12b (R3 = ethyl), and 12c (R3 = methyl) by analysis of 1H and 13C NMR spectra. Compounds 12d with unsubstituted amide (Entry 5, R3 = H, Table 1), 12e with bulky tert-butyl group (Entry 6, R3 = tBu, Table 1) and 12f with cyclopropyl group (Entry 7, R3 = cyclopropyl) did not show any atropisomers by 1H or 13C NMR spectra.
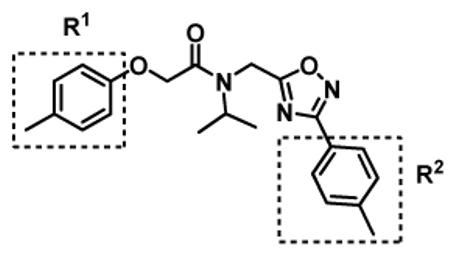
In the next generation of synthetic analogs, we retained the isopropyl R3 group (Figure 2) and modified the phenyl rings in 1 [i.e. library 11, (Scheme 1 and Table 3)]. Initial SAR studies demonstrated that substitutions on the para-position of the R1 and R2 with small hydrophobic groups are tolerable. For example, changing the para-methyl in R1 and R2 to trifluoromethyl or chlorine as in compounds 11a, 11c, 11d, 11e, 11f, 11g, 11i and 11n retained the in vitro CT-L inhibitory activities (Entries 14, 16-20, 22, 27, Table 3). Next we demonstrated that the R1 methyl is required whereas the R2 methyl is dispensable. Indeed, compounds 11b, 11h and 11m (Entries 15, 21 and 26, Table 3) with an unsubstituted phenyl ring as R2 showed slightly improved IC50 values around 0.3 to 0.5 μM indicating para-substitution of R2 phenyl in 1 is not required for inhibitory activity. However, compound 11j with both unsustituted phenyl rings resulted in 10 fold loss of CT-L activity (Entry 23, Table 3, IC50 = 6.22 μM). Removal of the R1 para-methyl group as in compounds 11k and 11l (Entries 24 and 25, Table 3) also led to 14- and 18-fold loss of activity with IC50 values of 8.5 and 11 μM, respectively. The loss of in vitro potency of compounds 11j, 11k and 11l suggest that para-substitution in R1 phenyl ring is critical to maintain CT-L proteasome activity. Changing the R1 para-methyl to meta- or ortho-positions as in compounds 11o and 11p (Entries 28 and 29, Table 3) was detrimental for in vitro potency further suggesting that R1 para-substitution is important for CT-L proteasome activity. Overall our SAR indicated that the para-methyl group in R1 but not in R2 and the R3 isopropyl amide are key features that are responsible for retaining the CT-L proteasome activity of compound 1 and loss of activity was consistent with unsubstituted R1 aromatic rings in this class of compounds.
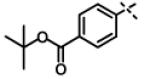
We next modified the chemical space between the amide moiety and R1/R2 in the parent compound 1 (Figure 2) using the synthetic routes and protocols described in Scheme 2 (also see supporting information). First, replacement of the ether-oxygen by methylene showed significant loss of CT-L activity (19a, IC50 53.48 μM, Entry 8, Table 2). Furthermore, substituting the amide group by urea as in 14 (Entry 9, IC50 > 10 μM, Table 2) also led to loss of activity. Replacement of the ether (H-bond acceptor) with NH (H-bond donor/acceptor) also decreased the in vitro CT-L activity (16, IC50 5.67 μM, Entry 10, Table 2). These modifications confirmed that the ether moiety, most likely, as H-bond acceptor, is critical for focused library synthesis and improving the CT-L inhibitory activity. Extending the spacer between the amide and the oxadiazole by one carbon as shown in 23 (Entry 11, IC50 > 10 μM, Table 2) was detrimental for CT-L inhibitory activity, probably due to the increased flexibility of the molecule. Compound 19b (IC50 0.37 μM, Entry 12, Table 2), a bulky R1 substituent with a rigid ether moiety and meta-pyridyl as R2 showed improved inhibitory activity compared to 1 (pyridyl moiety was chosen for the synthesis of 19b, based on the most potent analogs described in Table 3). Overall, synthetic modifications of the linkers between R1, R2 and the amide moiety in 1 were not tolerated.
Table 2.
Modifications of the spacer between the amide, R1 and R2 groups and SAR.
| Entry # | Compound Structure and ID |
IC50 (μM) CT-La |
|---|---|---|
| 8 |
|
53.48 ± 10.89 (n=3) |
| 9 |
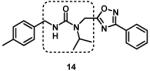
|
No inhibition @ 10 μM |
| 10 |
|
5.67 ± 0.96 (n=4) |
| 11 |
|
No inhibition @ 10 μM |
| 12 |
|
0.379 ± 0.06 (n=3) |
IC50 values are given as the average of 2 or more determinations; n = number of determinations.
To gain more insight in the interactions between R1 and the binding region, we modified the R1 via several synthetic modifications. For example, synthesis of 11q and 11r that possessed large hydrophobic groups such as phenyl and Br-naphthyl as para-R1 groups respectively further revealed that large hydrophobic groups are not tolerated in the binding region (Entries 30, 31, IC50 > 10 μM, Table 3). Replacement of the R1 methyl group in compound 1 by small hydrophobic fluorine as in 11s (Entry 32, Table 3) also displayed poor CT-L inhibitory activity. The 16-fold loss of CT-L inhibitory activity in compound 11s with para-fluorine compared to 11b with para-CF3 (Entry 15, IC50 = 0.43 μM) was not unexpected since fluorine is isosteric to hydrogen58 and as described above we have already observed the detrimental effects of unsubstituted R1 rings in compounds 11k, 11l and 11ac (Entries 24, 25 and 42, Table 3) toward the CT-L inhibitory activity. The hydrophilic COOH, COOtBu and OH groups in the para-position of the R1 as in compounds 11t, 11aq and 11v respectively also failed to maintain the CT-L inhibitory activity (Entries 33, 56 and 35, Table 3) indicating H-bond acceptor/donor moieties are not desirable. In contrast compound 11u (Entry 34, Table 3) with para-hydroxyphenyl as R1 and meta-pyridyl as R2 showed an IC50 value of 1 μM, and comparison of in vitro CT-L activities of 11u and 11v (Entries 34 and 35, Table 3) with para-hydoxyphenyl as R1 highlights the significance of the R2 meta-pyridyl group toward CT-L activity in this class of compounds.
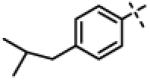
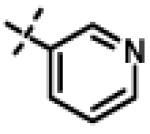
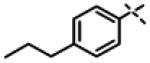
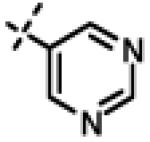
We next made 2 major observations that led us to our most potent compounds. First, we demonstrated that increasing the alkyl length from methyl in 1 to a propyl as in 11ap (IC50 = 0.27 μM) increased potency (Entry 55, Table 3). Second, replacing the tolyl R2 in 1 by a meta-pyridyl (as in 11x, Entry 37, Table 3) or para- pyridyl (as in 11y, Entry 38, Table 3) but not ortho-pyridyl (as in 11w, Entry 36, Table 3) improved potency with the meta-pyridyl (IC50 0.22 μM) being slightly better than the para-pyridyl (IC50 0.37 μ, Table 3). Substituting both, the R1 methyl with a propyl and the R2 tolyl with a meta-pyridyl as in 11ad (combined features of compounds 11ap and 11x) resulted in our most potent compound with an IC50 value of 27 nM. Furthermore, the length of the alkyl group in para-position of the R1-ring is critical with the propyl or butyl groups being the optimal size. Indeed, decreasing the size from propyl (or butyl) to ethyl, methyl and hydrogen as in 11ab, 11x and 11ac, resulted in progressive loss of inhibitory activity from 27 nM to 99 nM, 220 nM and 2710 nM, respectively. Increasing the size of the R1 para-hydrophobic moiety also resulted in progressive loss of potency from propyl or butyl to pentyl, hexyl, isobutyl, isopropyl, cyclohexyl and tert-butyl as in 11af (IC50 120 nM), 11ag (IC50 430 nM), 11ak (IC50 140 nM), 11ai (IC50 440 nM), 11ah (IC50 1356 nM) and 11aj (IC50 46.49 μM), respectively. Furthermore, substituting the pyridyl in 11ad with 3,5-pyrazine as in 11al did not alter the potency (IC50 32 nM) which further supports the importance of a meta-positioned nitrogen atom in the ring. In contrast, substituting the pyridyl in 11ad by 2,5-pyrazine as in 11am (Entry 52, IC50 105 nM, Table 3) with one meta-positioned nitrogen or 2,6-pyrimidine as in 11ao (Entry 54, IC50 1269 nM) which possess two ortho-positioned nitrogen atoms, led to 3-fold and 40-fold loss of CT-L activity respectively (Table 3) further highlighting the importance of the nitrogen in the meta-position of the ring. Compounds 11am and 11an that show CT-L inhibitory activities with IC50 values in the range of 100 nM also demonstrate propyl or butyl in the para-position of the R1 are equally tolerated in the binding region. Interestingly, compound 11ar and 11as with hydrophobic, electron withdrawing fluorine and chlorine in the para-position of the R1 and meta-pyridyl as R2 (Entries 57 and 58, Table 3) showed 3-5-fold improvement in inhibitory activities respectively comparing to its related analogs 11s (Entry 32, Table 3) 11h and (Entry 21, Table 3). Overall, our detailed SAR studies signify the importance of meta-pyridyl, isopropylamide and para-propyl/-butyl phenyl groups in the oxadiazole pharmacophore shown in 1 for inhibition of CT-L activity of the proteasome.
The parent compound 1 and 11ad were further evaluated for T-L and PGPH-L activities of the proteasome as shown in the Table 4. The in vitro data (IC50) indicated that this class of compounds shows excellent selectivity for CT-L inhibitory activity over both T-L and PGPH-L activities. This level of CT-L selectivity is impressive for a non-peptidic, small molecule as compared to the covalent proteasome inhibitors reported to date. Only a small number of compound classes have been reported with this level of activity. 11, 38
Table 4.
IC50 values of 1 and 11ad for CT-L, T-L and PGPH-L activities of the proteasome.
| Compound | CT-L (μM) | T-L (μM)a | PGPH-L (μM)a |
|---|---|---|---|
| 1 | 0.60 ± 0.18 ( n= 27) | >100 | >100 |
| 11ad | 0.027 ± 0.014 (n = 20) |
>100 | >100 |
The values given are the means of 3 experiments; n = number of determinations.
Lead compound 11ad is more potent than the parent compound 1 at inhibiting CT-L activity and survival/proliferation of intact human breast cancer cells
Our in vitro SAR studies demonstrated that substituting the methyl of ring A with a propyl and replacing ring B tolyl with meta-pyridine in 1 (IC50 600 nM) led to 22 fold more potent 11ad (IC50 27 nM). To determine whether these CT-L proteasome inhibitors are cell permeable and inhibit CT-L activity in intact cells, human breast cancer MDA-MB-468 cells were treated with increasing concentrations of 1 or 11ad for 2 hours and the cells were processed for CT-L activity41 determination as described under Methods. Figure 5A shows that 1 inhibited CT-L activity in a dose dependent manner with an IC50 value of 3.5 μM. Figure 5A also shows that consistent with the in vitro results, 11ad was more potent than the parent compound 1 and inhibited the CT-L activity with an IC50 value of 0.37 μM. We next determined whether compounds 1 and 11ad are capable of inhibiting tumor cell survival/proliferation. To this end, MDA-MB-468 cells were treated with parent compound 1 and 11ad and processed for MTT assay as described under Methods. Figure 5B shows that treatment with both compounds inhibited survival/proliferation but that 11ad was more potent than the parent compound 1.
Figure 5.
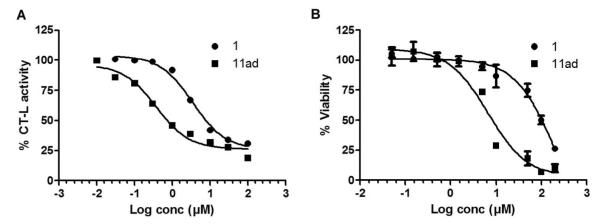
A: Lead 11ad is more potent at inhibiting proteasomal CT-L activity in intact human MDA-MB-468 cancer cells compared to the parent compound 1. B: Lead 11ad is more potent at inhibiting proliferation/survival of human MDA-MB-468 cells compared to the parent hit 1.
CONCLUSIONS
In this study we have extensively explored the SAR of oxadiazole-isopropylamide containing analogs as non-covalent CT-L proteasome inhibitors. The structure of the hit, 1 (IC50 = 0.60 ± 0.18 μM) served as a starting point for synthetic modifications and design and synthesis of novel, highly potent and non-covalent proteasome inhibitors. The LC/MS-MS and dialysis experiments confirmed that 1 binds to CT-L β-5 subunit of the proteasome in a non-covalent and rapidly reversible binding mode. Compound 11ad with an IC50 value of 27 nM was identified as one of the most potent and cell permeable proteasome inhibitors. Our detailed SAR analysis indicated that extending the R1-methyl in 1 to an optimal size with propyl or butyl in combination with meta-pyridyl as R2 significantly improves the CT-L proteasome inhibitory activity (11ad and 11ae, IC50 27 and 39 nM respectively). The isopropyl amide and the ether moieties in 1 are critical for inhibitory activity and modification of these moieties substantially reduced the CT-L inhibitory activity. The SAR demonstrated the importance of the length and composition of the linking chain between the amide moiety, R1 and R2 groups. The potent lead 11ad demonstrated efficacy in cellular assays and showed anti-proliferative activity at low micromolar concentrations. Our in vitro data also highlighted that this class of compounds shows high level of selectivity for CT-L proteasome inhibition over T-L and PGPH-L activities. Moreover, activities observed for some of the other analogs presented herein (such as 11al [IC50 32 nM], 11am [IC50 105 nM] and 11an [IC50 107 nM]) could also be optimized in the development of novel non-covalent CT-L proteasome inhibitors.
MATERIALS AND METHODS
CT-L, T-L, PGPH-L proteolytic activity assays
In the high-throughput screen, we used fluorogenic peptides as substrates to assay (at 10 μM) the 50,000 compounds library from ChemBridge for inhibitory activity against the CT-L proteolytic activity of the purified rabbit 20S proteasome, resulting in the identification of hit 1. To test for selectivity for CT-L over T-L and PGPH-L we used the corresponding fluorogenic peptides. Briefly, 1 nM of purified 20S rabbit proteasome was incubated with 20 μM Suc-Leu-Leu-Val-Tyr-AMC for the CT-L activity, Bz-Val-Gly-Arg-AMC for the T-L activity, and benzyloxycarbonyl Z-Leu-Leu-Glu-AMC for the PGPH-L activity for 1 h at 37 °C in 100 μl of assay buffer (50 mM Tris-HCl, pH 7.6) with or without compound. After incubation, production of hydrolyzed 7-amido-4-methyl-coumarin (AMC) groups was measured using a WALLAC Victor2 1420 Multilabel Counter with an excitation filter of 355 nm and an emission filter of 460 nm (Perkin Elmer Life Sciences, Turku, Finland). The amount of AMC released is within the linear range. Bortezomib was used as a positive control for IC50 determinations (IC50 of Bortezomib was typically around 10-30 nM). We used the following equation to calculate the in vitro IC50 values, A=1/1+([I]/IC50)n, where A is the fraction of activity remaining, [I] is the concentration of inhibitor, and n is the Hill Slope Coefficient. To determine proteasome activity in whole cell, extracts (5 μg) from cultured cell lysate was used instead of 20S rabbit proteasome, and followed the same assay mentioned above except using the following equation; Y = M + (L – M)/ (1+10^ ((X-logIC50)) from GraphPad software, where Y = % inhibition, X = inhibitor concentration, M= maximum % inhibition, L = lowest % inhibition.
Cell culture and cell lysate preparation
Human MDA-MB-468 breast cancer cells were cultured in DMEM medium containing 10% fetal bovine serum (FBS) supplemented with 100 units/ml of penicillin and 100 μg/ml of streptomycin. Cells were maintained at 37 °C in a humidified incubator in an atmosphere of 5% CO2.
Whole cell lysates were prepared as follows
Cells were harvested, washed with PBS twice, and homogenized in a lysis buffer (50 mM Tris-HCl, pH 8.0, 5 mM EDTA, 150 mM NaCl, 0.5% NP-40) for 30 min at 4 °C. Cell lysates were centrifuged at 12,000 g for 15 min, and the supernatants were collected as whole cell lysates.
Dialysis using purified rabbit 20S proteasome
To measure the effect of dialysis on CT-L activity, compounds 1 (10 μM), lactacystin (2.5 μM) or vehicle (DMSO) were added to rabbit 20S proteasome at a final concentration of 1 nM in proteasome assay buffer (50 mM Tris-HCl, pH 7.6) and incubated at room temperature for 30 min. After 30 min of incubation, proteasome-compound mixtures were added to 3500 MWCO Thermo Scientific Slide-A-Lyzer Mini Dialysis Units (Rockford, IL) and dialyzed against proteasome assay buffer. Immediately (t = 0) and 0.25, 1, 2, 4, and 18 hour of dialysis at 4 °C, samples were removed from the dialysis cassette and the CT-L activity of 20S proteasome was determined as described above under “CT-L, T-L, and PGPH-L proteolytic activity assays” section. Proteasome activity was normalized against proteasome activity of DMSO control.
