

Abstract
The current study tested the hypothesis that conditioned pain modulation is mediated by the release of endogenous opioids with a placebo-controlled (sugar pill) study of naltrexone (50 mg) in 33 healthy volunteers over two counter-balanced sessions. Pain modulation consisted of rating of heat pain (palm) during concurrent cold water immersion (foot). Compared to baseline heat pain ratings, concurrent foot immersion lowered pain intensity ratings, which suggests an inhibitory effect, was reduced with naltrexone, suggesting at least partial dependence of inhibition on endogenous opioids. An exploratory analysis revealed that individual differences in catastrophizing moderated the effects of naltrexone; endogenous opioid blockade abolished modulation in subjects lower in catastrophizing while modulation was unaffected by naltrexone among high catastrophizers. The results suggest a role of endogenous opioids in endogenous analgesia, but hint that multiple systems might contribute to conditioned pain modulation, and that these systems might be differentially activated as a function of individual differences in responses to pain.
Keywords: Conditioned pain modulation · Diffuse noxious inhibitory control · Endogenous opioids · Naltrexone · Heat pain · Catastrophizing
Introduction
Processing of pain by the nervous system is not passive; rather it is under active modulation by a number of endogenous mechanisms. Conditioned pain modulation (CPM) is a term that encompasses a variety of non-invasive methods to experimentally evaluate the capacity to inhibit pain (Villanueva, 2009; Willer et al., 1984) emphasizing the notion of “pain-inhibition-by-pain.” It is frequently characterized as a reduction of pain produced by a focal pain stimulus as a result of a second painful stimulus in a remote area of the body. Based on a recent consensus of pain researchers (Yarnitsky et al., 2010), the term CPM encompasses the previously described phenomenon termed diffuse noxious inhibitory control (DNIC). We have previously documented individual differences in conditioned pain modulation in which a cold water bath applied to the foot significantly diminishes pain produced by a heated probe applied to the hand (King et al., 2009; Riley et al., 2010a). Similar outcomes have been reported elsewhere (Price & McHaffie, 1988; Pud et al., 2009) with differences in the effectiveness of these pain inhibitory mechanisms as a function of age (Edwards et al., 2003; Riley et al., 2010a; Washington et al., 2000), gender (Popescu et al., 2010), and chronic pain (Heymen et al., 2010; Maixner et al., 1995; Pielsticker et al., 2005; Song et al., 2006; Staud et al., 2003; Wilder-Smith et al., 2009). Considering the fact that a number of clinical pain populations exhibit a reduced capacity to inhibit pain, understanding the mechanisms modulating pain transmission and perception could assist in the prevention and/or treatment of pain (Edwards, 2005).
Research is needed to evaluate the mechanisms mediating pain inhibition in CPM paradigms. Physiological correlates of conditioned pain modulation could be related to activity of endogenous opioids because these peptides, and their corresponding receptors are expressed in areas implicated in pain processing and transmission (Millan, 2002). Pharmacological studies have suggested a role of endogenous opioids through receptor blockade studies. Several animal studies have shown that opioid antagonists block the expression of stress-induced analgesia (Kelly & Franklin, 1987; King et al., 2007; Tricklebank et al., 1984) and DNIC (Kraus et al., 1981; Le Bars et al., 1981). In human studies of CPM (Table 1), however, the role of opioid peptides in endogenous analgesia has been inconsistent with some studies reporting blockade of pain inhibition (Pertovaara et al., 1982; Willer et al., 1990) while others report that opioid antagonists failed to affect inhibition (Edwards et al., 2004; Peters et al., 1992; Sprenger et al., 2011). In the recent study by Sprenger et al., reduction of pain inhibition was not influenced by naloxone but the relationships between cortical areas associated with pain inhibition were altered (Sprenger et al., 2011). Possible reasons for the mixed results and inconclusive support may be due to small sample sizes and the use of threshold-level test stimuli including heat/cold thresholds (Pertovaara et al., 1982), heat pain thresholds (Edwards et al., 2004), and nociceptive flexion reflex responses based on RIII responses to electrical stimulation (Peters et al., 1992; Willer et al., 1990), which is useful method to evaluate alterations in dorsal horn processing. However, other dynamics methods including temporal summation thresholds (Edwards et al., 2004) and a sustained heat pain pulse (Sprenger et al., 2011) have also resulted in negative findings.
Table 1. CPM studies reporting pharmacological blockade of endogenous opioids.
| Author | n | Pain outcome | Opioid antagonist | Route of administration | Effect on inhibition |
|---|---|---|---|---|---|
| Edwards et al. (2004) | 6 | TS, HPT | Naloxone | i.m. (6 mg/kg) | No effect |
| Peters et al. (1992) | 46a,b | NFR-RIII | Naloxone | i.v. (0.8 mg, 2 mL) | No effect |
| Pertovaara et al. (1982) | 12 | CT, HT | Naloxone | i.v. (2 mg) | Reduced |
| Sprenger et al. (2011) | 22a | HP | Naloxone | i.v. (0.15 mg/kg bolus, 0.2 mg/kg infusion) | No effect |
| Willer et al. (1990) | 9 | NFR-RIII | Naloxone | i.v. (0.4 mg, 4 mL) | Reduced |
CT cold thresholds, HT heat thresholds, HPT heat pain threshold, HP 47.5 °C heat pulse, NFR-RIII nociceptive flexion reflex threshold, TS temporal summation of heat pain, i.v. intravenous, i.m. intramuscular
Males only
Chronic pin patients
Considering the critical roles they play in many aspects of pain and analgesia, it is important to investigate further whether endogenous opioids contribute to CPM. The current study builds upon previous experimental human studies in our laboratory (King et al., 2009) by addressing several methodological shortcomings related to sample size and experimental pain assessments. Traditional psychophysical tests that have examined CPM have used brief, threshold-level test stimuli (Pertovaara et al., 1982; Peters et al., 1992; Willer et al., 1990) with the exception of one study that evaluated the effect of a noxious cold conditioning stimulus on temporal summation of pain (Edwards et al., 2004). The use of more dynamic supra-threshold pain stimuli, particularly with longer duration, has been gaining interest (Naert et al., 2008; Wong et al., 2010), because such stimuli are more phenomenologically similar to clinical pain (Tousignant-Laflamme et al., 2008).