MTT (3-(4,5-dimethythiazol-2-yl)-2,5-diphenyltetrazolium bromide) proliferation/survival assay
Cells were plated in 96-well plates in 100 μl medium and allowed to attach overnight. Cells were then incubated for 120 h with varying concentrations of inhibitors. Media was aspirated and replaced with 100 μl complete media containing 1 mg/ml MTT and incubated for three hours at 37 °C in 5% CO2 humidified incubator. Media was then aspirated and DMSO was added. Cells were incubated for 10 min at room temperature while shaking, and the absorbance was determined at 540 nm using a μQuant spectrophotometric plate reader (Bio-TEK, Winooski, VT). The IC50 values were determined using equation under CT-L, T-L, PGPH-L proteolytic activity assays.
Protein Digestion and Peptide Purification
Rabbit 20S Proteasome (1 nM), inhibitors and buffer (50 mM Tris-Hcl, pH 7.6) were incubated (450 μl total reaction volume) for 30 min at room temperature. After incubation, 112.5 μl of acetonitrile was added and trypsin was added to quench the reaction and denature the protein. Trypsin was added with an enzyme-to-substrate ratio of 1:50. The digestion was carried out for 4 hours at 37 °C. The digest was concentrated by vacuum centrifugation (ISS110, Speedvac, Thermo), and the peptides were extracted with C18 reversed phase pipette tip columns (Ziptip, Millipore). An aliquot (25%) of the total digest was injected into mass spectrometer. To assess LC/MS-MS performance, tryptic peptides from horse apomyoglobin (25 fmol) were spiked in each LC/MS-MS analysis.
LC/MS-MS Analysis
Liquid chromatography-tandem mass spectrometry (LC/MS-MS) peptide sequencing experiments were performed using a nanoflow liquid chromatograph (U3000, Dionex, Sunnyvale, CA) interfaced with an electrospray ion trap mass spectrometer (LTQ-Orbitrap, Thermo, San Jose, CA) in order to detect and localize modified peptides from the proteasome. The sample was first loaded onto a trap column (5mm × 300 μm ID packed with C18 reversed-phase resin, 5μm particle size, 100Å pore size) and washed for 8 minutes with aqueous 2% acetonitrile and 0.04% trifluoroacetic acid. The trapped peptides were eluted onto the analytical column, (C18 Pepmap 100, 75 μm ID × 15 cm, Dionex, Sunnyvale, CA). The 120-minute gradient was programmed as: 95% solvent A (2% acetonitrile + 0.1% formic acid) for 8 minutes, solvent B (90% acetonitrile + 0.1% formic acid) from 5% to 50% in 90 minutes, then solvent B from 50% to 90% B in 7 minutes and held at 90% for 5 minutes, followed by solvent B from 90% to 5% in 1 minute and re-equilibration for 10 minutes. The flow rate on analytical column was 300 nl/min. Five tandem mass spectra were collected in a data-dependent manner following each survey scan. The MS survey scans were performed in Orbitrap to obtain accurate peptide mass measurement and the MS/MS scans were performed in linear ion trap using 60 second exclusion for previously sampled peptide peaks.
Database Searching and Data Analysis
Sequest59 searches were performed against a database containing all rabbit proteasome protein sequences (n = 22) extracted from the UniProt (http://www.uniprot.org). Cleavage was set to fully tryptic, allowing up to two missed cleavages; the precursor mass tolerance was 1.08 Da and MS/MS mass tolerance was 0.8 Da. Dynamic modifications included oxidation (Met + 15.99492), and potential modifications on threonine (Thr +379.18957 for compound 1, Thr +394.20047 for compound 11ad and +213.10009 for β-lactone modification by lactacystin60). The search results were summarized in Scaffold 3.0. (www.proteomesoftware.com). The integrated peak areas for peptide quantification were calculated from extracted ion chromatograms (EIC) using QuanBrowser from Xcalibur 2.0. These values were restricted by m/z (+/− 0.02) and retention time (120 seconds). The accuracy of the m/z values and the fragmentation patterns of the target peptides were manually inspected to insure proper sequence assignment.
EXPERIMENTAL
General
All reagents were purchased from commercial suppliers and used without further purification. Melting points were determined using a Barnstead international melting point apparatus or Optimelt automated melting point system (Stanford Research Systems) and remain uncorrected. Proton NMR spectra were recorded on an Agilent-Varian Mercury 400 MHz spectrometer with CDCl3 or DMSO-d6 as the solvent. Carbon (13C) NMR spectra are recorded at 100 MHz. All coupling constants are measured in Hertz (Hz) and the chemical shifts (δH and δC) are quoted in parts per million (ppm) relative to TMS (δ 0), which was used as the internal standard. Formula guided High resolution mass spectroscopy (HRMS) was carried out on an Agilent 6210 LC-MS (ESI-TOF). HPLC analysis was performed using a JASCO HPLC system equipped with a PU-2089 Plus quaternary gradient pump and a UV-2075 Plus UV-VIS detector, using an Alltech Kromasil C-18 column (150 × 4.6 mm, 5 μm) and Agilent Eclipse XDB-C18 (150 × 4.6 mm, 5 μm). Thin layer chromatography was performed using silica gel 60 F254 plates (Fisher), with observation under UV when necessary. Anhydrous solvents (acetonitrile, dimethylformamide, ethanol, isopropanol, methanol and tetrahydrofuran) were used as purchased from Aldrich. Burdick and Jackson HPLC grade solvents (methanol, acetonitrile and water) were purchased from VWR for HPLC and high resolution mass analysis. HPLC grade TFA was purchased from Fisher.
N-Isopropyl-N-((3-p-tolyl-1,2,4-oxadiazol-5-yl)methyl)-2-(p-tolyloxy)acetamide (1)
To a solution of 10a (187 mg, 0.81 mmol) and triethylamine (164 mg, 1.62 mmol) in THF (15 ml) at r.t., was added 5a (179 mg, 0.97 mmol) in THF (3 mL) dropwise (1-2 min). Upon addition of the acetyl chloride 5a, a precipitate was formed and the reaction was completed in 15 min (monitored by tlc, Rf = 0.75, TLC, EtOAc/Hexane [1:1]). The THF was evaporated and the residue was dissolved in EtOAc (20 ml), washed with HCl (4M, 2 ×15 ml) and water (15 ml). The organic phase was dried (MgSO4), evaporated and the crude product obtained was purified by SiO2 chromatography (EtOAc/Hexane gradient elution) to obtain 1 (270 mg, 88%) as a white solid. mp 142.1-143.4 °C. HPLC 100% (tR = 11.8 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 8.0 Hz, 2H, [7.93 minor isomer]), 7.30-7.25 (m, 2H), 7.10 (d, J = 8.3 Hz, 2H, [7.05 minor isomer]), 6.87 (d, J = 8.5 Hz, 2H, [6.82 minor isomer]), 4.78 (s, 2H, [4.84 minor isomer]), 4.70 (s, 2H, [4.83 minor isomer]), 4.45-4.39 (m, 1H), 2.40 (s, 3H, [2.42 minor isomer]), 2.28 (s, 3H, [2.25 minor isomer]), 1.29 (d, J = 6.6 Hz, 6H, [1.15 minor isomer]); 1H NMR (400 MHz, d6-DMSO) δ 7.84 (d, J = 8.2 Hz, 2H, [7.87 minor isomer]), 7.36 (d, J = 7.9 Hz, 2H, [7.37 minor]), 7.01 (d, J = 8.6 Hz, 2H), 6.78 (d, J = 8.6 Hz, 2H, [6.75 minor isomer]), 4.88 (s, 2H, [4.98 minor isomer]), 4.71 (s, 2H, [4.82 minor isomer]), 4.31-4.21 (m, 1H, [4.62-4.52 minor isomer]), 2.37 (s, 3H), 2.18 (s, 3H), 1.26 (d, J = 6.6 Hz, 6H, [1.06 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.40 [176.54 minor isomer], 168.51, 168.56, 156.01 [155.81 minor isomer], 141.68, 131.23 [131.25 minor isomer shown], 130.29 [130.24 minor isomer], 129.68 [129.84 minor isomer shown], 127.65, 124.08, 114.68 [114.64 minor isomer], 67.99 [68.74 minor isomer], 48.96 [46.96 minor isomer], 37.20 [38.40 minor isomer], 21.49 [19.97 minor isomer], 20.81, 20.73; Anal. Calcd for C22H25N3O3: C, 69.64; H, 6.64; N, 11.07. Found: C, 69.51; H, 6.74; N, 11.13; LC-MS (ESI+) m/z 380.20 (M+H)+; HRMS (ESI+ve) m/z calculated for C22H26N3O3 (M+H)+ 380.1969, found 380.1975.
N-Isopropyl-N-((3-p-tolyl-1,2,4-oxadiazol-5-yl)methyl)-2-(4-(trifluoromethyl)phenoxy)acetamide (11a)
This compound was prepared from 5b (149 mg, 0.62 mmol) and 10a (120 mg, 0.52 mmol) using triethylamine (105 mg, 1.04 mmol) in a similar manner as described for compound 1. The crude product was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11a (196 mg, 87%) as a white solid. mp 76.6-78.5 °C. HPLC 99.5% (tR = 14.8 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 8.1 Hz, 2H), 7.57-7.49 (m, 2H), 7.25 (d, J = 7.9 Hz, 2H, [7.29 minor isomer]),7.04 (d, J = 8.5 Hz, 2H, [6.99 minor isomer]), 4.88 (s, 2H, [4.94 minor isomer]), 4.71 (s, 2H, [4.76 minor isomer]), 4.38-4.32 (m, 1H), 2.41 (s, 3H, [2.43 minor isomer]), 1.32 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100MHz, CDCl3) δ 176.16 [176.23 minor isomer], 168.57 [167.84 minor isomer], 167.80 [167.87 minor isomer], 160.48, 141.86 [142.43 minor isomer], 129.73, 127.58, 127.29 (q, J = 3.67 Hz), 124.47 (q, J = 270 Hz), 124.11 (q, J = 32.5 Hz), 123.88 [123.86 minor isomer], 114.96 [114.82 minor isomer], 67.48 [67.90 minor isomer], 49.00 [47.05 minor isomer], 37.23 [38.35 minor isomer], 21.81, 21.45 [19.96 minor isomer]; 19F NMR (376 MHz, CDCl3) δ −62.02 [−62.06 minor isomer]; LC-MS (ESI+) m/z 434.18 (M+H)+; HRMS (ESI+ve) m/z calculated for C22H23F3N3O3 (M+H)+ 434.1686, found 434.1711.
N-Isopropyl-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)-2-(4-(trifluoromethyl)phenoxy)acetamide (11b)
This compound was prepared from 5b (131 mg, 0.55 mmol) and 10d (100 mg, 0.46 mmol) using triethylamine (93 mg, 0.92 mmol) in a similar manner as described for compound 1. The crude product was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11b (172 mg, 89%) as a white solid. mp 97.9-99.3 °C. HPLC 100% (tR = 10.4 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.02-7.98 (m, 2H), 7.55-7.43 (m, 5H), 7.04 (d, J = 8.8 Hz, 2H, [7.01 minor isomer]), 4.88 (s, 2H, [4.94 minor isomer]), 4.72 (s, 2H, [4.79 minor isomer]), 4.39-4.35 (m, 1H), 1.32 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100MHz, CDCl3) δ 176.36 [176.47 minor isomer], 168.58 [168.54 minor isomer shown], 167.66 [166.71 minor isomer], 160.50 [160.47 minor isomer], 131.47 [131.92 minor isomer], 129.00 [129.20 minor isomer], 127.65, 127.29 (q, J = 3.74 Hz), 126.78 [126.17minor isomer], 124.46 (q, J = 270 Hz), 124.14 (q, J = 32.7 Hz), 114.97 [114.90 minor isomer], 67.56 [68.07 minor isomer], 48.96 [47.07 minor isomer], 37.21 [38.41 minor isomer], 21.48 [19.98 minor isomer]; 19F NMR (376 MHz, CDCl3) δ −62.03 [−62.07 minor isomer]; LC-MS (ESI+) m/z 420.16 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H21F3N3O3 (M+H)+ 420.1530, found 420.1547.
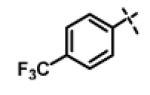
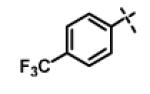
N-Isopropyl-2-(4-(trifluoromethyl)phenoxy)-N-((3-(4-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11c)
This compound was prepared from 5b (100 mg, 0.42 mmol) and 10b (100 mg, 0.35 mmol) using triethylamine (71 mg, 0.70 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11c (157 mg, 92%) as a white solid. mp 96.8-98.9 °C. HPLC 95% (tR = 19.0 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 4:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 8.3 Hz, 2H), 7.72 (d, J = 8.3 Hz, 2H), 7.54 (d, J = 8.8 Hz, 2H, [7.49 minor isomer]), 7.04 (d, J = 8.7 Hz, 2H, [6.97 minor isomer]), 4.88 (s, 2H, [4.91 minor isomer]), 4.72 (s, 2H, [4.84 minor isomer]), 4.42-4.38 (m, 1H), 1.34 (d, J = 6.6 Hz, 6H, [1.17 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.91, 167.87 [167.90 minor isomer], 167.60, 160.42 [160.02 minor isomer], 133.18 (q, J = 33 Hz), 130.16, 128.01 [128.12 minor isomer], 127.32 (q, J = 3.59 Hz), 126.03 (q, J = 3.81 Hz), 124.41 (q, J = 270 Hz), 124.23 (q, J = 32.59 Hz), 123.94 (q, J = 270 Hz), 114.94 [114.83 minor isomer], 67.51 [64.80 minor isomer], 49.08, 37.30, 29.94, 21.50 [19.97 minor isomer]; 19F NMR (376 MHz, CDCl3) δ −62.07 [−62.02 minor isomer], −62.13 [−62.14 minor isomer], −62.45 [−62.52 minor isomer]; LC−MS (ESI+) m/z 488.14 (M+H)+; HRMS (ESI+ve) m/z calculated for C22H20F6N3O3 (M+H)+ 488.1403, found 488.1419.
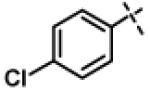
N-((3-(4-chlorophenyl)-1,2,4-oxadiazol-5-yl)methyl)-N-isopropyl-2-(4-(trifluoromethyl)phenoxy) acetamide (11d)
This compound was prepared from 5b (115 mg, 0.48 mmol) and 10c (101 mg, 0.40 mmol) using triethylamine (81 mg, 0.80 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11d (154 mg, 85%) as a white solid. mp 79.0-80.5 °C. HPLC 100% (tR = 16.5 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8.6 Hz, 2H), 7.54 (d, J = 8.7 Hz, 2H, [7.50 minor isomer]), 7.43 (d, J = 8.6 Hz, 2H, [7.46 minor isomer]), 7.04 (d, J = 8.6 Hz, 2H, [6.99 minor isomer]), 4.88 (s, 2H, [4.92 minor isomer]), 4.70 (s, 2H, [4.80 minor isomer]), 4.41-4.35 (m, 1H), 1.32 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.60 [176.74 minor isomer], 167.81, 167.70, 160.46 [160.40 minor isomer], 137.61, 129.35 [129.54 minor isomer], 128.96, 127.31 (q, J = 3.7 Hz), 125.28, 124.45 (q, J = 270 Hz), 124.17(q, J = 32 Hz), 114.95 [114.87 minor isomer], 67.54 [68.25 minor isomer], 49.00 [ 47.16 minor isomer], 37.25 [38.50 minor isomer], 21.50 [19.98 minor isomer]; 19F NMR (376 MHz, CDCl3) δ −62.03 [−62.08 minor isomer]; LC-MS (ESI+) m/z 454.12 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H20ClF3N3O3 (M+H)+ 454.1140, found 454.1149.
2-(4-Chlorophenoxy)-N-isopropyl-N-((3-p-tolyl 1,2,4-oxadiazol-5-yl)methyl)acetamide (11e)
This compound was prepared from 5d (106 mg, 0.52 mmol) and 10a (100 mg, 0.43 mmol) using triethylamine (87 mg, 0.86 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11e (151 mg, 88%) as a white solid. mp 133.1-136.5 °C. HPLC 99.6% (tR = 12.5 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 8.3 Hz, 2H, [7.91 minor isomer]), 7.30-7.26 (m, 2H), 7.23 (d, J = 9.0 Hz, 2H, [7.20 minor isomer]), 6.86 (d, J = 9.0 Hz, 2H, [6.87 minor isomer]), 4.81 (s, 2H, [4.86 minor isomer]), 4.70 (s, 2H, [4.79 minor isomer]), 4.42-4.35 (m, 1H), 2.41 (s, 3H, [2.41 minor isomer]), 1.30 (d, J = 6.6 Hz, 6H, [1.15 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.22 [176.34 minor isomer], 168.57, 168.01, 156.75, 141.75 [142.36 minor isomer], 129.90, 129.74 [129.72 minor isomer], 129.67, 127.61 [126.90 minor isomer], 123.98 [123.42 minor isomer], 116.24 [116.17 minor isomer], 67.95 [68.47 minor isomer], 48.93 [46.97 minor isomer], 37.17 [38.37 minor isomer], 21.82, 21.47 [19.98 minor isomer]; LC-MS (ESI+) m/z 400.15 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H23ClN3O3 (M+H)+ 400.1423, found 400.1448.
2-(4-Chlorophenoxy)-N-isopropyl-N-((3-(4-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)methyl) acetamide (11f)
This compound was prepared from 5d (86 mg, 0.42 mmol) and 10b (100 mg, 0.35 mmol) using triethylamine (71 mg, 0.70 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11f (129 mg, 81%) as a white solid. mp 106.4-108.9 °C. HPLC 99.8% (tR = 17.1 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 4:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 8.1 Hz, 2H, [8.13 minor isomer]), 7.73 (d, J = 8.2 Hz, 2H, [7.76 minor isomer]), 7.23 (d, J = 9.0 Hz, 2H, [7.18 minor isomer]), 6.91 (d, J = 9.0 Hz, 2H, [6.83 minor isomer]), 4.81 (s, 2H, [4.85 minor isomer]), 4.70 (s, 2H, [4.83 minor isomer]), 4.45-4.39 (m, 1H), 1.32 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 177.00 [177.20 minor isomer], 168.16, 167.64, 156.68, 133.11 (q, J = 32.6 Hz), 130.18, 129.76, 128.04, 126.96, 126.08 (q, J = 3.8 Hz), 123.96 (q, J = 270 Hz), 116.20 [116.08 minor isomer], 67.91 [68.88 minor isomer], 49.01 [47.09 minor isomer], 37.24 [38.52 minor isomer], 21.50 [19.88 minor isomer]; 19F NMR (376 MHz, CDCl3) δ −63.40 [−63.46 minor isomer]; LC-MS (ESI+) m/z 454.11 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H20ClF3N3O3 (M+H)+ 454.1140, found 454.1142.