The aim of this study was to examine whether CPM is attenuated following administration of an opioid antagonist. This aim was accomplished with a double-blind, placebo controlled, crossover study of an oral opioid antagonist, naltrexone prior to experimental pain testing. We examined subjective pain responses to noxious heat (test stimuli) at baseline, and then during a cold water bath (conditioning stimuli); testing was performed in separate sessions involving administration of naltrexone or placebo. We hypothesized that simultaneous exposure to the water bath would reduce the intensity of heat pain (e.g., endogenous analgesia) compared to the baseline session during placebo, and that this effect would be reversed by naltrexone. We also investigated individual difference factors that could influence pain inhibition, including catastrophizing (Edwards et al., 2006; Goodin et al., 2009), side effects of the medication (Riley et al., 2010b), and attention (Lautenbacher et al., 2007; Moont et al., 2010).
Methods
Subjects
The current study was approved by the institutional review board (IRB) at the University of Florida and consisted of a convenience sample of thirty-three healthy consenting subjects (n = 17 males; n = 16 females; average age: 23.5 ± 3.89 years) recruited through posted advertisements around the University of Florida campus. Most subjects identified themselves as Caucasian (n = 24). Other self-reported races included African American (4), Hispanic (3), and Asian (2). Subjects were compensated for participation. Subjects were provided a brief explanation of the general purpose and procedures of the study. If interested, they were informed about Health Insurance Portability and Account-ability Act (HIPAA) regulations and reviewed and signed an Informed Consent Form to grant authorization for collection of health data needed to determine eligibility. Then, subjects underwent a health screening, which consisted of a health questionnaire and clarification by interview.
Study exclusion criteria
While the use of naltrexone in experimental studies has been well tolerated (France et al., 2007; Ring et al., 2007), subjects were screened for a number of potential interactions including regular use of alcohol (>2 drinks/day), opioids, non-steroidal anti-inflammatory drugs, or a history of liver/kidney disease. In addition, subjects were ineligible for the study if they were unable to adequately communicate and provide informed consent, unable to reliably rate pain intensity, experienced uncontrolled hypertension, reported to have a serious systemic disease (e.g., diabetes, thyroid problems, etc.), neurological, cardiovascular/pulmonary, or serious psychiatric diseases (i.e., schizophrenia, bipolar disorder). Exclusion criteria also included any chronic pain conditions (e.g., low back pain, fibromyalgia) or any ongoing acute pain problem (e.g., headaches, injury-related pain). In addition, session specific exclusion criteria were maintained on the day of testing, which included the use of coffee (within the previous 2 h), alcohol (within the previous 24 h), and over-the-counter pain medications (within the previous 24 h) prior to the study. Subjects were rescheduled if they used these substances within the exclusion time. In addition, subjects were rescheduled if they had an active infectious disease or febrile condition (e.g., sinusitis, influenza).
Study protocol
Training session
After completing the health assessment, subjects completed a training session as described previously (King et al., 2009; Riley et al., 2010a) to orient them to the continuous pain rating system and to determine the temperature of the stimuli for use in subsequent experimental sessions. The goal was to determine temperatures that evoked mild-to-moderate pain for both a 30-s heat stimulus (Range: 46.0–50.0 °C) and cold water immersion (Range: 8.0–16.0 °C). A maximum temperature of 50.0 °C (n = 3) was used for heat and minimum temperature of 8.0 °C (n = 3) for cold. Subjects that did not reach the target pain ratings were assigned these temperatures. The temperatures were individually tailored to standardize the pain intensity range for all tests, which minimizes the risk of unacceptable discomfort for the subjects, avoids floor and ceiling effects, and reduces potential reporting biases. Many investigations have applied similar tailoring methods to evaluate group comparisons between pain patients (King et al., 2009) and older adults (Riley et al., 2010a) in addition to studies with baseline heat stimuli (Granot et al., 2006) and CPM (Granot et al., 2008; Moont et al., 2010). The average water temperature was 12.9 °C (±2.7), and the thermode temperature was 48.2 °C (±1.1). These temperatures were negatively correlated (r = −0.55, p < 0.01). No gender differences (All p > 0.10) were observed for heat (males: 48.4 ± 1.0 °C; females: 48.1 ± 1.2 °C) or cold (males: 12.1 ± 2.9 °C; females: 13.6 ± 2.3 °C).
Testing sessions
Following the training session, subjects participated in a control session and two experimental sessions with study medication on different days. The experimental sessions were scheduled on non-consecutive days to avoid potential carry-over effects from the medication sessions. The first session consisted of a control session in which subjects underwent the same heat pain testing procedure used in the medication sessions but without exposure to a conditioning stimulus (i.e., water bath) or medication. The purpose of the additional pre-experimental session was to confirm that the testing temperature resulted in the intended pain range, orient the subject to the actual timing of the testing conditions, and reduce across day inhibition (Quiton & Greenspan, 2008). The session timeline is illustrated in Fig. 1. Upon arrival at the testing laboratory, subjects were seated on a comfortable chair and asked to relax for several minutes before being asked about their health, alcohol and medication use, and filled out several questionnaires. Then, in double-blind fashion, subjects were given a non-descriptive pill of either naltrexone (50 mg) or placebo with a glass of water. An additional 45 min rest period was observed before commencement of the conditioned pain modulation procedure to allow naltrexone to reach peak concentrations in the body (Gonzalez & Brogden, 1988; Verebey, 1981). Next, CPM was evaluated: subjects were asked to attend to and rate the intensity of heat pain, which was produce by a focal heated stimulus (e.g., contact thermode, left palm). The rating of heat pain was continuously rated with an electronic visual analog scale (eVAS) during concurrent immersion of their right foot in a cold water bath (e.g., conditioning stimulus). For each trial, subjects placed their foot into the cold water bath for 40 s. Focal heat, which was set at the subject's individualized temperature, was delivered 10 s after placement of the foot into the bath for 30 s. Each psychophysical testing session consisted of five consecutive trials with a 3-min rest period in between trials. At the end of each session, subjects answered several questionnaires.
Fig. 1. Session timeline for evaluation of opioid involvement in conditioned pain modulation with a double-blind, placebo controlled crossover study of naltrexone.
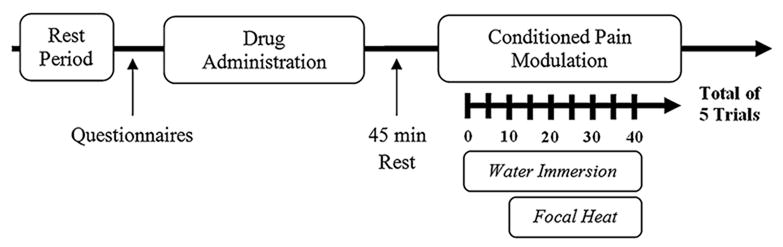
Study procedures
Study medication
Naltrexone hydrochloride (Revia, DuPont Pharmaceuticals Co; Wilmington, Delaware), is an orally administered opioid antagonist. Subjects were randomized to receive placebo medication (i.e., inert pill without active agents consisting of a gelatin empty capsule filled with approximately 450 mg of lactose) or naltrexone (50 mg) for their first conditioned pain modulation session. The randomization was provided by the Investigational Drug Service at the University of Florida, which handled encapsulation and medication dispensing. The number of subjects to receive placebo on the first medication session was eighteen (54.5 %) while fifteen (45.5 %) subjects received naltrexone.
Experimental stimulus (painful thermal contact)
As reported previously (King et al., 2009; Riley et al., 2010a), focal thermal stimuli (44.0–50.0 °C) were administered by a Peltier-based thermode (23 mm × 23 mm). Subjects were instructed to rate the pain produced by the thermode with the eVAS. For each trial, the thermode was brought to a neutral temperature (33.0 °C), and then brought into light skin contact with a solenoid. After a short period, the temperature was ramped (1.5 °C/s) to the desired temperature. After 30 s, the thermode remained in contact with the skin but was cooled back to 33.0 °C. Subsequent trials used widely spaced thermal pulses (3-min inter-stimulus interval) to minimize sensitization. Subjects provided continuous ratings of the pain intensity produced by the test stimulus (i.e., noxious heat) during the 30-s trial with an electronic visual analogue scale (eVAS). As described previously (King et al., 2009), the eVAS consists of a low-friction sliding potentiometer (100 mm travel) mounted to an inclined desk. Two anchors were provided for the 0–100 scale: the left endpoint (0) designated as ‘no pain’ and the right endpoint (100) designated as ‘most intense pain imaginable.’ Subjects were instructed to move the slider in proportion to their pain intensity in real time. The position of the slider (i.e., pain intensity ratings) was automatically converted into a percentage (0–100 %).
Conditioning stimulus (water immersion)
Cold-water immersion was used as the conditioning stimulus. The water bath was cooled by a refrigerated water circulator (Neslab, Portsmouth, NH). Water was maintained at a constant temperature throughout the water bath and constantly re-circulated to prevent local warming or cooling around the foot. Subjects were instructed to immerse their foot to the ankle in water set at a noxious cold temperature determined during baseline assessment (16–8 °C). The water level was set at a height of 7 cm to keep the stimulated area consistent.
Physiological and psychological measures
Blood pressure
Blood pressure was measured three times prior to (baseline) and immediately after pain testing. Blood pressure was recorded with a noninvasive electronic oscillometric sphygmomanometer with the cuff attached to the left arm.
Cognitive affective side effects
The cognitive affective side effects are a self-report measure designed to assess the impact of a study drug on cognitive and affective responses (Riley et al., 2010b). The Cognitive Affective Side Effects Questionnaire was initially designed to evaluate the effect of opioid analgesics, and the current study used the questionnaire to evaluate the side effects of naltrexone. The questionnaire consists of 44 items concerning physical symptoms subjects completed the cognitive affective side effects measure at the beginning (pre-drug) and end (post-drug) of each medication session to control for any pre-existing issues prior to testing. Change scores (Post–Pre) were used to determine potential effects of the medication. Grouping of side effects was performed based on data published by Riley et al. (2010b) including: (1) mental dulling; (2) relaxed; (3) unusual thought; (4) feelings of control; (5) confusion; and, (6) euphoria. Items regarding direct reference to “pain medication” (Item 35: “I like the mental effects of the pain medicine”; Item 39: “I feel a mental effect of the pain medicine”) were not included in the analysis.
Somatic side effects
The somatic side effects are a self-report questionnaire designed to measure side effects of drugs on physical well-being. The questionnaire consists of 28 items concerning physical symptoms. Subjects completed the somatic side effects at the beginning (pre-drug) and end (post-drug) of each medication sessions. Similar to the Cognitive Affective Side Effects Questionnaire, the Somatic Side Effects Questionnaire was used to evaluate the side effects of naltrexone. Response to each item was summed to obtain an overall score. Physical side effects (Riley et al., 2010b) were grouped by: (1) sedation; (2) tingling; (3) vasodilatation; (4) dry mouth; (5) poor balance; (6) nausea and (7) tremors.
Concentration and distraction
Two items focused on the effect of immersion on their perception of heat pain. These items were adapted from a post-testing questionnaire reported by Edwards and colleagues (Edwards et al., 2003). The following items were rated on a scale of 0 (not at all) to 10 (very much): (1) “Was it difficult to pay attention to the thermal stimulation?” and, (2) “When rating the thermal stimuli, were you distracted by the sensations in your foot?”
Pain catastrophizing
The Pain Catastrophizing Questionnaire is a thirteen item questionnaire that measures catastrophic thinking in response to pain (Sullivan et al., 1995), which specifically relates to a negative pain-related cognitive processes composed of rumination, helplessness, and magnification (Campbell et al., 2010a; Cano et al., 2005). The Pain Catastrophizing Questionnaire was administered before the training session.
Data analysis
The dependent variable for this study was the peak pain rating collected over the five thermal trials. Measures of area under the curve (AUC) was also determined by calculating the trapezoid formula to summarize heat pain intensity over time (Pruessner et al., 2003). Based on AUC with respect to increase (AUC), this parameter emphasis changes of pain and other physiological responses across time and is most related to the sensitivity of the system being studies, in this case pain intensity. For both peak pain and AUC, all five trials were collapsed to generate an overall score for the baseline (BASELINE) and conditioned pain modulation sessions with placebo (CPM + PLACEBO) or naltrexone (CPM + NALTREXONE). Differences among the different conditions were tested using repeated measures analysis of variance (ANOVA). Analyses include the partial η2 as a measure of effect size where appropriate. Following the conventions of Cohen (Cohen, 1988), partial η2 = 0.01 is considered a small effect, partial η2 = 0.06 a medium-sized effect and partial η2 = 0.14 a large effect. Pearson correlations were calculated between continuous variables. Paired t tests were used to compare means between condition pain modulation sessions. For exploratory analysis, pain catastrophizing was based on a tertile split into low, moderate, and high catastrophizing groups (Bartley & Rhudy, 2008; Picavet et al., 2002). Differences in outcomes based on Male–Female ratios were analyzed with a Chi-square. To determine the effects of naltrexone on cognitive/affective and physical functioning, the Cognitive Affective Side Effects and Somatic Side Effects Questionnaire were scored before (pre-drug) and after (post-drug) administration of study medication, which generated difference scores. Side effect profiles were rated on a five point scale with the following response choices: 1 = not at all, 2 = a little bit, 3 = somewhat, 4 = quite a bit, 5 = extremely. Paired-samples t tests were used to test for session differences on side-effects and psychological variables.