2-(4-Chlorophenoxy)-N-((3-(4-chlorophenyl)-1,2,4-oxadiazol-5 yl)methyl)-N-isopropylacetamide (11g)
This compound was prepared from 5d (98 mg, 0.48 mmol) and 10c (101 mg, 0.40 mmol) using triethylamine (81 mg, 0.80 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11g (133 mg, 79%) as a white solid. mp 133.4-135.8 °C. HPLC 99.9% (tR = 15.5 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 4:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 8.7 Hz, 2H, [7.96 minor isomer]), 7.44 (d, J = 8.6 Hz, 2H, [7.48 minor isomer]), 7.23 (d, J = 9.0 Hz, 2H, [7.19 minor isomer]), 6.91 (d, J = 9.1 Hz, 2H, [6.84 minor isomer]), 4.80 (s, 2H, [4.83 minor isomer]), 4.69 (s, 2H, [4.81 minor isomer]), 4.40-4.37 (m, 1H), 1.30 (d, J = 6.6 Hz, 6H, [1.15 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.66 [176.83 minor isomer], 168.10, 167.78, 156.69, 137.56, 129.75, 129.36 [129.54 minor isomer], 128.99, 126.93, 125.29, 116.20 [116.11 minor isomer], 67.90 [68.68 minor isomer], 48.96, 37.20, 21.49 [19.98 minor isomer]; LC-MS (ESI+) m/z 420.08 (M+H)+; HRMS (ESI+ve) m/z calculated for C2020Cl2N3O3 (M+H)+ 420.0876, found 420.0891.
2-(4-Chlorophenoxy)-N-isopropyl-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)acetamide (11h)
This compound was prepared from 5d (119 mg, 0.58 mmol) and 10d (104 mg, 0.48 mmol) using triethylamine (97 mg, 0.96 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11h (152 mg, 82%) as a white solid. mp 108.3-109.5 °C. HPLC 99.8% (tR = 9.5 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.03−7.99 (m, 2H), 7.56−7.41 (m, 3H), 7.23 (d, J = 9.0 Hz, 2H, [7.19 minor isomer]), 6.91 (d, J = 9.0 Hz, 2H, [6.87 minor isomer]), 4.80 (s, 2H, [4.85 minor isomer]), 4.70 (s, 2H), 4.43−4.36 (m, 1H), 1.30 (d, J = 6.6 Hz, 6H, [1.15 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.42, 168.56, 168.08, 156.73, 131.44 [131.87 minor isomer], 129.75 [129.68 minor isomer], 129.75 [129.02 minor isomer], 127.69, 126.91, 126.79, 116.23 [116.16 minor isomer], 67.92 [68.52 minor isomer], 48.97 [47.07 minor isomer], 37.19 [38.43 minor isomer], 21.47 [19.98 minor isomer]; LC-MS (ESI+) m/z 386.14 (M+H)+; HRMS (ESI+ve) m/z calculated for C20H21ClN3O3 (M+H)+ 386.1266, found 386.1269.
N-((3-(4-chlorophenyl)-1,2,4-oxadiazol-5-yl)methyl)-N-isopropyl-2-(p-tolyloxy)acetamide (11i)
This compound was prepared from 5a (89 mg, 0.48 mmol) and 10c (101 mg, 0.40 mmol) using triethylamine (81 mg, 0.80 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11i (131 mg, 82%) as a white solid. mp 133.5-134.3 °C. HPLC 99.7% (tR = 21.0 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.6 Hz, 2H), 7.43 (d, J = 8.6 Hz, 2H, [7.45 minor isomer]), 7.08 (d, J = 8.3 Hz, 2H, [7.03 minor isomer]), 6.87 (d, J = 8.6 Hz, 2H, [6.79 minor isomer]), 4.78 (s, 2H, [4.86 minor isomer]), 4.69 (s, 2H, [4.81 minor isomer]), 4.46-4.40 (m, 1H), 2.28 (s, 3H, [2.25 minor isomer]), 1.30 (d, J = 6.6 Hz, 6H, [1.15 minor isomer]); 13C NMR (101 MHz, CDCl3) δ 176.85, [176.00 minor isomer], 168.56, 167.78, 156.26, 137.34, 131.26, 130.28 [130.24 minor], 129.30 [129.45 minor isomer], 129.04, 125.42, 114.67 [114.59 minor isomer], 67.98 [68.95 minor isomer], 48.96 [47.05 minor isomer], 37.22 [38.48 minor isomer], 21.50 [19.99 minor isomer], 20.74; LC-MS (ESI+) m/z 400.14 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H23ClN3O3 (M+H)+ 400.1423, found 400.1423.
N-Isopropyl-2-phenoxy-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)acetamide (11j)
This compound was prepared from 5c (47 mg, 0.28 mmol) and 10d checked (50 mg, 0.23 mmol) using triethylamine (47 mg, 0.46 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11j (61 mg, 76%). as a white solid. mp 77.9-79.0 °C. HPLC 94.4% (tR = 11.6 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 4:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.02 (dd, J = 8.1, 1.6 Hz, 2H, partially overlapped, [8.05 minor isomer, overlapped]), 7.53-7.43 (m, 3H), 7.32-7.25 (m, 2H), 7.03-6.85 (m, 3H), 4.83 (s, 2H, [4.88 minor isomer]), 4.72 (s, 2H, [4.86 minor isomer]), 4.46-4.40 (m, 1H), 1.31 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.53 [176.65 minor isomer], 168.72 [168.80 minor isomer], 168.53, 158.01 [157.85 minor isomer], 156.40, 131.44 [131.81 minor isomer], 129.88 [129.82 minor isomer], 129.79, 129.00 [129.17 minor isomer], 127.73 [127.69 minor isomer], 127.04, 126.78, 122.00, 116.21, 114.86 [114.81 minor isomer shown], 67.66 [68.34 minor isomer shown], 49.09 [47.12 minor isomer], 37.28 [38.45 minor isomer], 21.43 [19.95 minor isomer]; LC-MS (ESI+) m/z 352.16 (M+H)+; HRMS (ESI+ve) m/z calculated for C20H22N3O3 (M+H)+ 352.1656, found 352.1662.
N-Isopropyl-2-phenoxy-N-((3-p-tolyl-1,2,4-oxadiazol-5-yl)methyl)acetamide (11k)
This compound was prepared from 5c (80 mg, 0.47 mmol) and 10a (90 mg, 0.39 mmol) using triethylamine (78 mg, 0.77 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11k (111 mg, 78%) as a white solid. mp 97.8-99.5 °C. HPLC 99.8% (tR = 8.5 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 8.2 Hz, 2H, [7.93 minor isomer shown]), 7.34-7.22 (m, 4H), 7.02-6.94 (m, 3H), 4.82 (s, 2H [4.87 minor isomer]), 4.71 (s, 2H, [4.83 minor isomer]), 4.45-4.39 (m, 1H), 2.41 (s, 3H [2.42 minor isomer]), 1.30 (d, J = 6.6 Hz, 6H, [1.15 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.49 [176.50 minor isomer], 168.34, 168.55, 158.08, 141.68 [142.23 minor isomer],129.86 [129.81 minor isomer],129.68, 127.65, 124.01, 121.95, 114.86 [114.83 minor isomer], 67.81 [68.49 minor isomer], 48.96 [46.89 minor isomer], 37.21 [38.37 minor isomer], 21.82, 21.48 [19.98 minor isomer]; LC-MS (ESI+) m/z 366.19 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H24N3O3 (M+H)+ 366.1812, found 366.1816.
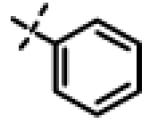
N-Isopropyl-2-phenoxy-N-((3-(4-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11l)
This compound was prepared from 5c (57 mg, 0.34 mmol) and 10b (80 mg, 0.28 mmol) using triethylamine (57 mg, 0.56 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11l (95 mg, 81%) as a white solid. mp 122.8-123.8 °C. HPLC 100% (tR = 11.6 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.13 (d, J = 8.1 Hz, 2H, [8.15 minor isomer]), 7.72 (d, J = 8.2 Hz, 2H, [7.75 minor isomer]), 7.35-7.26 (m, 2H), 7.03-6.94 (m, 3H, [6.90 minor isomer]), 4.83 (s, 2H, [4.89 minor isomer]), 4.72 (s, 2H, [4.86 minor isomer]), 4.48-4.43 (m, 1H), 1.32 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3 ) δ 177.15 [177.39 minor isomer], 168.45, 167.58, 158.04, 133.07 (q, J = 32.6 Hz), 130.29 [130.27 minor isomer shown], 129.87 [129.83 minor isomer],128.09, 125.97 (q, J = 3.7 Hz), 123.99 (q, J = 271 Hz), 121.99 [122.07 minor isomer], 114.83 [114.75 minor isomer], 67.75 [68.75 minor isomer], 49.01 [47.05 minor isomer], 37.27 [38.58 minor isomer], 21.50 [19.98 minor isomer]; 19F NMR (376 MHz, CDCl3) δ −63.37, [−63.43 minor isomer]; LC-MS (ESI+) m/z 420.15 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H21F3N3O3 (M+H)+ 420.1530, found 420.1530.
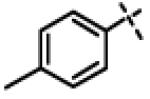
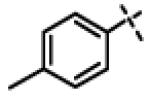
N-Isopropyl-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)-2-(p-tolyloxy)acetamide (11m)
This compound was prepared from 5a (102 mg, 0.55 mmol) and 10d (100 mg, 0.46 mmol) using triethylamine (93 mg, 0.92 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11m (153 mg, 91%). as a white solid. mp 134.7-136.5 °C. HPLC 96.3% (tR = 8.5 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.02 (dd, J = 8.0, 1.6 Hz, 2H overlapped, [8.04 minor isomer overlapped]), 7.55-7.41 (m, 3H), 7.08 (d, J = 8.3 Hz, 2H, [7.04 minor isomer]), 6.87 (d, J = 8.6 Hz, 2H, [6.81 minor isomer]), 4.78 (s, 2H, [4.85 minor isomer]), 4.71 (s, 2H, [4.83 minor isomer]), 4.45-4.40 (m, 1H), 2.28 (s, 3H, [2.25 minor isomer]), 1.30 (d, J = 6.6 Hz, 6H, [1.15 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.62, 168.55, 168.53, 156.00 [155.80 minor isomer], 131.37 [131.74 minor isomer], 131.22 [131.25 minor isomer], 130.28 [130.24 minor isomer], 128.97 [129.13 minor isomer], 127.73, 126.89, 114.68 [114.62 minor isomer], 67.97 [68.75 minor isomer], 48.93 [46.94 minor isomer], 37.20 [38.44 minor isomer], 21.48, [19.98 minor isomer], 20.74; LC-MS (ESI+) m/z 366.18 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H24N3O3 (M+H)+ 366.1812, found 366.1828.
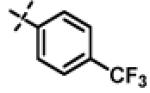
N-Isopropyl-2-(p-tolyloxy)-N-((3-(4-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11n)
This compound was prepared from 5a (78 mg, 0.42 mmol) and 10b (100 mg, 0.35 mmol) using triethylamine (71 mg, 0.70 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11n (127 mg, 84%) as a white solid. mp 146.9-148.6 °C. HPLC 99% (tR = 16.1 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 4:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 8.1 Hz, 2H), 7.72 (d, J = 8.2 Hz, 2H, overlapped, [7.75 minor isomer, overlapped]), 7.08 (d, J = 8.3 Hz, 2H, [7.02 minor isomer]), 6.87 (d, J = 8.6 Hz, 2H, [6.77 minor isomer]), 4.79 (s, 2H, [4.89 minor isomer]), 4.70 (s, 2H, [4.81 minor isomer]), 4.48-4.42 (m, 1H), 2.28 (s, 3H, [2.23 minor isomer]), 1.31 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 177.20 [177.43 minor isomer], 168.63, 167.57, 155.98 [155.73 minor isomer], 133.05 (q, J = 32.5 Hz), 131.27 [131.37 minor isomer], 130.29 [130.24 minor isomer], 128.09, 125.97 (q, J = 3.8 Hz), 124.0 (q, J = 270.6 Hz), 114.65 [114.54 minor isomer], 67.94 [68.85 minor isomer], 48.97 [47.08 minor isomer], 37.26 [38.63 minor isomer], 21.50 [19.62 minor isomer], 20.74; 19F NMR (376 MHz, CDCl3) δ −63.39 [−63.45 minor isomer]; LC-MS (ESI+) m/z 434.18 (M+H)+; HRMS (ESI+ve) m/z calculated for C22H23F3N3O3 (M+H)+ 434.1686, found 434.1693.
N-Isopropyl-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)-2-(m-tolyloxy)acetamide (11o)
This compound was prepared from 5h (66 mg, 0.36 mmol) and 10d (65 mg, 0.30 mmol) using triethylamine (61 mg, 0.60 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11o (83 mg, 76%) as a colorless viscous compound; HPLC 98.8% (tR = 8.6 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.01 (dd, J = 8.0, 1.5 Hz, 2H, [8.08 minor isomer]), 7.52-7.42 (m, 4H), 7.17 (t, J = 7.6 Hz, 1H, overlapped, [7.15, minor isomer])), 6.82-6.69 (m, 2H), 4.79 (s, 2H, [4.85 minor isomer]), 4.71 (s, 2H, [4.84 minor isomer]), 4.46-4.40 (m, 1H), 2.31 (s, 3H, [2.25 minor isomer]), 1.31 (d, J = 6.6 Hz, 6H, [1.16 minor isomer shown]); 13C NMR (100 MHz, CDCl3) δ 176.58, 168.56, 168.43, 158.09, 139.99, 131.36 [131.74 minor isomer], 129.58 [129.53 minor isomer], 128.96 [129.13 minor isomer], 127.72, 126.90, 122.80 [122.84 minor isomer], 115.64 [115.74 minor isomer], 111.70 [111.46 minor isomer], 67.79 [68.59 minor isomer], 48.97 [46.97 minor isomer shown], 37.22 [38.52 minor isomer], 21.76, 21.49 [19.98 minor isomer shown]; LC-MS (ESI+) m/z 366.19 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H24N3O3 (M+H)+ 366.1812, found 366.1817.
N-Isopropyl-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)-2-(o-tolyloxy)acetamide (11p)
This compound was prepared from 5i (51 mg, 0.28 mmol) and 10d (50 mg, 0.23 mmol) using triethylamine (57 mg, 0.56 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11p (71 mg, 85%) as a white solid. mp 97.3-98.0 °C. HPLC 98.0% (tR = 9.5 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 7.95 (broad dd, J = 7.9, 1.6 Hz, 2H), 7.51-7.43 (m, 3H), 7.17-7.09 (m, 2H), 6.92-6.87 (m, 2H), 4.82 (s, 2H, [4.81 minor isomer]), 4.71 (s, 2H), 4.48-4.38 (m, 1H), 2.28 (s, 3H, [2.20 minor isomer]), 1.29 (d, J = 6.6 Hz, 6H, [1.14 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.57 [176.68 minor isomer], 168.57, 168.50, 156.23, 131.38 [131.77 minor isomer], 131.18, 128.98 [129.15 minor isomer], 127.73, 127.25, 126.87, 126.84, 121.61 [121.69 minor isomer], 111.28 [111.63 minor isomer], 68.01 [68.86 minor isomer], 48.94 [48.90 minor isomer], 37.22 [38.26 minor isomer], 21.51 [20.07 minor isomer], 16.62; LC-MS (ESI+) m/z 366.18 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H24N3O3 (M+H)+ 366.1812, found 366.1821.
2-(Biphenyl-4-yloxy)-N-isopropyl-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)acetamide (11q)
This compound was prepared from 5e (116 mg, 0.47 mmol) and 10d (85 mg, 0.39 mmol) using triethylamine (79 mg, 0.78 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11q (145 mg, 87%) as a white solid. mp 147.8-148.7 °C. HPLC 99.6% (tR = 14.6 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.02 (dd, J = 8.2, 1.4 Hz, 2H), 7.55-7.36 (m, 9H), 7.31 (t, J = 7.4 Hz, 1H), 7.05 (d, J = 8.8 Hz, 2H, [7.00 minor isomer]), 4.87 (s, 2H, [4.91 minor isomer]), 4.73 (s, 2H), 4.48-4.42 (m, 1H), 1.33 (d, J = 6.6 Hz, 6H, [1.17 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.55 [176.69 minor isomer], 168.58, 168.30 [168.38 minor isomer], 157.63 [157.49 minor isomer], 140.78, 135.04 [135.07 minor isomer], 131.36 [131.77 minor isomer], 128.98 [129.16 minor isomer], 128.94, 128.53, 127.71, 127.04, 127.01, 126.88, 115.17 [115.10 minor isomer], 67.86 [68.62 minor isomer], 48.98 [47.02 minor isomer], 37.22 [38.50 minor isomer], 21.51 [20.00 minor isomer shown]; LC-MS (ESI+) m/z 428.19 (M+H)+; HRMS (ESI+ve) m/z calculated for C26H26N3O3 (M+H)+ 428.1969, found 428.1968.