Results
Psychophysical effects of placebo and naltrexone
Individuals' mean peak eVAS heat pain ratings for the baseline, placebo, and naltrexone conditions are provided in Fig. 2. Results of repeated measures ANOVA with a Greenhouse-Geisser correction revealed a significant main effect of study drug on CPM (F2,62 = 3.40, p = 0.05, η2 = 0.10), such that, relative to their own baseline peak heat pain ratings, individuals' peak heat pain ratings reported during concurrent application of cold pressor pain were significantly lower following the administration of placebo (t31 = 2.79, p < 0.01 with Bonferroni adjustment). Conversely, individuals' peak heat pain ratings reported during concurrent application of cold pressor pain following administration of naltrexone were not statistically different from their peak baseline heat pain ratings (t31 = 1.24, p = 0.23). These results suggest that the efficacy of endogenous pain modulation was not disrupted following the administration of a placebo (i.e., significant endogenous analgesia). However, administration of the opioid antagonist naltrexone reversed the reduction of pain during CPM, which could be attributed to changes in endogenous opioid activity.
Fig. 2. Mean (±SD) peak pain intensity ratings collapsed over five testing trials during baseline (heat only) and conditioned pain modulation trials with administration of study medication. *p < 0.05.
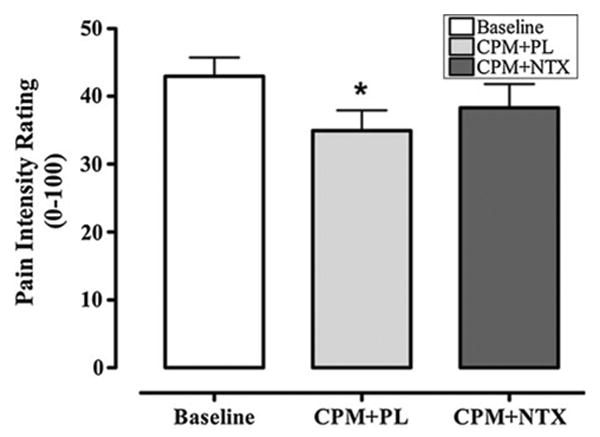
Similar to results obtained with peak pain scores, differences in CPM were also observed when AUC was used as the primary endpoint (data not presented). Results of repeated measures ANOVA with a Greenhouse-Geisser correction revealed a significant main effect of the study drug on CPM (F2,62 = 4.594, p = 0.02). AUC scores were lower during CPM with the control medication compared to baseline (t31 = 2.86, p < 0.01), but AUC scores did not significantly differ between baseline and naltrexone (t31 = 1.63, p = 0.11).
Effects of placebo and naltrexone on physiological responses
At the beginning of the sessions, there were no differences among control, placebo or naltrexone in systolic and diastolic blood pressure (all p's>0.10). Comparing blood pressures immediately preceding pain testing and following each pain testing trial, there were no differences in SBP or DBP (all p's>0.10). No relationships were detected between SBP and DBP with any of the pain outcome measures (r range = 0.05–0.14; all p's>0.10).
Effects of placebo and naltrexone on concentration and distraction
The post-testing psychological measures were comparable between placebo and naltrexone. Subjects were able to concentrate equally (t33 = 0.28, p > 0.05) on the heat stimulus during conditioning pain modulation trials with placebo (8.2 ± 1.4) and naltrexone (8.1 ± 1.7). Similar results were observed with the difficulty to attend to the heat stimulus (t33 = 0.14, p > 0.05) during sessions with placebo (2.3 ± 2.0) and naltrexone (2.3 ± 2.3). Correlations between concentration and distraction were significant for placebo (r = −0.52, p = 0.002) and naltrexone (r = −0.66, p < 0.001).
Interaction of pain catastrophizing with conditioned pain modulation
The use of a tertile split (Table 2) to categorize pain catastrophizing into low, moderate, and high catastrophizing groups was successful for producing three distinct clusters of pain catastrophizing (F2,29 = 54.51, p < 0.001, η2 = 0.79). Tukey's post hoc tests showed that the low catastrophizing group reported significantly less catastrophizing on the standard Pain Catastrophizing Questionnaire than did the moderate (p < 0.001) and high (p < 0.001) catastrophizing groups. Similarly, the moderate catastrophizing group reported significantly less catastrophizing than the high catastrophizing group (p < 0.001). Results from a series of ANOVAs demonstrated that the three catastrophizing groups did not significantly differ in their reports of baseline peak heat pain ratings (F2,29 = 0.26, p = 0.77, η2 = 0.02), nor were there significant catastrophizing group differences for the temperatures of the tailored heat pain test stimulus (F2,29 = 0.31, p = 0.74, η2 = 0.02) or the tailored cold pain conditioning stimulus (F2,29 = 2.06, p = 0.13, η2 = 0.05).
Table 2. Means (±SD) for low, moderate, and high catastrophizing groups based on a tertile split.
| Low | Moderate | High | p value | |
|---|---|---|---|---|
| Demographics | ||||
| Male/female ratio | 6/5 | 6/5 | 4/6 | 0.75 |
| Age | 23.5 ± 2.6 | 23.0 ± 2.2 | 22.4 ± 1.3 | 0.49 |
| Testing temperatures | ||||
| Heat | 48.1 ± 1.3 | 48.4 ± 0.9 | 48.1 ± 1.1 | 0.74 |
| Cold | 12.2 ± 3.6 | 12.1 ± 2.0 | 14.2 ± 1.9 | 0.13 |
| Concentration | ||||
| Placebo | 8.2 ± 1.5 | 8.8 ± 1.1 | 7.7 ± 1.6 | 0.21 |
| Naltrexone | 8.2 ± 1.7 | 8.4 ± 1.4 | 7.6 ± 1.4 | 0.62 |
| Difficulty to attend | ||||
| Placebo | 2.1 ± 2.5 | 2.1 ± 1.8 | 2.9 ± 1.9 | 0.52 |
| Naltrexone | 2.2 ± 2.8 | 2.3 ± 2.2 | 2.4 ± 1.7 | 0.98 |
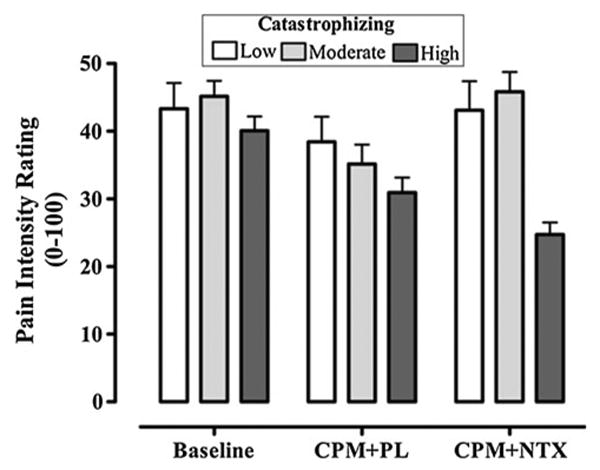
An additional exploratory repeated measures ANOVA with a Greenhouse-Geisser correction was completed to determine whether pain catastrophizing significantly interacted with the administration of study drug in relation to CPM. Results of this analysis were suggestive of a marginal interactive effect between catastrophizing and the study drug on endogenous analgesia (F4,58 = 1.96, p = 0.07, η2 = 0.12). It is noteworthy that the effect size for this interaction was large; however, the observed power for detecting this effect was sub-optimal (observed power = 0.48). As can be seen in Fig.3, the reduction from baseline of peak heat pain ratings following adminstration of placebo was not significantly different across the three catastrophizing groups (F2,29 = 0.51, p = 0.61, η2 = 0.03), suggesting that the low, moderate, and high catastrophizing groups produced significant and comparable amounts of CPM in the placebo condition. Interestingly, there was a significant difference in the pattern of CPM responses as a function of pain catastrophizing following administration of naltrexone (F2,29 = 4.16, p = 0.03, η2 = 0.22). Specifically, in response to naltrexone, the high catastrophizing group continued to report significantly reduced peak heat pain ratings compared to their baseline ratings (t9 = 3.75, p < 0.01 with Bonferroni correction), whereas heat pain ratings following adminstration of naltrexone were not significantly different from baseline ratings for either the low (t10 = 0.03, p = 0.97) or moderate (t10 = −0.10, p = 0.93) catatrophizing groups. These results suggest that the opioid-blockade produced by naltrexone was successful in reversing the CPM responses of the low and moderate catastrophizing groups; however, naltrexone did not reduce the CPM of the high pain catastrophizing group.
Fig. 3. Mean (±SD) scores of peak pain intensity ratings as a function of catastrophizing during baseline (heat only) and conditioned pain modulation trials with placebo and naltrexone.
Side effect profiles of placebo and naltrexone
Overall, naltrexone produced mild side effects, which were indicated by differences in the cognition/affect (Cognitive Affective side effects) and physical (Somatic side effects) subscales (Fig. 4). While naltrexone produced higher overall scores for the Cognitive-Affective and Somatic side effects than placebo, the difference was significant with the Somatic Side Effects (t33 = −2.94, p = 0.02) but not the Cognitive-Affective side effects (t33 = −1.82, p = 0.08). As for the Cognitive-Affective side effects subscales, when compared to placebo, naltrexone produced significant increases in Mental Dulling (t33 = −4.47, p < 0.001) and Confusion (t33 = −17.05, p < 0.001), and produced significant decreases in Euphoria (t33 = −27.77, p < 0.001). The Relaxed subscale was reduced in the naltrexone session but this effect was not significant (t33 = −1.63, p = 0.11). The other Cognitive-Affective side effects subscales (Unusual thoughts, Feelings of control) showed no changes between medication sessions (p > 0.10). The impact of these cognitive side effects on endogenous analgesia was not supported (All p > 0.10). On the Somatic side effects subscales, subjects treated with naltrexone also reported more Sedation (t33 = −3.71, p = 0.01) and Poor balance (t33 = −2.02, p = 0.05) compared to placebo. The other Somatic side effects sub-scales (Vasodilatation, Tingling, Dry mouth, Tremors, Nausea) showed no changes between medication sessions (p > 0.10).). No Somatic side effects subscale showed a significant correlation with CPM (All p > 0.10). No differences in Cognitive-Affective and Somatic side effects subscales as a function of pain catastrophizing were observed for placebo (p > 0.10, η2 < 0.13) or naltrexone (All p > 0.10; η2 < 0.11).
Fig. 4.
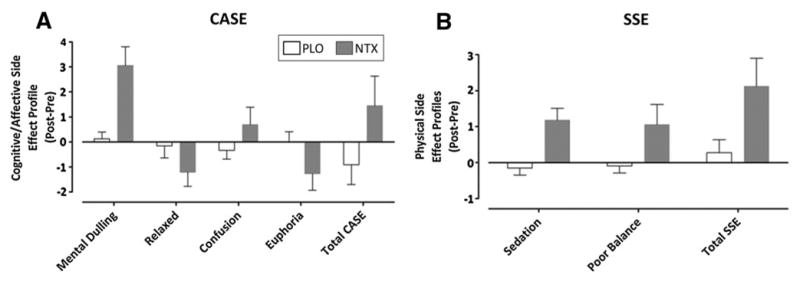
Mean (±SD) difference scores of cognitive/affective (Cognitive Affective Side Effects, CASE; a) and physical (Somatic Side Effects, SSE; b) side effects during conditioned pain modulation trials with placebo (open bar) and naltrexone (closed bar)
Discussion
The current study tested the hypothesis that endogenous opioids are involved in pain inhibition associated with CPM in which a painful conditioning stimulus (foot immersion) was paired with heat pain elicited by a focal stimulus to the palm. We found that subjects experienced less pain during a conditioned pain modulation protocol when compared to the baseline session with heat only. In contrast to placebo, pain inhibition was blocked by naltrexone leading to a comparable level of pain intensity to the baseline session. The inability to fully reverse pain inhibition by naltrexone was not expected since other mechanisms may also involved, but the results suggest that endogenous analgesia observed in the current protocol is mediated, at least in part, by the release of endogenous opioids. Furthermore, most of the psychological and biological factors were not related to endogenous analgesia with the exception of pain catastrophizing, which was not associated with the magnitude of endogenous analgesia but with sensitivity to opioid blockade. In particular, pain inhibition was abolished for low and moderate catastrophizers following naltrexone (opioid mediated endogenous analgesia); however, pain inhibition remains significant for high catastrophizers (non-opioid mediated endogenous analgesia).