2-(6-Bromonaphthalen-2-yloxy)-N-isopropyl-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)acetamide (11r)
This compound was prepared from 5g (140 mg, 0.47 mmol) and 10d (85 mg, 0.39 mmol) using triethylamine (79 mg, 0.78 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11r (141 mg, 75%) as a white solid. mp 97.7-98.4 °C. HPLC 98.1% (tR = 19.6 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 7.99-7.86 (m, 3H), 7.67 (d, J = 9.0 Hz, 1H), 7.56 (d, J = 8.9 Hz, 1H), 7.53-7.36 (m, 4H), 7.24 (dd, J = 9.0, 2.6 Hz, 1H), 7.19 (d, J = 2.3 Hz, 1H), 4.93 (s, 2H, [4.98 minor isomer]), 4.72 (s, 2H, [4.86 minor isomer]), 4.52-4.46 (m, 1H), 1.33 (d, J = 6.6 Hz, 6H, [1.18 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.47, 168.59, 168.14, 156.26, 133.02, 131.80, 131.36 [131.40 minor isomer], 130.63, 130.00 [129.94 minor isomer], 129.82, 129.03 [129.14 minor isomer], 128.94, 127.63 [127.68 minor isomer], 126.75, 119.65 [119.49 minor isomer], 117.90, 107.72, 67.89 [68.50 minor isomer], 49.03 [47.10 minor isomer], 37.23 [38.65 minor isomer], 21.53 [19.99 minor isomer]; LC-MS (ESI+) m/z 480.09 and 482.09 (M+H)+; HRMS (ESI+ve) m/z calculated for C24H23BrN3O3 (M+H)+ 480.0917, found 480.0914.
2-(4-Fluorophenoxy)-N-isopropyl-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)acetamide (11s)
This compound was prepared from 5f (95 mg, 0.50 mmol) and 10d (91 mg, 0.42 mmol) using triethylamine (85 mg, 0.84 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11s (127 mg, 82%) as a white solid. mp 87.9-89.9 °C. HPLC 99.5 % (tR = 16.00 min, 50% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.00 (dd, J = 8.1, 1.6 Hz, 2H, [8.04 minor isomer]), 7.54-7.44 (m, 3H), 7.02-6.88 (m, 4H), 4.80 (s, 2H, [4.85 minor isomer]), 4.72 (s, 2H, [4.83 minor isomer]), 4.44-4.38 (m, 1H), 1.31 (d, J = 6.7 Hz, 6H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.49 [177.63 minor isomer], 168.82 [168.56 minor isomer], 168.24, 157.98 (d, J = 238 Hz), 154.24 (d, J = 2.07 Hz), 131.44 [131.86 minor], 129.00 [129.19 minor isomer], 127.69, 126.82, 116.34 (d, J = 23 Hz), 116.03 (d, J = 8.11 Hz), 68.30 [68.92 minor isomer], 48.91 [46.94 minor isomer], 37.18 [38.39 minor isomer], 21.46 [19.97 minor isomer]; 19F NMR (376 MHz, CDCl3) δ −123.13; LC-MS (ESI+) m/z 370.15 (M+H)+; HRMS (ESI+ve) m/z calculated for C20H21FN3O3 (M+H)+ 370.1562, found 370.1567.
N-Isopropyl-(3-phenyl-1,2,4-oxadiazol-5-yl)methylamino-2-oxoethoxy)benzoic acid (11t)
A solution of 11aq (50 mg, 0.11 mmol) in trifluoroacetic acid (3 ml) and dichloromethane (5 ml) were stirred at room temperature for 2 h. Acetone (5 ml) was added to the reaction mixture and the solvents were evaporated under vacuum to give the pure compound 11t (41 mg, 95%) as a white compound. mp 211.3-213.8 °C. HPLC 100 % (tR = 14.8 min, 40% MeOH in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, DMSO) δ 12.58 (brs, 1H), 7.97-7.90 (m, 2H, [8.03 -7.98 minor isomer]), 7.82 (d, J = 8.8 Hz, 2H), 7.58-7.53 (m, 3H), 6.96 (d, J = 8.8 Hz, 2H, [6.99 minor isomer]), 5.09 (s, 2H, [5.02 minor isomer]), 4.75 (s, 2H, [5.00 minor isomer]), 4.32-4.20 (m, 1H, [4.66-4.60 minor isomer]), 1.28 (d, J = 6.5 Hz, 6H, [1.07 minor isomer]). 13C NMR (100 MHz, CDCl3) δ 176.32, 171.41, 168.58, 167.80, 162.38, 132.70 [132.63 minor isomer], 131.48 [131.94 minor isomer], 129.22, 129.01 [129.22 minor isomer], 127.68 [126.71 minor isomer], 122.97 [122.90 minor isomer], 114.71 [114.68 minor isomer], 67.46 [67.96 minor isomer], 49.07 [47.24 minor isomer], 37.24 [38.43 minor isomer], 21.46 [19.96 minor isomer]; LC-MS (ESI+) m/z 396.15 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H22N3O5 (M+H)+ 396.1554, found 396.1566.
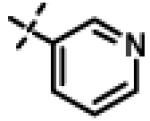
2-(4-Hydroxyphenoxy)-N-isopropyl-N-((pyridin-3-yl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11u)
To a solution of 10f (59 mg, 0.27 mmol) and triethylamine (55 mg, 0.54 mmol) in THF (10 ml) at r.t., was added 2-(4-hydroxyphenoxy)acetyl chloride (5s) (60 mg, 0.32 mmol) in THF (1 ml, dropwise 3-4 min). Upon addition of acetyl chloride, a precipitate was formed and the reaction was completed in 15 min. (monitored by tlc, Rf = 0.50, EtOAc/Hexane [2:1]). The THF was evaporated and the crude product obtained was purified by SiO2 chromatography (EtOAc/Hexane gradient elution) to obtain pure 11u (83 mg, 83%) as a white solid. mp 140.0-142.4 °C. HPLC 95.5% (tR = 5.47 min, 45% MeOH in 0.1% TFA water 20 min); The 1H NMR showed 5:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 9.11 (s, 1H, [9.23 minor isomer]), 8.70 (dd, J = 4.8, 1.4 Hz, 1H, [8.74 minor isomer]), 8.30 (dt, J = 8.0, 1.9 Hz, 1H), 7.43 (dd, J = 7.9, 4.8 Hz, 1H), 6.88 (d, J = 9.1 Hz, 2H), 6.80 (d, J = 9.1 Hz, 2H), 4.78 (s, 2H, [4.88 minor isomer]), 4.69 (s, 2H), 4.60-4.52 (m, 1H), 1.29 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 177.39, 169.00, 166.13, 152.05, 151.05, 148.10, 135.85, 124.36, 117.00 [116.45 minor isomer] 115.97, 68.37, 48.88, 37.08, 21.49 [19.99 minor isomer]; LC-MS (ESI+) m/z 369.15 (M+H)+; HRMS (ESI+ve) m/z calculated for C19H21N4O4 (M+H)+ 369.1557, found 369.1571.
2-(4-Hydroxyphenoxy)-N-isopropyl-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)acetamide (11v)
This compound was prepared from 5s (52 mg, 0.28 mmol) and 10d (50 mg, 0.23 mmol) using triethylamine (47 mg, 0.46 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11v (68 mg, 80%) as a white solid. mp 155.5-158.5 °C. HPLC 100% (tR = 13.1 min, 40% CH3CN in 0.1% TFA water 20 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.01 (dd, J = 7.8, 1.7 Hz, 2H, [8.04 minor isomer]), 7.51-7.43 (m, 3H), 6.85 (d, J = 9.0 Hz, 2H, [6.80 minor isomer]), 6.73 (d, J = 9.0 Hz, 2H, [6.70 minor isomer]), 4.75 (s, 2H, [4.85 minor isomer]), 4.71 (s, 2H, [4.80 minor isomer]), 4.48-4.41 (m, 1H), 1.30 (d, J = 6.6 Hz, 6H, [1.15 minor isomer]); 13C NMR (100 MHz, DMSO) δ 178.76 [178.92 minor isomer], 168.57 [168.67 minor isomer], 168.12 [168.25 minor isomer], 152.16 [152.02 minor isomer], 151.45, 150.38, 132.27 [132.45 minor isomer], 129.97 [130.02 minor isomer], 127.61 [127.69 minor isomer], 126.78 [126.55 minor isomer], 116.21 [116.30minor isomer], 116.16 [116.13 minor isomer], 67.33 [67.42 minor isomer], 48.31, 37.77, 21.34 [19.94 minor isomer]. LC-MS (ESI+) m/z 368.16 (M+H)+; HRMS (ESI+ve) m/z calculated for C20H22N3O4 (M+H)+ 368.1605, found 368.1615.
N-Isopropyl-N-((3-(pyridin-2-yl)-1,2,4-oxadizaol-5-yl)methyl)-2-(p-tolyloxy)acetamide (11w)
This compound was prepared from 5a (43 mg, 0.23 mmol) and 10e (42 mg, 0.19 mmol) using triethylamine (39 mg, 0.38 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11w (52 mg, 75%) as a white solid. mp 125.7-126.9 °C; HPLC 96.2% (tR = 15.7 min, 40% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.79 (d, J = 4.7 Hz, 1H), 8.06 (d, J = 7.9 Hz, 1H), 7.82 (td, J = 7.8, 1.7 Hz, 1H), 7.42 (appdd, J = 6.7, 4.9 Hz, 1H), 7.09 (d, J = 8.4 Hz, 2H, [7.03 minor isomer]), 6.87 (d, J = 8.5 Hz, 2H, [6.80 minor isomer]), 4.79 (s, 2H, [4.93 minor isomer]), 4.77 (s, 2H, [4.81 minor isomer]), 4.46-4.38 (m, 1H), 2.29 (s, 3H, [2.24 minor isomer]), 1.29 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 177.38, 168.57, 168.37, 155.96, 150.60 [150.73 minor isomer], 146.46, 137.19, 131.25, 130.30, 125.72, 123.51, 114.66, 67.98 [68.99 minor isomer], 49.01, 37.24, 21.44 [19.95 minor isomer], 20.72; LC-MS (ESI+) m/z 367.17 (M+H)+; HRMS (ESI+ve) m/z calculated for C20H23N4O3 (M+H)+ 367.1765, found 367.1774.
N-Isopropyl-N-((3-(pyridin-3-yl)-1,2,4-oxadizaol-5-yl)methyl)-2-(p-tolyloxy)acetamide 11x
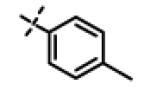
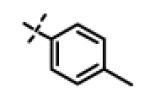
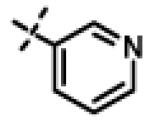
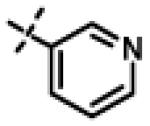
This compound was prepared from 5a (51 mg, 0.28 mmol) and 10f (50 mg, 0.23 mmol) using triethylamine (47 mg, 0.46 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11x (69 mg, 82%) as a white solid. mp 126.5-128.3 °C. HPLC 98.3% (tR = 10.6 min, 35% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 4:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 9.24 (s, 1H), 8.72 (appdd, J = 4.8, 1.2 Hz, 1H, [8.75 minor isomer]), 8.27 (dt, J = 7.9, 1.6 Hz, 1H), 7.39 (dd, J = 8.0, 4.9 Hz, 1H, [7.43 minor isomer]), 7.08 (d, J = 8.6 Hz, 2H, [7.02 minor isomer]), 6.86 (d, J = 8.5 Hz, 2H, [6.77 minor isomer]), 4.78 (s, 2H, [4.88 minor isomer]), 4.70 (s, 2H, [4.78 minor isomer]), 4.49-4.39 (m, 1H), 2.27 (s, 3H, [2.22 minor isomer]), 1.31 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 177.23, 168.60, 166.65, 155.94 [155.72 minor isomer], 152.24 [152.52 minor isomer], 148.92 [148.87 minor isomer], 134.97, 131.30, 130.28, 123.79 [123.86 minor isomer], 123.20, 114.64 [114.53 minor isomer], 67.91 [69.02 minor isomer], 48.99 [48.96 minor isomer], 37.27 [38.66 minor isomer], 21.52 [19.99 minor isomer], 20.72; LC-MS (ESI+) m/z 367.17 (M+H)+; HRMS (ESI+ve) m/z calculated for C20H23N4O3 (M+H)+ 367.1765, found 367.1774.
N-Isopropyl-N-((3-(pyridin-4-yl)-1,2,4-oxadizaol-5-yl)methyl)-2-(p-tolyloxy)acetamide (11y)
This compound was prepared from 5a (21 mg, 0.12 mmol) and 10g (22 mg, 0.10 mmol) using triethylamine (19 mg, 0.19 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11y (29 mg, 78%) as a white solid. mp 150.6-151.7 °C. HPLC 96.6% (tR = 14.7 min, 30% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.75 (d, J = 4.8 Hz, 2H), 7.87 (d, J = 4.6 Hz, 2H), 7.09 (d, J = 8.1 Hz, 2H, [7.02 minor isomer]), 6.87 (d, J = 8.5 Hz, 2H, [6.76 minor isomer]), 4.80 (s, 2H, [4.90 minor isomer]), 4.70 (s, 2H, [4.79 minor isomer]), 4.51-4.40 (m, 1H), 2.29 (s, 3H, [2.23 minor isomer]), 1.32 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 177.57, 168.65, 167.05, 155.94, 150.79 [150.91 minor isomer], 134.35, 131.30, 130.29, 121.57, 114.64 [114.50 minor isomer], 67.91 [69.08 minor isomer], 49.01[48.97 minor isomer], 37.27 [38.61 minor isomer], 21.52 [19.98 minor isomer], 20.72; LC-MS (ESI+) m/z 367.18 (M+H)+; HRMS (ESI+ve) m/z calculated for C20H23N4O3 (M+H)+ 367.1765, found 367.1780.
N-Isopropyl-2-(6-methylpyridin-3-yloxy)-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)acetamide (11z)
A solution of 6-methyl-3-hydroxypyridin (80 mg, 0.73 mmol), N-isopropyl-2-chloro-N-((3-phenyl-1,2,4-oxodiazol-5-yl)methyl)acetamide (15) (214 mg, 0.73 mmol) and potassium carbonate (510 mg, 3.67 mmol) in acetonitrile (25 ml) were refluxed for 14 h. The solvent was evaporated; and the residue was dissolved in ethyl acetate (20 ml), washed with water (2 × 20 ml). The organic phase was dried (MgSO4), evaporated and the crude compound obtained was purified by SiO2 chromatography (EtOAc/Hexane gradient elution) to afford 11z as (193 mg, 72%) a white solid. mp 130.3-132.2 °C. HPLC 97.26% (tR = 8.3 min, 30% CH3CN in 0.1% TFA water 30 min); 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 2.9 Hz, 1H, [8.26 minor isomer]), 8.00 (dd, J = 7.9, 1.6 Hz, 1H, [8.04 minor isomer]), 7.52-7.44 (m, 4H), 7.20 (d, J = 8.5 Hz, 1H), 4.93 (s, 2H, [5.01 minor isomer]), 4.89-4.80 (m, 1H), 4.72 (s, 2H, [4.78 minor isomer]), 4.30-4.23 (m, 1H, [4.89-4.80 minor isomer]), 2.63 (s, 3H), 1.35 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.25, 168.58, 166.98, 153.61, 149.71, 131.50 [131.94 minor isomer], 129.26, 129.04, 127.69, 126.73, 125.54, 67.68, 48.90 [47.05 minor isomer], 37.21, 21.61, 21.51 [19.99 minor isomer]; LC-MS (ESI+) m/z 367.18 (M+H)+; HRMS (ESI+ve) m/z calculated for C20H23N4O3(M+H)+ 367.1765, found 367.1759.
2-(4-Ethylphenoxy)-N-isopropyl-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)acetamide (11aa)
This compound was prepared from 5j (93 mg, 0.47 mmol) and 10d (85 mg, 0.39 mmol) using triethylamine (79 mg, 0.78 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11aa (123 mg, 83%) as a white solid. mp 106.7-109.2 °C. HPLC 95.5% (tR = 11.5 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 2:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.03 (dd, J = 7.9, 1.4 Hz, 2H), 7.55-7.38 (m, 3H), 7.11 (d, J = 8.4 Hz, 2H, [7.07 minor isomer]), 6.90 (d, J = 8.5 Hz, 2H, [6.84 minor isomer]), 4.79 (s, 2H, [4.86 minor isomer]), 4.71 (s, 2H, [4.83 minor isomer]), 4.46-4.40 (m, 1H), 2.59 (q, J = 7.5 Hz, 2H, [2.55 minor isomer]), 1.31 (d, J = 6.6 Hz, 6H, [1.16 minor isomer partially overlapped]), 1.20 (t, J = 7.6 Hz, 3H, partially overlapped with the doublet at 1.16); 13C NMR (100 MHz, CDCl3) δ 176.62 [176.77 minor isomer], 168.56 [168.66 minor isomer], 168.55 [168.65 minor isomer], 156.14 [155.94 minor isomer], 137.73, 131.38 [δ 131.75 minor isomer], 129.11, 128.98, 127.73, 126.90, 114.72 [114.65 minor isomer], 67.97 [68.74 minor isomer], 48.98, 37.23 [38.44 minor isomer], 28.21, 21.48 [19.99 minor isomer], 16.01: LC-MS (ESI+) m/z 380.21 (M+H)+; HRMS (ESI+ve) m/z calculated for C22H26N3O3 (M+H)+ 380.1969, found 380.1964.