Opioid mediated endogenous analgesia
A number of potential supraspinal (Bouhassira et al., 1993) and spinal (Price & McHaffie, 1988) sites, each of which express opioid receptors (Millan, 2002), have been shown to mediate diffuse noxious inhibitory controls in animal models, and these sites may also be involved in endogenous analgesic processes observed here in humans. The study's demonstration of the involvement of opioid peptides in endogenous analgesia is consistent with other animal (Kraus et al., 1981; Le Bars et al., 1981) studies of DNIC and CPM, respectively. Psychophysical tests like the cold pressor elicit β-endorphin release (Casale et al., 1985; Suzuki et al., 2007); which may bind to mu-opioid receptors in the central nervous system thereby reducing pain (Rosa et al., 1988; Suzuki et al., 2007). The results suggest involvement of non-opioid pathways as well. For example, given the role that norepinephrine and serotonin play in descending modulation of pain (Basbaum & Fields, 1984), it is possible that bulbospinal norepinephrine and serotonin pathways play a role in endogenous analgesia produced during conditioned pain modulation. In several animal studies, complete or partial reduction in diffuse noxious inhibitory controls was demonstrated by administration of serotonin (Chitour et al., 1982; Kraus et al., 1982) and α-adrenergic (Gjerstad et al., 2000; Wen et al., 2010) receptor antagonists, respectively. Additionally, complete blockade has been accomplished by opioid and adrenergic receptor antagonism (Wen et al., 2010). Further research in humans is needed to clarify the role serotonin and norepinephrine in endogenous analgesia and how these pathways interact with endogenous opioids.
Several studies have employed a pharmacological blockade design to evaluate the role of endogenous opioid-mediated CPM; however, the pattern of results is inconsistent (Table 1). For example, Willer et al. (1990) found that the inhibition of the RIII reflex, a spinal nociceptive flexion reflex, during concurrent exposure to a thermal conditioning was blocked by naloxone 5 min post injection. Pertovaara et al. (1982) reported that naloxone blocked the inhibition of thermal thresholds of the upper lip during ischemic conditioning stimulus at the arm. However, other studies have failed to support this hypothesis. Edwards et al. (Edwards et al., 2004) found that naloxone failed to block inhibition of temporal summation at 49.0 °C, or to affect thermal heat thresholds during concurrent cold water immersion. In addition, Peters et al. (1992) reported that naloxone failed to affect the RIII reflex and pain thresholds, but the experimental design focused on the presence of clinical pain in patients as a potential conditioning stimulus, and the design also involved administration of naloxone following saline administration. A recent fMRI study (Sprenger et al., 2011) reported that while naloxone failed to reverse endogenous analgesia (i.e., reduction of heat pain by ice packs), it abolished the relationship between the strength of the conditioning stimulus and the magnitude of endogenous analgesia. In addition, connectivity analyses of areas involved in pain modulation were disrupted by naloxone. The inconsistent results may be attributed to variability in sample size (Edwards et al., 2004), sample selection (Peters et al., 1992), or choice of test stimuli.
A number of additional reasons could explain support for or against the opioid mediation of CPM. First, all of the studies used naloxone, which circumvents first-pass metabolism, and body weight was used to determine the dosing in some studies. In terms of dosing, it is unclear if higher doses of naloxone, which are commonly, used in other blockade studies (i.e., 8 mg in 20 mL of saline; Bruehl et al., 2002) would offer a better methodology to evaluate opioid functioning. Based on a number of studies in animals suggesting a partial role of the endogenous opioids, it was also suggested that a more parametric study of naloxone be conducted to evaluate different doses (Edwards et al., 2004). Given the short half-life of naloxone, most of the studies engaged experimental testing within the time of peak plasma concentrations with the exception of Sprenger et al. In order to maintain stable plasma concentrations of naloxone, the study used an initial bolus (0.15 mg/kg, intravenous) followed by a constant infusion (0.2 mg/kg/h) of naloxone over the experiment based on a previous study in rats (Tepperman et al., 1983). Second, the subject groups may also complicate the observation of opioid blockade. The study by Peters and colleagues measured CPM in a chronic pain cohort, with an idea that the conditioning stimulus could be the experience of clinical pain (Peters et al., 1992). Furthermore, the dose of naloxone was not adjusted for body weight and was given following placebo assessment. Sprenger used male subject to avoid gender related differences in pain modulation. In the Sprenger study, male subjects were recruited to undergo CPM in the presence of naloxone. Interestingly, while Sprenger found no evidence of the effect of naloxone on perceptual changes in pain during CPM, naloxone did interfere with the relationship between the conditioning stimulus and the magnitude of pain inhibition. Finally, the conditioning stimulus could also be a factor. The CPM paradigm used a modified cold pressor test, which consisted of covering the leg with several bags of crushed ice (∼0.0 °C) or neutral water (25.0 °C).
Impact of pain catastrophizing opioid mediated endogenous analgesia
While measures of attention failed to show any relationship with pain inhibition, pain catastrophizing was associated with differential sensitivity to opioid blockade. Pain catastrophizing represents a maladaptive cognitive response to pain. Higher levels of catastrophizing are associated with greater pain sensitivity (Edwards et al., 2006), but conflicting reports have been reported regarding pain inhibition (van Wijk & Veldhuijzen, 2010), with some studies reporting a reduction (Goodin et al., 2009; Weissman-Fogel et al., 2008) or an enhancement (Granot et al., 2008) of endogenous analgesia as a function of catastrophizing. The current study found no evidence of a differential effect of catastrophizing on baseline pain sensitivity since temperature for heat and cold was comparable among the three groups or the magnitude of endogenous pain inhibition. While parameters explaining these discrepancies are unknown, the ability to modulate endogenous analgesia by catastrophizing may depend on the methods to induce endogenous analgesia and situational (Campbell et al., 2010a; Edwards et al., 2005, 2008; Goodin et al., 2009) versus dispositional (Campbell et al., 2010a) assessment of catastrophizing. Despite these conflicting observations, the relationship between catastrophizing and the functioning of the endogenous opioid system remains unknown.
While catastrophizing was not associated with the magnitude of endogenous analgesia, our findings suggest that catastrophizing interfered with its opioid-mediation, which has not been previously reported While previous studies have shown catastrophizing to predict lower levels of CPM (Campbell et al., 2010b; Edwards et al., 2006; Goodin et al., 2009), we found that catastrophizing was not associated with the magnitude of pain inhibition, rather with resistance to opioid blockade. One mechanism whereby catastrophizing may influence the opioid mediation of CPM is by interfering with opioid-rich pain-inhibitory circuits (Campbell & Edwards, 2009; Gracely et al., 2004) activated during CPM, such as the periaqueductal gray (Piche et al., 2009; Wilder-Smith et al., 2004). Our findings suggest that in high catastrophizing individuals, pain inhibition may be mediated through other neurochemical mechanisms. For example, non-opioid mechanisms are involved in stress-induced analgesia, which may be relevant in the current study.
Further research is needed to elucidate the neurophysiological mechanisms underlying the effects of a variety of pain-coping strategies. For example, in one study, individuals with more positive expectations regarding an experimental pain stimulus showed greater reductions in pain tolerance after naloxone administration (Frid et al., 1979). Similarly, perceived self-efficacy, which could be considered the opposite of catastrophizing, was associated with opioid-mediated endogenous analgesia (Bandura et al., 1987), such that subjects with greater self-efficacy showed greater sensitivity to opioid blockade by naloxone. The present findings suggest that those who are low in catastrophizing may show similar effects, while high catastrophizers may be less able to recruit endogenous opioid pain-inhibitory systems. Future research should replicate and examine the observation of non-opioid mediated endogenous analgesia in high catastrophizers with other pharmacological probes and a more detailed evaluation of the pain-coping strategies that participants employ.
Additional factors do not account for opioid or non-opioid mediated endogenous analgesia
To evaluate potential factors related to endogenous analgesia, several psychological and side effect measures were evaluated following the CPM trials with placebo and naltrexone. Analysis of side effect profiles suggests naltrexone produces mild side effects, which did not appear to influence the magnitude of endogenous analgesia. Since pain is an attention-demanding experience, the ability to concentrate on the heat thermal stimulus may have been reduced by the conditioning stimulus. Based on the conclusions of other studies (Lautenbacher et al., 2007; Moont et al., 2010), CPM/DNIC effects are not attributable to distraction including methods using continuous ratings of temporal summation (Staud et al., 2003). However, the most important finding is that subjects were able to concentrate on the heat thermal stimulus during both naltrexone and placebo sessions suggesting that naltrexone did not alter concentration/distraction, which could be used to explain the lack of inhibition during the placebo session.
Limitations
A number of potential limitations merit consideration. First, a single administration of a 50 mg dose of naltrexone, which is the current available form of the medication, is a standard procedure because this dose should bind to and block μ-opioid receptors (Lee et al., 1988; Walsh et al., 1996). It might be possible that a single dose was not sufficient to completely saturate these receptors and higher or multiple dose regimens might be warranted as reported by Sprenger et al. (2011). It is possible that larger doses may have resulted in greater overall drug differences. While we did not collect data related to body weight or body mass index (BMI), it is also possible that opioid blockade was less effective in heavier subjects (Bruehl et al., 2010) although none of the current subjects appeared to exceed 100 kg body weight. Naloxone, which can be administered parenterally, and naltrexone are potent opioid antagonists, which are able to exert its blockade effects due to the competitive blockade of μ-opioid receptors by medication and its active metabolite including 6-β-naltrexol for naltrexone (Crabtree, 1984; Meyer et al., 1984). While some investigators have used intravenous naloxone to investigate the opioid-mediation of endogenous pain inhibition (Sprenger et al., 2011), we chose naltrexone for several reasons. Naltrexone has better oral bioavailability compared to naloxone, which is commonly given intravenously. Naltrexone has greater potency and duration of action compared to naloxone (Zimmerman & Leander, 1990). Based on antagonistic evaluations of opioid analgesia in rodent preparations, naltrexone was more potent than naloxone (Jiang et al., 1977; Pert et al., 1973; Takemori & Portoghese, 1984; Zimmerman & Leander, 1990) including its active metabolite (Porter et al., 2002). For naloxone, plasma concentrations are dependent on the route of administration (∼10 min for intravenous) while concentrations peak around 45–60 min for orally administered naltrexone. Furthermore, the half-life of the medications also differs with naloxone lasting 30–81 min while naltrexone is up to 14 h. Both medications are opioid antagonists are metabolized through hepatic mechanisms (Crabtree, 1984; Gonzalez & Brogden, 1988). A disadvantage of naltrexone is that it is difficult to dose by body weight; therefore, we administered 50 mg in order to achieve adequate blockade in the vast majority of subjects. Most experimental studies have used this dose to evaluate physiological and subjective responses to a cold pressor task (al'Absi et al., 2004; Kotlyar et al., 2008) and other pain studies (Burns et al., 2009) without any discussion of possible weight issues, nor did they control for BMI. In addition, we did not include a condition to evaluate the effects of repeated heat pain trials in the absence of water immersion with naltrexone, which may reduce the ability to determine the full impact of the endogenous opioids on pain inhibition since naltrexone could influence the intensity of heat pain particularly over repeated trials. Future studies will be designed to address this issue. Finally, placebo is an important factor in the development of analgesic responses and may influence the magnitude of CPM. One of the active factors in placebo (i.e., expectation) has been shown to differentially mediate CPM (Goffaux et al., 2007, 2009). However, the placebo response is less likely a factor in the current study considering that (1) each CPM session was conducted in the same manner, and (2) no instructions were given regarding the medications effects on pain or pain inhibition due to the cold water during the informed consent process. Therefore, it is unlikely that the observed CPM effect is attributable to placebo.
Conclusions
The present study addressed several limitations of previous studies including small sample size (Edwards et al., 2004) and the use of short, static pain stimuli (i.e., threshold and tolerance; Pertovaara et al., 1982; Willer et al., 1990). The current study used a prolonged test stimulus to examine the mechanisms of endogenous analgesia, a novel method that has not been previously studied. More importantly, the present study demonstrated a role of the endogenous opioid system in mediating endogenous analgesia. While oral administration of naltrexone blocked the inhibition of focal heat pain during cold water immersion, opioid blockade was not explained by increased side effects, decreases in concentration, or increases in difficulty attending to focal heat. However, based on an exploratory analysis, endogenously released opioids contribute to pain inhibition in subjects with low to moderate catastrophizing but not high catastrophizing, suggesting an interesting implication that negative cognitive processing of pain may influence endogenous analgesia through different mechanisms. Because a number of mechanisms mediate inhibition, further research is required to tease out the potential interactions of multiple factors.
Acknowledgments
This research was supported by a University of Florida College of Dentistry (UFCD) Student Summer Research Fellowship, UFCD Seed Grant (Riley), NIH-NIDCR grant T32 DE007200 (King), UF CTSI UL1TR000064/KL2TR000065 (King), American Pain Society Future Leaders in Pain Research Grant Program (King), AG033906-06S1 (Fillingim), and the UF Comprehensive Center for Pain Research (CCPR). The authors of this study have no conflict of interests. This material was also supported by the North Florida/South Georgia Veterans Health System, Gainesville, FL, USA.
Contributor Information
Christopher D. King, Email: cking@dental.ufl.edu, Department of Community Dentistry and Behavioral Science, College of Dentistry, University of Florida, 1329 SW 16th St, Suite 5180, PO Box 103628, Gainesville, FL 32610-3628, USA.