2-(4-Ethylphenoxy)-N-isopropyl-N-((3-pyridin-3-yl)-1,2,4-oxadiazol-5-yl)methylacetamide (11ab)
This compound was prepared from 5j (50 mg, 0.25 mmol) and 10f (46 mg, 0.21 mmol) using triethylamine (43 mg, 0.42 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11ab (65 mg, 81%) as a white solid. mp 104.4-106.3 °C. HPLC 95.7% (tR = 5.3 min, 50% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 4:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 9.21 (s, 1H), 8.67 (brs, 1H), 8.23 (d, J = 7.7 Hz, 1H), 7.42-7.29 (broad m, 1H), 7.04 (d, J = 8.5 Hz, 2H, [6.98 minor isomer]), 6.82 (d, J = 8.5 Hz, 2H, [6.73 minor isomer]), 4.72 (s, 2H, [4.82 minor isomer]), 4.64 (s, 2H, [4.74 minor isomer]), 4.40-4.32 (m, 1H), 2.51 (q, J = 7.6 Hz, 2H partially overlapped, [2.47 minor isomer]), 1.25 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]), 1.11 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 177.26, [ 177.46 minor isomer], 168.59, [168.65 minor isomer], 166.65, [166.87 minor isomer], 156.09, 152.11, 148.82, 137.79, 135.02, 129.10, 114.68 [114.57 minor isomer], 67.97 [69.01 minor isomer], 49.00 [46.98 minor isomer], 37.29 [38.61 minor isomer], 28.19 [29.93 minor isomer], 21.49 [19.99 minor isomer], 16.00; LC-MS (ESI+) m/z 381.19 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H25N4O3(M+H)+ 381.1921, found 381.1912.
N-Isopropyl-2-phenoxy-N-((3-(pyridine-3-yl)-1,2,4-oxadiazol-5-yl)methyl)acetamide-(11ac)
This compound was prepared from 5c (55 mg, 0.32 mmol) and 10f (59 mg, 0.27 mmol) using triethylamine (55 mg, 0.54 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11ac (81 mg, 85%) as a white solid. mp 83.0-85.5 °C. HPLC 97.5 % (tR = 11.4 min, 30% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 4:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 9.29 (s, 1H), 8.79 (brs, 1H), 8.29 (d, J = 8.0 Hz, 1H), 7.45 (m, 1H), 7.33-7.22 (m, 2H), 7.02-6.87 (m, 3H), 4.82 (s, 2H, [4.89 minor isomer]), 4.71 (s, 2H, [4.84 minor isomer]), 4.51-4.37 (m, 1H), 1.32 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 177.21 [177.41 minor isomer]), 168.43, 166.62, [166.87 minor isomer], 158.01 [157.83 minor isomer], 152.20 [152.53 minor isomer], 148.85, 134.99, 129.85, 123.83, 123.19, 121.98, 114.81 [114.71 minor isomer], 67.65 [68.66 minor isomer], 48.97 [47.08 minor isomer], 37.28 [38.61 minor isomer], 21.49 [19.97 minor isomer]; LC-MS (ESI+) m/z 353.16 (M+H)+; HRMS (ESI+ve) m/z calculated for C19H21N4O3(M+H)+ 353.1608, found 353.1614.
N-Isopropyl-2-(4 propylphenoxy)-N-((3-pyridin-3-yl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11ad)
This compound was prepared from 5k (84 mg, 0.40 mmol) and 10f (72 mg, 0.33 mmol) using triethylamine (67 mg, 0.66 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11ad (109 mg, 84%) as a white solid. mp 103.2-105.5 °C. HPLC 100% (tR = 7.58 min, 50% CH3CN in 0.1% TFA water 20 min); The 1H NMR showed 4:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 9.26 (s, 1H), 8.78-8.70 (m, 1H), 8.29 (dt, J = 8.0 Hz, J = 2.0 Hz, 1H), 7.42 (appdd, J = 8.0, 4.9 Hz, 1H), 7.08 (d, J = 8.6 Hz, 2H, [ 7.03 minor isomer]), 6.87 (d, J = 8.6 Hz, 2H, [6.79 minor isomer]), 4.78 (s, 1H), 4.79 (s, 2H, [4.89 minor isomer]), 4.70 (s, 2H, [4.81 minor isomer shown]), 4.48-4.38 (m, 1H), 2.51 (t, J = 7.7 Hz, 2H, [2.47 minor isomer]), 1.63-1.52 (m, 2H), 1.32 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]), 0.91 (t, J = 7.3 Hz, 3H, [0.89 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 177.06 [177.24 minor isomer], 168.34 [168.40 minor isomer], 166.33 [166.57 minor isomer], 155.86 [155.62 minor isomer], 151.74 [152.16 minor isomer], 148.44 [148.50 minor isomer]), 135.98 [136.05 minor isomer], 134.93[134.83 minor isomer], 129.45 [129.41 minor isomer], 123.66 [123.09 minor isomer], 114.34 [114.24 minor isomer], 67.62 [68.73 minor isomer], 48.74 [46.81 minor isomer], 37.11 [38.41 minor isomer], 37.06, 24.69 [23.47 minor isomer], 21.26 [19.74 minor isomer], 13.78; LC-MS (ESI+) m/z 395.21 (M+H)+; HRMS (ESI+ve) m/z calculated for C22H27N4O3 (M+H)+ 395.2078, found 395.2080.
2-(4-Butylphenoxy)-N-isopropyl-N-((3-(pyridine-3-yl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11ae)
This compound was prepared from 5l (76 mg, 0.34 mmol) and 10f (61 mg, 0.28 mmol) using triethylamine (57 mg, 0.56 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11ae (94 mg, 82%) as a white solid. mp 94.5-95.2 °C. HPLC 98.13% (tR = 10.2 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 4:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 9.29 (s, 1H, [9.26 minor isomer overlapped]), 8.80-8.73 (m, 1H), 8.46 (d, J = 7.7 Hz, 1H, [8.33 minor isomer]), 7.56-7.52 (m, 1H [7.50 minor isomer]), 7.09 (d, J = 8.6 Hz, 2H, [7.02 minor isomer]), 6.87 (d, J = 8.6 Hz, 2H, [6.78 minor isomer]), 4.78 (s, 2H, [4.90 minor isomer]), 4.70 (s, 2H, [4.80 minor isomer]), 4.47-4.40 (m, 1H), 2.54 (t, J = 7.3 Hz, 2H, [2.48 minor isomer]), 1.59-1.46 (m, 2H), 1.36-1.29 [m, 8H and around 1.32 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 177.23, 168.58, 166.66, 156.07, 152.24 [152.54 minor isomer]), 148.93, 136.46, 134.97, 129.64, 123.80, 123.21, 114.59 [114.50 minor isomer]), 67.91, 48.97, 37.29, 34.95, 34.03, 22.53, 21.50 [19.99 minor isomer], 14.20; LC-MS (ESI+) m/z 409.23 (M+H)+; HRMS (ESI+ve) m/z calculated for C23H29N4O3(M+H)+ 409.2234, found 409.2238.
N-Isopropyl-2-(4-pentylphenoxy)-N-((3-pyridin-3-yl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11af)
This compound was prepared from 5m (78 mg, 0.32 mmol) and 10f (59 mg, 0.27 mmol) using triethylamine (55 mg, 0.54 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11af (95 mg, 83%) as a white solid. mp 93.0-95.9 °C. HPLC 96.5 % (tR = 17.1 min, 70% MeOH in 0.1% TFA water 20 min); 1H NMR (400 MHz, CDCl3) δ 9.19 (s, 1H, [9.24 minor isomer]), 8.67 (appdd, J = 4.9, 1.6 Hz, 1H), 8.24-8.22 (m, 1H, [8.30 minor isomer]), 7.37-7.28 32 (m, 1H), 7.02 (d, J = 8.4 Hz, 2H, [6.95 minor isomer]), 6.81 (d, J = 8.4 Hz, 2H, [6.72 minor isomer]), 4.72 (s, 2H, [4.83 minor isomer]), 4.64 (s, 2H, [4.74 minor isomer]), 4.40-4.33 (m, 1H), 2.46 (t, J = 7.8 Hz, 2H, partially overlapped [2.41 minor isomer partially overlapped]), 1.52-1.41 (m, 2H), 1.36-1.13 [broad m, 10H, and around 1.25 (d, J = 6.8 Hz, 6H)] ), 0.81 (t, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 177.29 [177.50 minor isomer], 168.62, [168.67 minor isomer], 166.56 [ 166.79 minor isomer], 156.07 [155.82 minor isomer], 152.19, 151.88 [152.19 minor isomer], 148.60, 136.51 [136.33 minor isomer], 135.25, 129.63 [129.59 minor isomer], 123.97, 114.60 [114.50 minor isomer], 67.90 [69.04 minor isomer], 49.00 [47.08 minor isomer], 37.30 [38.65 minor isomer], 35.24, 31.69, 31.57 [29.93 minor], 22.77, 21.51 [19.99 minor isomer], 14.29; LC-MS (ESI+) m/z 423.24 (M+H)+; HRMS (ESI+ve) m/z calculated for C24H31N4O3(M+H) 423.2391, found 423.2393.
2-(4-Hexylphenoxy)-N-isopropyl-N-((3-(pyridin-3-yl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11ag)
This compound was prepared from 5n (98 mg, 0.38 mmol) and 10f (70 mg, 0.32 mmol) using triethylamine (65 mg, 0.64 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11ag (122 mg, 87%) as a white solid. mp 94.4-96.1 °C. HPLC 93.8 % (tR = 7.7 min, 80% MeOH in 0.1% TFA water 20 min); The 1H NMR showed 4:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 9.28 (s, 1H), 8.75 (brs, 1H), 8.33 (appd, J = 7.9 Hz, 1H, [8.29 minor isomer]), 7.50-7.43 (m, 1H), 7.09 (d, J = 8.6 Hz, 2H, [7.03 minor isomer]), 7.87 (d, J = 8.7 Hz, 2H, [6.79 minor isomer]), 4.79 (s, 2H, [4.90 minor isomer]), 4.71 (s, 2H, [4.81 minor isomer]), 4.49-4.36 (m, 1H), 2.53 (t, J = 7.8 Hz, 2H partially overlapped, [2.48 minor isomer partially overlapped]), 1.59-1.51 (m, 2H), 1.33-1.27 (m, 12H and around 1.32 [d, J = 6.6 Hz, 6H, [1.16 minor isomer]), 0.91-0.83 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 177.32 [177.51 minor isomer], 168.58, [168.64 minor isomer], 166.54, 156.07 [155.82 minor isomer], 151.91 [152.38 minor isomer], 148.59 [148.68 minor isomer], 136.48 [136.54 minor isomer], 135.25 [135.13 minor isomer], 129.62 [129.58 minor isomer], 123.99 [123.40 minor isomer], 114.59 [114.50 minor isomer], 67.86 [68.96 minor isomer], 48.97 [47.05 minor isomer], 37.30 [38.64 minor isomer], 35.27 [35.22 minor isomer], 31.96, 31.85, 29.17, 22.84, 21.49 [19.97 minor isomer], 14.35; LC-MS (ESI+) m/z 437.25 (M+H)+; HRMS (ESI+ve) m/z calculated for C25H33N4O3(M+H)+ 437.2547, found 437.2548.
N-Isopropyl-2-(4-cyclohexylphenoxy)-N-((3-pyridin-3-yl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11ah)
This compound was prepared from 5o (79 mg, 0.31 mmol) and 10f (57 mg, 0.26 mmol) using triethylamine (53 mg, 0.52 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11ah (88 mg, 78%) as a sticky solid. HPLC 97.00 % (tR = 17.0 min, 70% MeOH in 0.1% TFA water 30 min); 1H NMR (400 MHz, CDCl3) δ 9.27 (s, 1H), 8.78-8.72 (m, 1H), 8.30 (appdt, J = 8.0, 2.0 Hz, 1H), 7.42-7.39 (m, 1H), 7.12 (d, J = 8.7 Hz, 2H, [7.06 minor isomer]), 6.88 (d, J = 8.7 Hz, 2H, [6.80 minor isomer]), 4.78 (s, 2H, [4.89 minor isomer]), 4.71 (s, 2H, [4.81 minor isomer]), 4.46-4.39 (m, 1H), 2.48-2.38 (m, 2H), 1.83-1.71 (m, 4H), 1.38-1.24 (brm, 11H, and around 1.32 [d, J = 6.6 Hz, 6H, [1.17 (minor isomer]); 13C NMR (100 MHz, CDCl3 ) δ 177.26 [177.47 minor isomer], 168.57, 166.66 [166.88 minor isomer ], 156.11 [155.86 minor isomer], 152.20 [152.49 minor isomer], 148.91 [148.86 minor isomer],141.82, 141.75, 134.96, 128.03, 123.86, 114.60 [114.50 minor isomer], 67.88 [69.03 minor isomer], 48.98 [47.07 minor isomer], 43.88, 37.30 [38.66 minor isomer], 34.85, 27.13 [29.93 minor isomer], 26.36, 21.51 [19.99 minor isomer]; LC-MS (ESI+) m/z 435.24 (M+H)+; HRMS (ESI+ve) m/z calculated for C25H31N4O3(M+H)+ 435.2391, found 435.2395.
N-Isopropyl-2-(4-isopropylphenoxy)-N-((3-(pyridin-3-yl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11ai)
This compound was prepared from 5p (61mg, 0.29 mmol) and 10f (52 mg, 0.24 mmol) using triethylamine (49 mg, 0.48 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11ai (77 mg, 81%) as a sticky white solid. HPLC 99.1% (tR = 6.7 min, 50% CH3CN in 0.1% TFA water 20 min); 1H NMR (400 MHz, CDCl3) δ 9.27 (s, 1H), 8.77-8.70 (m, 1H), 8.30 (dt, J = 8.0,1.8 Hz, 1H), 8.40 (appdd, J = 7.7, 5.0 Hz, 1H), 7.14 (d, J = 8.6 Hz, 2H, [7.08 minor isomer]), 6.89 (d, J = 8.6 Hz, 2H, [6.81 minor isomer]), 4.79 (s, 2H, [4.89 minor isomer]), 4.71 (s, 2H, [4.81 minor isomer]), 4.45-4.39 (m, 1H), 2.8-82.78 (m, 1H), 1.32 (d, J = 6.6 Hz, 6H, [1.17 minor isomer]), 1.20 (d, J = 6.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 177.26 [177.47 minor isomer], 168.56, 166.65 [166.88 minor isomer], 156.11 [155.88 minor isomer], 152.22 [152.52 minor isomer], 148.91, 142.44, 134.96, 127.66, 123.81 [123.91 minor isomer], 123.25, 114.64 [114.53 minor isomer], 67.89 [69.01 minor isomer], 48.98 [47.06 minor isomer], 37.30 [38.65 minor isomer], 33.49, 24.37, 21.50 [19.99 minor isomer]; LC-MS (ESI+) m/z 395.20 (M+H)+; HRMS (ESI+ve) m/z calculated for C22H27N4O3(M+H)+ 395.2078, found 395.2074.
N-Isopropyl-2-(4-tert-butylphenoxy)-N-((3-(pyridin-3-yl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11aj)
This compound was prepared from 5r (63 mg, 0.28 mmol) and 10f (50 mg, 0.23 mmol) using triethylamine (47 mg, 0.46 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11aj (78 mg, 83%) as a sticky colorless solid. HPLC 97.96% (tR = 9.1 min, 50% CH3CN in 0.1% TFA water 20 min); 1H NMR (400 MHz, CDCl3) δ 9.28 (s, 1H), 8.73 (brs, 1H), 8.20 (appd, J = 8.0 Hz, 1H), 7.40 (appdd, J = 7.7, 4.9 Hz, 1H), 7.29 (d, J = 8.7 Hz, 2H, [7.24 minor isomer shown]), 6.89 (d, J = 8.7 Hz, 2H, [6.81 minor isomer]), 4.78 (s, 2H, [4.89 minor isomer]), 4.71 (s, 2H, [4.83 minor isomer]), 4.43-4.37 (m, 1H), 1.32 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]), 1.27 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 177.27 [177.48 minor isomer], 168.62, [168.53 minor isomer], 166.64 [166.86 minor isomer], 155.76 [155.53 minor isomer], 152.23 [152.52 minor isomer], 148.89, 144.69, 134.97, 126.64 [126.60 minor isomer], 123.87, 123.26, 114.26 [114.15 minor isomer], 67.75 [68.86 minor isomer], 48.98 [47.00 minor isomer], 37.32 [38.62 minor isomer], 34.35, 31.70 [31.67 minor isomer], 21.52 [20.01 minor isomer]; LC-MS (ESI+) m/z 409.22 (M+H)+; HRMS (ESI+ve) m/z calculated for C23H29N4O3(M+H)+ 409.2234, found 409.2233.
N-Isopropyl-2-(4-isobutylphenoxy)-N-((3-(pyridin-3-yl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11ak)
This compound was prepared from 5q (74 mg, 0.32 mmol) and 10f (59 mg, 0.27 mmol mmol) using triethylamine (55 mg, 0.54 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11ak (97 mg, 88%) as a white solid. mp 121.5-123.7 °C. HPLC 98.3% (tR = 10.8 min, 50% CH3CN in 0.1% TFA water 20 min); 1H NMR (400 MHz, CDCl3) δ 9.27 (s, 1H), 8.74 (brs, 1H), 8.29 (d, J = 7.8 Hz, 1H), 7.41(brm, 1H), 7.05 (d, J = 8.6 Hz, 2H, [7.00 minor isomer]), 6.87 (d, J = 8.6 Hz, 2H, [6.79 minor isomer]), 4.79 (s, 2H, [4.90 minor isomer]), 4.71 (s, 2H, [4.81 minor isomer]), 4.47-4.40 (m, 1H), 2.40 (d, J = 7.2 Hz, 2H, partially overlapped, [2.36 minor isomer partially overlapped ]), 1.85-1.73 (m, 1H), 1.31 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]), 0.87 (d, J = 6.6 Hz, 6H, [0.85 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 177.24, 168.59, 166.72, 156.15 [155.92 minor isomer], 152.14, 148.89, 135.26 [135.34 minor isomer], 134.93, 130.35, 114.46 [114.37 minor isomer], 67.93 [69.06 minor isomer], 48.98 [47.06 minor isomer], 44.73, 37.30 [38.65 minor isomer], 30.53 [30.49 minor isomer], 22.52, 21.51 [19.98 minor isomer]; LC-MS (ESI+) m/z 409.23 (M+H)+; HRMS (ESI+ve) m/z calculated for C23H29N4O3(M+H)+ 409.2234, found 409.2231.
N-Isopropyl-2-(4-propylphenoxy)-N-((3-(pyrimidin-2-yl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11al)
This compound was prepared from 5k (46 mg, 0.22 mmol) and 10h (40 mg, 0.18 mmol) using triethylamine (36 mg, 0.36 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11al (58 mg, 81%) as a white solid. mp 92.7-94.0 °C. HPLC 97.2 % (tR = 12.1 min, 50% CH3CN in 0.1% TFA water 20 min); 1H NMR (400 MHz, CDCl3) δ 9.39-9.28 (m, 3H), 7.09 (d, J = 8.5 Hz, 2H, [7.01 minor isomer]), 6.87 (d, J = 8.6 Hz, 2H, [6.75 minor isomer]), 4.78 (s, 2H, [4.92 minor isomer]), 4.70 (s, 2H), 4.49-4.41 (m, 1H), 2.52 (t, J = 7.6 Hz, 2H, [2.45 minor isomer]), 1.64-1.47 (m, 2H), 1.32 (d, J = 6.8 Hz, 6H, [1.17 minor isomer]), 0.91 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 177.87, 168.67, 164.52, 160.62 [160.82 minor isomer], 155.72 [156.04 minor isomer], 136.33, 129.71, 121.78, 114.54 [114.40 minor isomer], 67.83, 49.03 [49.00 minor isomer], 37.34 [38.87 minor isomer], 24.92, 21.53 [20.05 minor isomer],14.02; LC-MS (ESI+) m/z 396.20 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H26N5O3(M+H)+ 396.2030, found 396.2031.
N-Isopropyl-2-(4-propylphenoxy)-N-((3-(pyrazin-2-yl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11am)
This compound was prepared from 5k (77 mg, 0.36 mmol) and 10j (66 mg, 0.30 mmol) using triethylamine (61 mg, 0.60 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11am (101 mg, 85%) as a colorless sticky solid. HPLC 97.3 % (tR = 11.1 min, 50% MeOH in 0.1% TFA water 30 min); 1H NMR (400 MHz, CDCl3) δ 9.29 (appd, J = 1.4 Hz, 1H, [9.26 minor isomer]), 8.73-8.70 (m, 2H), 7.08 (d, J = 8.6 Hz, 2H, [7.01 minor isomer]), 6.86 (d, J = 8.6 Hz, 2H, [6.77 minor isomer]), 4.78 (s, 2H, [4.94 minor isomer]), 4.76 (s, 2H, [4.79 minor isomer]), 4.46-4.36 (m, 1H), 2.50 (t, J = 7.8 Hz, 2H, [2.45 minor isomer]), 1.6381.51 (m, 2H), 1.30 (d, J = 6.6 Hz, 6H, [1.15 minor isomer]), 0.90 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 178.00, 168.64, 166.65 [166.86 minor isomer], 156.08 [155.82 minor isomer], 146.96, [146.67 minor isomer], 145.02 [145.09 minor isomer], 144.62, 142.47, 136.29, 129.72 [129.67 minor isomer], 114.58 [114.50 minor isomer], 67.92 [ 69.16 minor isomer], 49.02 [47.08 minor isomer], 37.35 [38.85 minor isomer], 24.92, 21.49 [19.96 minor isomer], 14.02; LC-MS (ESI+) m/z 396.19 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H26N5O3(M+H)+ 396.2030, found 396.2028.
2(-4-Butylphenoxy)-N-isopropyl-N-((3-(pyrazin-2-yl)-1,2,4-oxadiazol-5-yl)methylecetamide (11an)
This compound was prepared from 5l (54 mg, 0.24 mmol) and 10j (44 mg, 0.20 mmol) using triethylamine (41 mg, 0.40 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11an (64 mg, 78%) as a yellow/green sticky solid. HPLC 97.97 % (tR = 9.7 min, 70% MeOH in 0.1% TFA water 30 min); 1H NMR (400 MHz, CDCl3) δ 9.30 (d, J = 1.4 Hz, 1H), 9.27 (brs, 1H), 8.74-8.71 (m, 1H), 7.09 (d, J = 8.6 Hz, 2H, [7.02 minor isomer]), 6.87 (d, J = 8.6 Hz, 2H, [6.78 minor isomer]), 4.80 (s, 2H, [4.95 minor isomer]), 4.76 (s, 2H, [4.80 minor isomer]), 4.48-4.39 (m, 1H), 2.53 (t, J = 7.7 Hz, 2H, [2.47 minor isomer])), 1.63-1.48 (m, 3H), 1.37-1.14 [m, 10H and around 1.30 (d, J = 6.7 Hz, 6H)] 1.16 minor isomer]), 0.90 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 178.00, 168.64, 166.64, 156.05 [155.78 minor isomer], 146.65 [146.94 minor isomer], 145.00 [145.08 minor isomer], 144.61, 142.47 [142.08 minor isomer], 136.49 [136.57 minor isomer], 129.65 [129.80 minor isomer], 114.60 [114.53 minor isomer], 67.93 [69.17 minor isomer], 49.01 [47.13 minor isomer], 37.34 [38.85 minor isomer], 34.94, 34.00, 22.52 [21.11 minor isomer], 21.47 [19.95 minor isomer shown], 14.17; LC-MS (ESI+) m/z 410.222 (M+H)+; HRMS (ESI+ve) m/z calculated for C22H28N5O3(M+H)+ 410.2187, found 410.2185.
N-Isopropyl-2-(4-propylphenoxy)-N-((3-(pyrimidin-2-yl)-1,2,4-oxadiazol-5-yl)methyl)acetamide (11ao)
This compound was prepared from 5k (46 mg, 0.22 mmol) and 10i (40 mg, 0.18 mmol) using triethylamine (36 mg, 0.36 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11ao (63 mg, 88%) as a colorless sticky solid; HPLC 97.9 % (tR = 7.1 min, 50% CH3CN in 0.1% TFA water 20 min); 1H NMR (400 MHz, CDCl3) δ 8.96 (d, J = 4.9 Hz, 2H), 7.45 (t, J = 4.9 Hz, 1H, [7.48 minor isomer]), 7.10 (d, J = 8.6 Hz, 2H, [7.04 minor isomer]), 6.88 (d, J = 8.6 Hz, 2H, [6.81 minor isomer]), 4.83 (s, 2H, [4.99 minor isomer]), 4.80 (s, 2H, [4.81 minor isomer]), 4.44-4.34 (m, 1H), 2.52 (t, J = 7.7 Hz, 2H, partially overlapped [2.48 minor isomer, partially overlapped]), 1.66-1.51 (m, 2H, [1.46-1.38 minor isomer]), 1.28 (d, J = 6.6 Hz, 2H, [1.14 minor isomer]), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 178.04, 168.62 [167.47 minor isomer], 167.85, 158.19 [158.27 minor isomer], 156.27, 156.09, 136.24, 129.73, 122.37, 114.59, 67.97 [69.02 minor isomer], 49.02 [47.00 minor isomer], 37.36 [38.85 minor isomer], 37.34, 24.93, 21.42 [19.90 minor isomer], 14.02; LC-MS (ESI+) m/z 396.19 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H26N5O3(M+H)+ 396.2030, found 396.2025.
N-Isopropyl-2-(4-propylphenoxy)-N-((3-p-tolyl-1,2,4-oxadiazol-5-yl)methyl)acetamide (11ap)
This compound was prepared from 5k (140 mg, 0.66 mmol) and 10a (127 mg, 0.55 mmol) using triethylamine (111 mg, 1.10 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11ap (195 mg, 87%) as a white solid. mp 121.7-122.3 °C. HPLC 99.3 % (tR = 11.1 min, 45% MeOH in 0.1% TFA water 30 min); 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 8.2 Hz, 2H), 7.29-7.20 (m, 2H), 7.07 (d, J = 8.6 Hz, 2H, [7.03 minor isomer]), 6.87 (d, J = 8.6 Hz, 2H, [6.82 minor isomer]), 4.77 (s, 2H, [4.83 minor isomer]), 4.69 (s, 2H, [4.82 minor isomer]), 4.45-4.34 (m, 1H), 2.54-2.46 (m, 2H), 2.39 (s, 3H, [2.40 minor isomer]), 1.60-1.50 (m, 2H), 1.29 (d, J = 6.6 Hz, 6H, [1.13 minor isomer]), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 176.43 [δ 176.57 minor isomer]), 168.56 [168.80 minor isomer]), 168.50, 156.18 [155.99 minor isomer]), 141.66 [142.14 minor isomer]), 136.16, 129.69 [129.83 minor isomer]), 127.65, 124.09 [123.58 minor isomer]), 114.64 [114.59 minor isomer]), 67.99 [68.74 minor isomer]), 48.94 [46.89 minor isomer]), 37.37 [38.41 minor isomer]), 37.22, 24.92, 21.82, 21.47 [19.98 minor isomer]), 14.03; LC-MS (ESI+) m/z 408.22 (M+H)+; HRMS (ESI+ve) m/z calculated for C24H30N3O3(M+H)+ 408.2282, found 408.2279.
tert-Butyl-4-(2-(isopropyl((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)amino)-2-oxoethoxy)benzoate (11aq)
A solution of tert-Butyl-4-hydroxybenzoate (27, see supporting information for 27) (27 mg, 0.14 mmol), 15 (41 mg, 0.14 mmol) and potassium carbonate (97 mg, 0.70 mmol) in acetonitrile (20 ml) was heated under reflux overnight. Acetonitile was evaporated and the residue was dissolved in ethyl acetate (20 ml) and washed with water (20 ml × 2). The organic phase was dried (MgSO4) and the product obtained was purified by SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11aq (51 mg, 80% as a white solid). mp 143.0-144.4 °C. HPLC 99.7% (tR = 14.1 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.8 Hz, 2H, [8.01 minor isomer]), 7.94 (d, J = 8.8 Hz, 2H, [7.88 minor isomer]), 7.50-7.38 (m, 3H), 6.96 (d, J = 8.9 Hz, 2H, overlapped, [6.93 minor isomer, overlapped]), 4.86 (s, 2H, [4.91 minor isomer]), 4.69 (s, 2H, [4.78 minor isomer]), 4.41-4.31 (m, 1H), 1.55 (s, 9H), 1.29 (d, J = 6.6 Hz, 6H, [1.14 minor isomer]); 13C NMR (100 MHz, CDCl3) 176.14 [176.24 minor isomer], 168.32, 167.60 [167.68 minor isomer], 165.40 [165.65 minor isomer],161.11 [161.06 minor isomer], 159.73, 131.60 [131.64 minor isomer], 131.51 [131.45 minor isomer],128.77 [128.96 minor isomer], 127.45 [126.50 minor isomer], 125.51 [125.46 minor isomer], 124.25, 114.96, 114.11 [114.06 minor isomer], 80.74 [80.51 minor isomer], 67.30 [67.84 minor isomer], 48.78 (46.83 minor isomer), 36.98 [38.22 minor isomer], 28.24 [29.70 minor isomer], 21.24 [19.75 minor isomer]; LC-MS (ESI+) m/z 469.26 (M+NH )+ 4 ; HRMS (ESI+ve) m/z calculated for C25H30N3O5 (M+H)+ 452.2180, found 452.2191.
2-(4-Fluorophenoxy)-N-isopropyl-N-((3-(pyridin-3-yl)1,2,4-oxadiazol-5-yl)methyl)acetamide (11ar)
This compound was prepared from 5f (93 mg, 0.49 mmol) and 10f (90 mg, 0.41 mmol) using triethylamine (83 mg, 0.82 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11ar (132 mg, 87%) as a white solid. mp 83.8-84.6 °C. HPLC 100% (tR = 10.8 min, 30% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 4.5:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 9.25 (s, 1H), 8.73 (d, J = 4.1 Hz, 1H, [8.76 minor isomer]), 8.25 (dt, J = 8.0, 1.9 Hz, 1H), 7.40 (dd, J = 7.9, 4.9 Hz, 1H, partially overlapped, [7.43 minor isomer]), 7.05-6.83 (m, 4H, [6.88-6.78 minor isomer]), 4.79 (s, 2H, [4.81 minor isomer]), 4.71 (s, 2H, [4.86 minor isomer]), 4.47-4.35 (m, 1H), 1.32 (d, J = 6.6 Hz, 6H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3) 176.97 [177.10 minor isomer], 168.06, 166.25 [166.62 minor isomer], 157. 72 (d, J = 239 Hz), 153.93 (d, J = 2.06 Hz), 151.39 [152.19 minor isomer], 148.07 [148.41 minor isomer], 135.18 [134.86 minor isomer], 123.80, 123.16, 116.03 (d, J = 23 Hz), 115.75 (d, J = 8 Hz), 67.91 [68.67 minor isomer], 48.72 [46.89 minor isomer], 37.03 [38.34 minor isomer], 21.26 [19.75 minor isomer]; 19F NMR (376 MHz, CDCl3) δ −122.60 [−122.53 minor isomer]; LC-MS (ESI+) m/z 371.15 (M+H)+; HRMS (ESI+ve) m/z calculated for C19H20FN4O3 (M+H)+ 371.1514, found 371.1517.
2-(4-Chlorophenoxy)-N-isopropyl-N-((3-(pyridin-3-yl)1,2,4-oxadiazol-5-yl)methyl)acetamide (11as)
This compound was prepared from 5d (66 mg, 0.32 mmol) and 10f (59 mg, 0.27 mmol) using triethylamine (55 mg, 0.54 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 11as (91 mg, 87%) as a white solid. mp 118.1-119.9 °C. HPLC 99.0% (tR = 10.4 min, 35% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 4:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 9.25 (s, 1H), 8.73 (dd, J = 4.8, 1.4 Hz, 1H, overlapped [8.76 minor isomer, overlapped]), 8.24 (dt, J = 7.9, 1.9 Hz, 1H), 7.41 (dd, J = 8.0, 4.9 Hz, 1H), 7.22 (d, J = 9.0 Hz, 2H, [7.18 minor isomer]), 6.90 (d, J = 9.0 Hz, 2H, [6.83 minor isomer]), 4.80 (s, 2H, [4.85minor isomer]), 4.70 (s, 2H, [4.83 minor isomer]), 4.48-4.31 (m, 1H), 1.32 (d, J = 6.6 Hz, 6H, [1.17 minor isomer]); 13C NMR (100 MHz, CDCl3) 176.74, 167.82, 166.35, 156.39, 152.39 [152.03 minor isomer], 148.59 [148.55 minor isomer], 134.59, 129.51 [129.47 minor isomer], 126.68, 123.63 [122.89 minor isomer], 115.89 [115.83 minor isomer], 67.59 [68.57 minor isomer], 48.75 [46.93 minor isomer], 37.03 [38.38 minor isomer], 21.24 [18.67 minor isomer]; LC-MS (ESI+) m/z 387.03 (M+H)+; HRMS (ESI+ve) m/z calculated for C19H20ClN4O3 (M+H)+ 387.1218, found 387.1212.
N-Isobutyl-N-((3-p-tolyl-1,2,4-oxadiazol-5-yl)methyl)-2-(p-tolyloxy)acetamide (12a)
This compound was prepared from 5a (95 mg, 0.52 mmol) and 10m (106 mg, 0.43 mmol) using triethylamine (87 mg, 0.86 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 12a (147 mg, 87%) as a white solid. mp 78.4-79.8 °C. HPLC 99.1 % (tR = 11.8 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed approximately 2:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 8.2 Hz, 2H), 7.30-7.25 (m, 2H), 7.08 (d, J = 8.3 Hz, 2H, [7.04 minor isomer]), 6.86 (d, J = 8.6 Hz, 2H, [6.81 minor isomer]), 4.85 (s, 2H, [4.93 minor isomer]), 4.79 (s, 2H, [4.86 minor isomer]), 3.36-3.34 (m, 2H), 2.41 (s, 3H, [2.42 minor isomer]), 2.27 (s, 3H, [2.24 minor isomer]), 2.06-1.94 (m, 1H), 1.01 (d, J = 6.6 Hz, 6H, [0.86 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 175.43 [175.48 minor isomer], 169.22 [169.08 minor isomer], 168.61 [168.79 minor isomer], 156.04 [155.63 minor isomer], 141.82 [142.15 minor isomer], 131.21 [131.33 minor isomer], 130.24, 129.74 [129.84 minor isomer], 127.66 [127.67 minor isomer], 123.92 [123.53 minor isomer], 114.82 [114.54 minor isomer], 67.23 [68.55 minor isomer], 55.50 [54.51 minor isomer], 42.10 [43.59 minor isomer], 27.73 [26.86 minor isomer], 21.85, 20.75 [20.70 minor isomer], 20.28 [20.15 minor isomer]; LC-MS (ESI+) m/z 394.20 (M+H)+; HRMS (ESI+ve) m/z calculated for C23H28N3O3 (M+H)+ 394.2125, found 394.2127.
N-Ethyl-N-((3-p-tolyl-1,2,4-oxadiazol-5-yl)methyl)-2-(p-tolyloxy)acetamide (12b)
This compound was prepared from 5a (51 mg, 0.28 mmol) and 10l (50 mg, 0.23 mmol) using triethylamine (47 mg, 0.46 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 12b (69 mg, 82%) as a white solid. mp 97.4-100.1 °C. HPLC 99.6% (tR = 9.0 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 2:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 8.1 Hz, 2H, partially overlapped, [7.89 minor isomer]), 7.31-7.27 (m, 2H), 7.09 (d, J = 8.2 Hz, 2H, [7.03 minor isomer]), 6.87 (d, J = 8.6 Hz, 2H, [6.79 minor isomer]), 4.84 (s, 2H, [4.91 minor isomer]), 4.78 (s, 2H, [4.82 minor isomer]), 3.63 (q, J = 7.1 Hz, 2H, partially overlapped, [3.59 minor isomer overlapped]), 2.41 (s, 3H, [2.42 minor isomer]), 2.29 (s, 3H, [2.24 minor isomer]), 1.29 (t, J = 7.1 Hz, 3H, [1.16 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 175.56 [175.49 minor isomer], 168.86 [168.80 minor isomer], 168.66 [168.48 minor isomer], 156.00 [155.65 minor isomer], 141.85 [142.12 minor isomer], 131.27 [131.32 minor isomer], 130.30 [130.27 minor isomer], 129.75 [129.82 minor isomer], 127.66, 123.89 [123.56 minor isomer], 114.72 [114.52 minor isomer], 67.61 [68.52 minor isomer], 43.31 [42.97 minor isomer], 41.30 [42.72 minor isomer], 21.83, 20.74 [20.68 minor isomer], 14.14 [12.56 minor isomer]; LC-MS (ESI+) m/z 366.19 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H24N3O3 (M+H)+ 366.1812, found 366.1810.
N-Methyl-N-((3-p-tolyl-1,2,4-oxadiazol-5-yl)methyl)-2-(p-tolyloxy)acetamide (12c)
This compound was prepared from 5a (67 mg, 0.36 mmol) and 10k (61 mg, 0.30 mmol) using triethylamine (61 mg, 0.60 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 12c (100 mg, 95%) as a white solid. mp 99.1-100.9 °C. HPLC 96.7% (tR = 7.4 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 2:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 8.3 Hz, 2H, overlapped, [7.90 minor isomer, overlapped]), 7.30-7.26 (m, 2H), 7.08 (d, J = 8.1 Hz, 2H, [7.03 minor isomer]), 6.87 (d, J = 8.8 Hz, 2H, [6.78 minor isomer]), 4.87 (s, 2H [4.94 minor isomer]), 4.78 (s, 2H, [4.82 minor isomer]), 3.28 (s, 3H, [3.12 minor isomer]), 2.41 (s, 3H, [2.42 minor isomer)], 2.28 (s, 3H, [2.23 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 175.13 [175.03 minor isomer], 169.08 [168.92 minor isomer shown], 168.80 [168.68 minor isomer shown], 155.58 [155.89 minor isomer], 141.91 [142.12 minor isomer], 131.32 [131.38 minor isomer], 130.29, 129.77 [129.82 minor isomer], 127.65, 123.82 [123.54 minor isomer], 114.76, [114.44 minor isomer], 67.53 [67.49 minor isomer], 43.98 [45.45 minor isomer], 35.77 [35.22 minor isomer], 21.85 [21.83 minor isomer], 20.75 [20.74 minor isomer]; LC-MS (ESI+) m/z 352.17 (M+H)+; HRMS (ESI+ve) m/z calculated for C20H22N3O3 (M+H)+ 352.1657, found 352.1658.
N-((3-p-tolyl-1,2,4-oxadiazol-5-yl)methyl)-2-(p-tolyloxy)acetamide (12d)
This compound was prepared from 5a (58 mg, 0.31 mmol) and 10n (49 mg, 0.26 mmol) using triethylamine (54 mg, 0.53 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 12d (69 mg, 79%) as a white solid. mp 113.7-115.5 °C. HPLC 99.9% (tR = 6.5 min, 60% CH3CN in 0.1% TFA water 30 min); 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 7.12 (d, J = 8.2 Hz, 2H), 6.87 (d, J = 8.6 Hz, 2H), 4.84 (d, J = 5.9 Hz, 2H), 4.59 (s, 2H), 2.42 (s, 3H), 2.31 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 175.46, 169.14, 168.66, 155.20, 142.02, 131.98, 130.49, 129.82, 127.64, 123.67, 114.82, 67.70, 35.46, 21.85, 20.77; LC-MS (ESI+) m/z 360.14 (M+Na)+; HRMS (ESI+ve) m/z calculated for C19H20N3O3 (M+H)+ 338.1499, found 338.1505.
N-tert-Butyl-N-((3-p-tolyl-1,2,4-oxadiazol-5-yl)methyl-2-(p-tolyloxy)acetamide (12e)
This compound was prepared from 5a (40 mg, 0.22 mmol) and 10o (44 mg, 0.18 mmol) using triethylamine (36 mg, 0.36 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 12e (53 mg, 75%) as a white solid. mp 129.1-130.0 °C. HPLC 96.2% (tR = 18.3 min, 60% CH3CN in 0.1% TFA water 30 min); 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 8.2 Hz, 2H), 7.27 (m, 2H), 7.02 (m, 2H), 6.78 (d, J = 8.6 Hz, 2H), 4.95 (s, 2H), 4.76 (s, 2H), 2.42 (s, 3H), 2.24 (s, 3H), 1.47 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 176.97, 169.45, 168.78, 155.88, 142.11, 131.07, 130.17, 129.81, 127.66, 123.62, 114.55, 69.81, 59.15, 40.71, 28.47, 21.86, 20.72; LC-MS (ESI+) m/z 394.22 (M+H)+; HRMS (ESI+ve) m/z calculated for C23H28N3O3 (M+H)+ 394.2125, found 394.2114.
N-Cyclopropyl-N-((3-p-tolyl-1,2,4-oxadiazol-5-yl)methyl)-2-(p-tolyloxy)acetamide (12f)
This compound was prepared from 5a (74 mg, 0.40 mmol) and 10p (76 mg, 0.33 mmol) using triethylamine (67 mg, 0.66 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 12f (98 mg, 79%) as a white solid. mp 113.8-115.2 °C. HPLC 94.8% (tR = 6.2 min, 70% MeOH in 0.1% TFA water 20 min); 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8.0 Hz, 2H), 7.31-7.23 (m, 2H), 7.06 (d, J = 8.2 Hz, 2H), 6.86 (d, J = 8.3 Hz, 2H), 4.97 (s, 2H), 4.88 (s, 2H), 3.10-3.02 (m, 1H), 2.42 (s, 3H), 2.26 (s, 3H), 1.05-0.91 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 175.92, 171.51, 168.61, 156.24, 141.86, 131.02, 130.19, 129.75, 127.65, 123.91, 114.81, 67.14, 43.44, 29.96, 21.82, 20.72, 9.29; LC-MS (ESI+) m/z 378.18 (M+H)+; HRMS (ESI+ve) m/z calculated for C22H24N4O3(M+H)+ 378.1812, found 378.1811.
1-Isopropyl-3-(4-methylbenzyl)-1-((3 phenyl-1,2,4-oxadiazol-5-yl)methyl)urea (14)
A solution of 10d (70 mg, 0.32 mmol), 4-methylbenzyl isocyanate 13 (47 mg, 0.32 mmol) and triethylamine (40 mg, 0.39 mmol) were heated under reflux in benzene overnight (14-15h). The organics were evaporated and the residue was dissolved in ethyl acetate (20 ml) and washed with HCl (4M, 3×10 mL) and water (2×20 mL). The organic phase was dried (MgSO4), evaporated, and the crude product obtained was purified by SiO2 chromatography (EtOAc/Hexane gradient elution) to obtain pure 14 (91 mg, 78%) as a white solid. mp 87.6-89.1 °C. HPLC 96.26% (tR = 14.98 min, 50% CH3CN in 0.1% TFA water 30 min); 1H NMR (400 MHz, CDCl3) δ 7.98-7.95 (m, 2H), 7.44-7.49 (m, 1H), 7.49-7.44 (m, 2H), 7.24 (d, J = 8.0 Hz, 2H), 7.13 (d, J = 7.8 Hz, 2H), 5.52 (apparent t, 1H), 4.63 (s, 2H), 4.43 (d, J = 4.7 Hz, 2H), 4.40-4.34 (m, 1H), 2.33 (s, 3H), 1.22 (d, J = 6.8 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 177.63, 168.47, 158.01, 137.13, 136.32, 131.58, 129.52, 129.03, 128.03, 127.71, 126.53, 47.12, 45.27, 37.62, 21.36, 20.96; LC-MS (ESI+) m/z 365.20 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H25N4O2 (M+H)+ 365.1972, found 365.1988.
N-Isopropyl-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)-2-(p-tolylamino)acetamide (16)
A solution of p-toluidine (19 mg, 0.18 mmol), 15 (65 mg, 0.22 mmol) and sodium acetate (18 mg, 0.22 mmol) in ethanol (20 ml) were refluxed for 15 h. Ethanol was evaporated and the product was purified by SiO2 chromatography (EtOAc/Hexane gradient elution) to obtain 16 as a yellow-brown sticky solid (51 mg, 78%); HPLC 96.59% (tR = 12.2 min, 45% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 4:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.07-8.04 (m, 2H), 7.54-7.44 (m, 3H), 7.01 (d, J = 8.1 Hz, 2H), 6.59 (d, J = 8.3 Hz, 2H), 4.76 (s, 2H, [4.71 minor isomer]), 4.29-4.16 (m, 1H, [4.98-4.90 minor isomer]), 4.05 (s, 2H), 2.25 (s, 3H, [2.23 minor isomer]), 1.34 (d, J = 6.6 Hz, 6H, [1.18 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 176.66 [176.22 minor isomer], 169.91 [169.69 minor isomer], 168.65 [168.92 minor isomer], 145.14, 131.43 [131.83 minor isomer], 130.01 [130.14 minor isomer], 129.00 [129.18 minor isomer], 127.74 [127.70 minor isomer], 127.33, 126.82, 113.51 [113.65 minor isomer], 47.98 [49.59 minor isomer], 46.18 [46.74 minor isomer], 37.25 [37.44 minor isomer], 21.32 [19.96 minor isomer], 20.65 [20.14 minor isomer]; LC-MS (ESI+) m/z 365.19 (M+H)+; HRMS (ESI+ve) m/z calculated for C21H25N4O2(M+H)+ 365.1972, found 365.1978.
N-Isopropyl-N-((3-phenyl-1,2,4-oxadiazol-5-yl)methyl)-3-(4-(trifluoromethyl)phenyl)propanamide (19a)
This compound was prepared from 18a (131 mg, 0.55 mmol) and 10d (100 mg, 0.46 mmol) using triethylamine (93 mg, 0.92 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 19a (169 mg, 88%) as a white solid. mp 96.1-97.7 °C. HPLC 98.5% (tR = 5.27 min, 60% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.06 (dd, J = 7.9, 1.7 Hz, 2H, overlapped [8.03 minor isomer overlapped]), 7.56-7.42 (m, 5H), 7.37 (d, J = 8.1 Hz, 2H, [7.33 minor isomer]), 4.70 (s, 2H, [4.61 minor isomer]), 4.25-4.18 (m, 1H, [4.97 minor isomer]), 3.08 (t, J = 7.6 Hz, 2H, [3.02 minor isomer]), 2.79 (t, J = 7.6 Hz, 2H, [2.71minor isomer]), 1.24 (d, J = 6.7 Hz, 6H, [1.12 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 177.07 [176.66 minor isomer], 171.92 [172.02 minor isomer], 168.58, 145.48, 131.40 [131.84 minor isomer], 129.19, 129.07, 129.00 [128.88 minor isomer], 127.70 [127.67 minor isomer], 126.94, 125.64 (q, J = 3.76 Hz), 124.50 (q, J = 271 Hz), 48.78 [45.82 minor isomer], 37.16 [38.72 minor isomer], 34.81 [35.30 minor isomer], 31.09 [31.17 minor isomer], 21.33 [20.20 minor isomer]; 19F NMR (376 MHz, CDCl3) δ −62.75,[−62.78 minor isomer]; LC-MS (ESI+) m/z 418.18 (M+H)+; HRMS (ESI+ve) m/z calculated for C22H23F3N3O2 (M+H)+ 418.1737, found 418.1745.
Benzofuran-2-carboxylic acid isopropyl-(3-pyridin-3-yl-[1,2,4]oxadiazol-5-yl)methyl) amide (19b)
This compound was prepared from 18b (63 mg, 0.35 mmol) and 10f (63 mg, 0.29 mmol using triethylamine (59 mg, 0.58 mmol) in a similar manner as described for compound 11u. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 19b (86 mg, 82%) as a sticky colorless solid. HPLC 96.1% (tR = 4.1 min, 70% MeOH in 0.1% TFA water 20 min); 1H NMR (400 MHz, CDCl3) δ 9.27 (s, 1H), 8.71 (appd, J = 4.0 Hz, 1H), 8.32 (d, J = 7.9 Hz, 1H), 7.63 (d, J = 7.7 Hz, 1H), 7.46-7.38 (m, 3H, [7.57-7.46 minor isomer]), 7.31-7.25 (m, 2H), 5.03-4.93 (m, 1H), 4.89 (brs, 2H), 1.37 (d, J = 3.1 Hz, 6H, [7.50 minor isomer]). 13C NMR (100 MHz, CDCl3) δ 166.69, 161.31, 154.93, 152.05, 148.69, 135.19, 127.05 [126.98 minor isomer], 123.98 [123.95 minor isomer], 122.63 [123.26 minor isomer] 113.13, 112.13, 49.89, 38.21, 22.78, 21.66: LC-MS (ESI+) m/z 363.16 (M+H)+; HRMS (ESI+ve) m/z calculated for C20H19N4O3(M+H)+ 363.1452, found 363.1455.
N-Isopropyl-N-(2-(3-phenyl-1,2,4-oxadiazol-5-yl)ethyl)-2-(p-tolyloxy)acetamide (23)
This compound was prepared from 5a (56 mg, 0.55 mmol) and 22 (106 mg, 0.46 mmol, see supporting information for the synthesis of 22) using triethylamine (93 mg, 0.92 mmol) in a similar manner as described for compound 1. The crude product obtained was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to afford pure 23 (152 mg, 87%) as a sticky colorless solid. HPLC 97.51% (tR = 22.7 min, 50% CH3CN in 0.1% TFA water 30 min); The 1H NMR showed 3:1 ratio of atropisomers: 1H NMR (400 MHz, CDCl3) δ 8.07-8.02 (m, 2H), 7.52-7.44 (m, 3H), 7.09 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 4.69 (s, 2H), 4.29-4.20 (m, 2H, [4.55-4.44 minor isomer]), 3.72 (t, J = 7.4 Hz, 2H, [3.89 minor isomer]), 3.28 (t, J = 7.4 Hz, 2H), 2.28, (s, 3H, [2.26 minor isomer]), 1.28 (d, J = 6.7 Hz, 6H, [1.20 minor isomer]); 13C NMR (100 MHz, CDCl3) δ 177.76, 168.50, 168.40, 155.99, 131.37 [131.18 minor isomer], 130.29, 129.05, 127.66 [126.99 minor isomer], 114.59 [114.67 minor isomer], 68.31 [68.99 minor isomer], 48.80 [47.87 minor isomer shown], 38.56 [41.31 minor isomer], 26.18 [28.64 minor isomer], 21.36 [20.45 minor isomer]; 20.72. LC-MS (ESI+) m/z 380.20 (M+H)+; HRMS (ESI+ve) m/z calculated for C22H26N3O3(M+H)+ 380.1969, found 380.1965.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NCI grant 1PO1 CA118210-03 (PI-Saïd M. Sebti). We acknowledge Dr. Kenyon Daniel (Mofffiit Chemical Biology Core-screening and modeling unit) for helpful discussions and Nan Sun (Chemical Biology Core-screening unit) for technical assistance. We also thank the Chemistry unit of the Moffitt Chemical Biology Core for technical assistance and maintenance of the NMR, Mass Spectrometry, HPLC instruments. The Moffitt Proteomics Facility is supported in part by the US Army Medical Research and Materiel Command under Award No. W81XWH-08-2-0101 for a National Functional Genomics Center. The Moffitt Proteomics and Chemical Biology Core facilities are supported by the National Cancer Institute under Award No. P30-CA076292 as a Cancer Center Support Grant, and the Moffitt Foundation.
ABBREVIATIONS USED
- CT-L
chymotrypsin like
- T-L
trypsin like
- SAR
structure activity relationship
- PGPH-L
postglutamylpeptidase hydrolysis-like
- DCM
dichloromethane
- THF
tetrahydrofuran
- DMF
dimethylformamide
- DMSO
dimethylsulfoxide
- TFA
trifluoroacetic acid
- LC/MS-MS
Liquid chromatography-tandem mass spectrometry
- LC-MS
Liquid chromatography mass spectrometry
- HRMS
High resolution mass spectroscopy
- ESI
Electrospray ionization
- HPLC
High performance liquid chromatography
- AMC
7-amino-4-methyl-coumarin
Footnotes
Supporting information available: (i) Synthetic protocols and characterization for libraries 5 and 10, and compounds 15, 18a-b, 20-27 (except 23) (ii) Scanned NMR spectra, HPLC, HRMS and LC-MS reports for 1, 11x, 11aa, 11ab, 11ac, 11ad, 11ae, 11af, 11ah, 11al, 11am, 11an, 11ao, 11ap, 12d, 12e. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Hochstrasser M. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr. Opin. Cell Biol. 1995;7:215–223. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- 2.Yamasaki L, Pagano M. Cell cycle, proteolysis and cancer. Curr. Opin. Cell Biol. 2004;16:623–628. doi: 10.1016/j.ceb.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 4.Ciechanover A, Orian A, Schwartz AL. The ubiquitin-mediated proteolytic pathway: mode of action and clinical implications. J. Cell. Biochem. 2000:40–51. doi: 10.1002/(sici)1097-4644(2000)77:34+<40::aid-jcb9>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 5.Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell (Cambridge, Mass.) 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 6.Baumeister W, Walz J, Zuhl F, Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–880. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 7.Groll M, Ditzel L, Loewe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 .ANG. resolution. Nature (London) 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 8.Groll M, Huber R. Inhibitors of the eukaryotic 20S proteasome core particle: a structural approach. Biochim. Biophys. Acta, Mol. Cell Res. 2004;1695:33–44. doi: 10.1016/j.bbamcr.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 9.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem. Biol. 2001;8:739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 10.Borissenko L, Groll M. 20S Proteasome and Its Inhibitors: Crystallographic Knowledge for Drug Development. Chem. Rev. (Washington, DC, U. S.) 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- 11.Blackburn C, Gigstad KM, Hales P, Garcia K, Jones M, Bruzzese FJ, Barrett C, Liu JX, Soucy TA, Sappal DS, Bump N, Olhava EJ, Fleming P, Dick LR, Tsu C, Sintchak MD, Blank JL. Characterization of a new series of non-covalent proteasome inhibitors with exquisite potency and selectivity for the 20S b5-subunit. Biochem. J. 2010;430:461–476. doi: 10.1042/BJ20100383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Genin E, Reboud-Ravaux M, Vidal J. Proteasome inhibitors: recent advances and new perspectives in medicinal chemistry. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2010;10:232–256. doi: 10.2174/156802610790725515. [DOI] [PubMed] [Google Scholar]
- 13.Groll M, McArthur KA, Macherla VR, Manam RR, Potts BC. Snapshots of the Fluorosalinosporamide/20S Complex Offer Mechanistic Insights for Fine Tuning Proteasome Inhibition. J. Med. Chem. 2009;52:5420–5428. doi: 10.1021/jm900559x. [DOI] [PubMed] [Google Scholar]
- 14.Groll M, Kim KB, Kairies N, Huber R, Crews CM. Crystal structure of epoxomicin:20S proteasome reveals a molecular basis for selectivity of a’,b’-poxyketone proteasome inhibitors. J. Am. Chem. Soc. 2000;122:1237–1238. [Google Scholar]
- 15.Kupperman E, Lee Edmund C, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010;70:1970–1980. doi: 10.1158/0008-5472.CAN-09-2766. [DOI] [PubMed] [Google Scholar]
- 16.Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. [Erratum to document cited in CA152:517050] Cancer Res. 2010;70:3853. doi: 10.1158/0008-5472.CAN-09-2766. [DOI] [PubMed] [Google Scholar]
- 17.Dick Lawrence R, Fleming Paul E. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010;15:243–249. doi: 10.1016/j.drudis.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Dorsey BD, Menta E, Bernardini R, Bernareggi A, Casara PG, D’Arasmo G, Ferretti E, De Munari S, Oliva A, Iqbal M, Chatterjee S, Ruggeri B, Ator MA, Williams M, Mallamo JP. CEP-18770: Discovery of a Potent, Selective and Orally Active Proteasome Inhibitor for the Treatment of Cancer. Frontiers in CNS and Oncology Medicinal Chemistry, ACS-EFMC; Siena, Italy. October 7-9 2007; COMC-027. [DOI] [PubMed] [Google Scholar]
- 19.Piva R, Ruggeri B, Williams M, Costa G, Tamagno I, Ferrero D, Giai V, Coscia M, Peola S, Massaia M, Pezzoni G, Allievi C, Pescalli N, Cassin M, di Giovine S, Nicoli P, de Feudis P, Strepponi I, Roato I, Ferracini R, Bussolati B, Camussi G, Jones-Bolin S, Hunter K, Zhao H, Neri A, Palumbo A, Berkers C, Ovaa H, Bernareggi A, Inghirami G. CEP-18770: a novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood. 2008;111:2765–2775. doi: 10.1182/blood-2007-07-100651. [DOI] [PubMed] [Google Scholar]
- 20.Sterz J, von Metzler I, Hahne J-C, Lamottke B, Rademacher J, Heider U, Terpos E, Sezer O. The potential of proteasome inhibitors in cancer therapy. Expert Opin. Invest. Drugs. 2008;17:879–895. doi: 10.1517/13543784.17.6.879. [DOI] [PubMed] [Google Scholar]
- 21.Dorsey BD, Iqbal M, Chatterjee S, Menta E, Bernardini R, Bernareggi A, Cassara PG, D’Arasmo G, Ferretti E, De MS, Oliva A, Pezzoni G, Allievi C, Strepponi I, Ruggeri B, Ator MA, Williams M, Mallamo JP. Discovery of a Potent, Selective, and Orally Active Proteasome Inhibitor for the Treatment of Cancer. J. Med. Chem. 2008;51:1068–1072. doi: 10.1021/jm7010589. [DOI] [PubMed] [Google Scholar]
- 22.Fuchs O. Proteasome inhibition as a therapeutic strategy in patients with multiple myeloma. Mult. Myeloma. 2009:101–125. [Google Scholar]
- 23.Lam KS, Lloyd GK, Neuteboom STC, Palladino MA, Sethna KM, Spear MA, Potts BC. From natural products to clinical trials: NPI-0052 (salinosporamide A), a marine actinomycete-derived anticancer agent. Nat. Prod. Chem. Drug Discovery. 2010:355–373. [Google Scholar]
- 24.Zhou H-J, Aujay MA, Bennett MK, Dajee M, Demo SD, Fang Y, Ho MN, Jiang J, Kirk CJ, Laidig GJ, Lewis ER, Lu Y, Muchamuel T, Parlati F, Ring E, Shenk KD, Shields J, Shwonek PJ, Stanton T, Sun CM, Sylvain C, Woo TM, Yang J. Design and Synthesis of an Orally Bioavailable and Selective Peptide Epoxyketone Proteasome Inhibitor (PR-047) J. Med. Chem. 2009;52:3028–3038. doi: 10.1021/jm801329v. [DOI] [PubMed] [Google Scholar]
- 25.Groll M, Berkers CR, Ploegh HL, Ovaa H. Crystal Structure of the Boronic Acid-Based Proteasome Inhibitor Bortezomib in Complex with the Yeast 20S Proteasome. Structure (Cambridge, MA, U. S.) 2006;14:451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 26.Groll M, Huber R, Potts BCM. Crystal Structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in Complex with the 20S Proteasome Reveal Important Consequences of b-Lactone Ring Opening and a Mechanism for Irreversible Binding. J. Am. Chem. Soc. 2006;128:5136–5141. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]
- 27.Schmidtke G, Holzhutter H-G, Bogyo M, Kairies N, Groll M, De Giuli R, Emch S, Groettrup M. How an inhibitor of the HIV-I protease modulates proteasome activity. J. Biol. Chem. 1999;274:35734–35740. doi: 10.1074/jbc.274.50.35734. [DOI] [PubMed] [Google Scholar]
- 28.Furet P, Imbach P, Fuerst P, Lang M, Noorani M, Zimmermann J, Garcia-Echeverria C. Structure-Based optimization of 2-aminobenzylstatine derivatives: potent and selective inhibitors of the chymotrypsin-Like activity of the human 20S proteasome. Bioorg. Med. Chem. Lett. 2002;12:1331–1334. doi: 10.1016/s0960-894x(02)00178-6. [DOI] [PubMed] [Google Scholar]
- 29.Furet P, Imbach P, Noorani M, Koeppler J, Laumen K, Lang M, Guagnano V, Fuerst P, Roesel J, Zimmermann J, Garcia Echeverria C. Entry into a New Class of Potent Proteasome Inhibitors Having High Antiproliferative Activity by Structure-Based Design. J. Med. Chem. 2004;47:4810–4813. doi: 10.1021/jm049660v. [DOI] [PubMed] [Google Scholar]
- 30.Basse N, Papapostolou D, Pagano M, Reboud-Ravaux M, Bernard E, Felten A-S, Vanderesse R. Development of lipopeptides for inhibiting 20S proteasomes. Bioorg. Med. Chem. Lett. 2006;16:3277–3281. doi: 10.1016/j.bmcl.2006.03.033. [DOI] [PubMed] [Google Scholar]
- 31.Kohno J, Koguchi Y, Nishio M, Nakao K, Kuroda M, Shimizu R, Ohnuki T, Komatsubara S. Structures of TMC-95A-D: Novel proteasome inhibitors from Apiospora montagnei Sacc. TC 1093. J. Org. Chem. 2000;65:990–995. doi: 10.1021/jo991375+. [DOI] [PubMed] [Google Scholar]
- 32.Formicola L, Marechal X, Basse N, Bouvier-Durand M, Bonnet-Delpon D, Milcent T, Reboud-Ravaux M, Ongeri S. Novel fluorinated pseudopeptides as proteasome inhibitors. Bioorg. Med. Chem. Lett. 2009;19:83–86. doi: 10.1016/j.bmcl.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 33.Marechal X, Pujol A, Richy N, Genin E, Basse N, Reboud-Ravaux M, Vidal J. Noncovalent inhibition of 20S proteasome by pegylated dimerized inhibitors. Eur. J. Med. Chem. 2012;52:322–327. doi: 10.1016/j.ejmech.2012.02.052. [DOI] [PubMed] [Google Scholar]
- 34.Basse N, Montes M, Marechal X, Qin L, Bouvier-Durand M, Genin E, Vidal J, Villoutreix BO, Reboud-Ravaux M. Novel Organic Proteasome Inhibitors Identified by Virtual and in Vitro Screening. J. Med. Chem. 2010;53:509–513. doi: 10.1021/jm9011092. [DOI] [PubMed] [Google Scholar]
- 35.Groll M, Koguchi Y, Huber R, Kohno J. Crystal Structure of the 20 S Proteasome:TMC-95A Complex: A Non-covalent Proteasome Inhibitor. J. Mol. Biol. 2001;311:543–548. doi: 10.1006/jmbi.2001.4869. [DOI] [PubMed] [Google Scholar]
- 36.Groll M, Gallastegui N, Marechal X, Le RV, Basse N, Richy N, Genin E, Huber R, Moroder L, Vidal J, Reboud-Ravaux M. 20S proteasome inhibition: designing noncovalent linear peptide mimics of the natural product TMC-95A. ChemMedChem. 2010;5:1701–1705. doi: 10.1002/cmdc.201000293. [DOI] [PubMed] [Google Scholar]
- 37.Kaiser M, Groll M, Siciliano C, Assfalg-Machleidt I, Weyher E, Kohno J, Milbradt AG, Renner C, Huber R, Moroder L. Binding mode of TMC-95A analogues to eukaryotic 20S proteasome. ChemBioChem. 2004;5:1256–1266. doi: 10.1002/cbic.200400096. [DOI] [PubMed] [Google Scholar]
- 38.Gallastegui N, Beck P, Arciniega M, Huber R, Hillebrand S, Groll M. Hydroxyureas as Noncovalent Proteasome Inhibitors. Angew. Chem., Int. Ed. 2012;51:247–249. doi: 10.1002/anie.201106010. [DOI] [PubMed] [Google Scholar]
- 39.Meiners S, Heyken D, Weller A, Ludwig A, Stangl K, Kloetzel P-M, Krueger E. Inhibition of Proteasome Activity Induces Concerted Expression of Proteasome Genes and de Novo Formation of Mammalian Proteasomes. J. Biol. Chem. 2003;278:21517–21525. doi: 10.1074/jbc.M301032200. [DOI] [PubMed] [Google Scholar]
- 40.Lawrence HR, Kazi A, Luo Y, Kendig R, Ge Y, Jain S, Daniel K, Santiago D, Guida WC, Sebti SM. Synthesis and biological evaluation of naphthoquinone analogs as a novel class of proteasome inhibitors. Bioorg. Med. Chem. 2010;18:5576–5592. doi: 10.1016/j.bmc.2010.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ge Y, Kazi A, Marsilio F, Luo Y, Jain S, Brooks W, Daniel KG, Guida WC, Sebti SM, Lawrence HR. Discovery and Synthesis of Hydronaphthoquinones as Novel Proteasome Inhibitors. J. Med. Chem. 2012;55:1978–1998. doi: 10.1021/jm201118h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ozcan S, Aslamuzzaman K, Marsilio F, Daniel K, Brooks W, Guida W, Lawrence H, Sebti S. Abstract 1359: Identification of a novel class of compounds as proteasome inhibitors: Synthesis and structure activity relationship studies of PI-1833 library; American Association of Cancer Research, 102nd annual meeting; Orlando, Florida. April 15 2011; Orlando, Florida: Cancer Research; [Google Scholar]
- 43.Villoutreix B, Reboud-Ravaux M, Basse N, Vidal J, Montes M. Nitrogen Heterocyclic Derivatives as Proteasome Modulators. Oct 20, 2011. US 2011/0257176A1.
- 44.Villoutreix B, Reboud-Ravaux M, Basse N, Vidal J, Montes M. Nitrogen heterocycle derivatives as proteasome modulators. Jan 7, 2010. WO2010001365A1.
- 45.Blackburn C, Barrett C, Blank JL, Bruzzese FJ, Bump N, Dick LR, Fleming P, Garcia K, Hales P, Jones M, Liu JX, Nagayoshi M, Sappal DS, Sintchak MD, Tsu C, Xia C, Zhou X, Gigstad KM. Optimization of a series of dipeptides with a P3 [small beta]-neopentyl asparagine residue as non-covalent inhibitors of the chymotrypsin-like activity of human 20S proteasome. MedChemComm. 2012;3:710–719. doi: 10.1016/j.bmcl.2010.09.032. [DOI] [PubMed] [Google Scholar]
- 46.Baciocchi E, Fabbri C, Lanzalunga O. Lignin peroxidase-catalyzed oxidation of nonphenolic trimeric lignin model compounds: Fragmentation reactions in the intermediate radical cations. J. Org. Chem. 2003;68:9061–9069. doi: 10.1021/jo035052w. [DOI] [PubMed] [Google Scholar]
- 47.Spurg A, Waldvogel SR. High-yielding cleavage of (aryloxy) acetates. Eur. J. Org. Chem. 2008:337–342. [Google Scholar]
- 48.Shah MR, Arfan M, Amin H, Hussain Z, Qadir MI, Iqbal Choudhary M, VanDerveer D, Ahmed Mesaik M, Soomro S, Jabeen A, Khan IU. Synthesis of new bergenin derivatives as potent inhibitors of inflammatory mediators NO and TNF-a. Bioorg. Med. Chem. Lett. 2012;22:2744–2747. doi: 10.1016/j.bmcl.2012.02.096. [DOI] [PubMed] [Google Scholar]
- 49.Wrzesien J, Graham D. Synthesis of SERS active nanoparticles for detection of biomolecules. Tetrahedron. 2012;68:1230–1240. [Google Scholar]
- 50.Joseph R, Ramanujam B, Acharya A, Rao CP. Lower Rim 1,3-Di{bis(2-picolyl)}amide Derivative of Calix[4]arene (L) as Ratiometric Primary Sensor toward Ag+ and the Complex of Ag+ as Secondary Sensor toward Cys: Experimental, Computational, and Microscopy Studies and INHIBIT Logic Gate Properties of L. J. Org. Chem. 2009;74:8181–8190. doi: 10.1021/jo901676s. [DOI] [PubMed] [Google Scholar]
- 51.Gezginci MH, Martin AR, Franzblau SG. Antimycobacterial Activity of Substituted Isosteres of Pyridine- and Pyrazinecarboxylic Acids. 2. J. Med. Chem. 2001;44:1560–1563. doi: 10.1021/jm000350w. [DOI] [PubMed] [Google Scholar]
- 52.Ji J, Lee C-L, Sippy KB, Li T, Gopalakrishnan M. Oxadiazole derivatives as neuronal nicotinic acetylcholine receptor ligands and a4b2 pos. allosteric modulators and their preparation, pharmaceutical compositions and use in the treatment of diseases. 2008. 2008-134678 20080269236, 20080606.
- 53.Sindkhedkar MD, Desai VN, Loriya RM, Patel MV, Trivedi BK, Bora RO, Diwakar SD, Jadhav GR, Pawar SS. Preparation of erythromycin macrolides and ketolides having antimicrobial activity. 2008. 2007-IB2405 2008023248, 20070822.
- 54.Romeiro LAS, Ferreira M. d. S., da Silva LL, Castro HC, Miranda ALP, Silva CLM, Noel F, Nascimento JB, Araujo CV, Tibirica E, Barreiro EJ, Fraga CAM. Discovery of LASSBio-772, a 1,3-benzodioxole N-phenylpiperazine derivative with potent alpha 1A/D-Adrenergic receptor blocking properties. Eur. J. Med. Chem. 2011;46:3000–3012. doi: 10.1016/j.ejmech.2011.04.032. [DOI] [PubMed] [Google Scholar]
- 55.Weingarth M, Raouafi N, Jouvelet B, Duma L, Bodenhausen G, Boujlel K, Schollhorn B, Tekely P. Revealing molecular self-assembly and geometry of non-covalent halogen bonding by solid-state NMR spectroscopy. Chem. Commun. (Cambridge, U. K.) 2008:5981–5983. doi: 10.1039/b813237b. [DOI] [PubMed] [Google Scholar]
- 56.Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science (Washington, D. C.) 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 57.Fenteany G, Schreiber SL. Lactacystin, proteasome function, and cell fate. J. Biol. Chem. 1998;273:8545–8548. doi: 10.1074/jbc.273.15.8545. [DOI] [PubMed] [Google Scholar]
- 58.Meanwell NA. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 59.Eng JK, McCormack AL, Yates JR., III An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 60.Dick LR, Cruikshank AA, Destree AT, Grenier L, McCormack TA, Melandri FD, Nunes SL, Palombella VJ, Parent LA, Plamondon L, Stein RL. Mechanistic studies on the inactivation of the proteasome by lactacystin in cultured cells. J. Biol. Chem. 1997;272:182–188. doi: 10.1074/jbc.272.1.182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.