Burel Goodin, Department of Community Dentistry and Behavioral Science, College of Dentistry, University of Florida, 1329 SW 16th St, Suite 5180, PO Box 103628, Gainesville, FL 32610-3628, USA.
Lindsay L. Kindler, Department of Community Dentistry and Behavioral Science, College of Dentistry, University of Florida, 1329 SW 16th St, Suite 5180, PO Box 103628, Gainesville, FL 32610-3628, USA
Robert M. Caudle, Department of Oral and Maxillary Surgery, College of Dentistry, University of Florida, Gainesville, FL, USA
Robert R. Edwards, Department of Anesthesiology, Brigham and Women's Hospital and Harvard Medical School, Chestnut Hill, MA, USA
Nikolaus Gravenstein, Department of Anesthesiology, College of Medicine, University of Florida, Gainesville, FL, USA.
III Joseph L. Riley, Department of Community Dentistry and Behavioral Science, College of Dentistry, University of Florida, 1329 SW 16th St, Suite 5180, PO Box 103628, Gainesville, FL 32610-3628, USA.
Roger B. Fillingim, North Florida/South Georgia Veterans Health System, Gainesville, FL, USA
References
- al'Absi M, Wittmers LE, Ellestad D, Nordehn G, Kim SW, Kirschbaum C, et al. Sex differences in pain and hypothalamic-pituitary-adrenocortical responses to opioid blockade. Psychosomatic Medicine. 2004;66:198–206. doi: 10.1097/01.psy.0000116250.81254.5d. [DOI] [PubMed] [Google Scholar]
- Bandura A, O'Leary A, Taylor CB, Gauthier J, Gossard D. Perceived self-efficacy and pain control: Opioid and nonopioid mechanisms. Journal of Personality and Social Psychology. 1987;53:563–571. doi: 10.1037//0022-3514.53.3.563. [DOI] [PubMed] [Google Scholar]
- Bartley EJ, Rhudy JL. The influence of pain catastrophizing on experimentally induced emotion and emotional modulation of nociception. J Pain. 2008;9:388–396. doi: 10.1016/j.jpain.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Fields HL. Endogenous pain control systems: Brainstem spinal pathways and endorphin circuitry. Annual Review of Neuroscience. 1984;7:309–338. doi: 10.1146/annurev.ne.07.030184.001521. [DOI] [PubMed] [Google Scholar]
- Bouhassira D, Bing Z, Lebars D. Studies of brain structures involved in diffuse noxious inhibitory controls in the rat—the rostral ventromedial medulla. Journal of Physiology-London. 1993;463:667–687. doi: 10.1113/jphysiol.1993.sp019616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruehl S, Burns JW, Chung OY, Magid E, Chont M, Gilliam W, et al. Hypoalgesia associated with elevated resting blood pressure: Evidence for endogenous opioid involvement. Journal of Behavioral Medicine. 2010;33:168–176. doi: 10.1007/s10865-009-9241-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruehl S, Chung OY, Ward P, Johnson B, McCubbin JA. The relationship between resting blood pressure and acute pain sensitivity in healthy normotensives and chronic back pain sufferers: The effects of opioid blockade. Pain. 2002;100:191–201. doi: 10.1016/s0304-3959(02)00295-6. [DOI] [PubMed] [Google Scholar]
- Burns JW, Bruehl S, Chung OY, Magid E, Chont M, Goodlad JK, et al. Endogenous opioids may buffer effects of anger arousal on sensitivity to subsequent pain. Pain. 2009;146:276–282. doi: 10.1016/j.pain.2009.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CM, Edwards RR. Mind-body interactions in pain: The neurophysiology of anxious and catastrophic pain-related thoughts. Translational Research. 2009;153:97–101. doi: 10.1016/j.trsl.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CM, Kronfli T, Buenaver LF, Smith MT, Berna C, Haythornthwaite JA, et al. Situational versus dispositional measurement of catastrophizing: Associations with pain responses in multiple samples. The Journal of Pain. 2010a;11:443–e442–453. e442. doi: 10.1016/j.jpain.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CM, Witmer K, Simango M, Carteret A, Loggia ML, Campbell JN, et al. Catastrophizing delays the analgesic effect of distraction. Pain. 2010b;149:202–207. doi: 10.1016/j.pain.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano A, Leonard MT, Franz A. The significant other version of the Pain Catastrophizing Scale (PCS-S): Preliminary validation. Pain. 2005;119:26–37. doi: 10.1016/j.pain.2005.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casale G, Pecorini M, Cuzzoni G, de Nicola P. Beta-endorphin and cold pressor test in the aged. Gerontology. 1985;31:101–105. doi: 10.1159/000212687. [DOI] [PubMed] [Google Scholar]
- Chitour D, Dickenson AH, Lebars D. Pharmacological evidence for the involvement of serotonergic mechanisms in diffuse noxious inhibitory controls (DNIC) Brain Research. 1982;236:329–337. doi: 10.1016/0006-8993(82)90718-1. [DOI] [PubMed] [Google Scholar]
- Cohen J. Statistical power analysis for the behavioral sciences. 2. Hillsdale, NJ: L. Erlbaum Associates; 1988. [Google Scholar]
- Crabtree BL. Review of naltrexone, a long-acting opiate antagonist. Clin Pharm. 1984;3:273–280. [PubMed] [Google Scholar]
- Edwards RR. Individual differences in endogenous pain modulation as a risk factor for chronic pain. Neurology. 2005;65:437–443. doi: 10.1212/01.wnl.0000171862.17301.84. [DOI] [PubMed] [Google Scholar]
- Edwards RR, Campbell CM, Fillingim RB. Catastrophizing and experimental pain sensitivity: Only in vivo reports of catastrophic cognitions correlate with pain responses. The Journal of Pain. 2005;6:338–339. doi: 10.1016/j.jpain.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Edwards RR, Fillingim RB, Ness TJ. Age-related differences in endogenous pain modulation: A comparison of diffuse noxious inhibitory controls in healthy older and younger adults. Pain. 2003;101:155–165. doi: 10.1016/s0304-3959(02)00324-x. [DOI] [PubMed] [Google Scholar]
- Edwards RR, Kronfli T, Haythornthwaite JA, Smith MT, McGuire L, Page GG. Association of catastrophizing with interleukin-6 responses to acute pain. Pain. 2008;140:135–144. doi: 10.1016/j.pain.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RR, Ness TJ, Fillingim RB. Endogenous opioids, blood pressure, and diffuse noxious inhibitory controls: A preliminary study. Perceptual and Motor Skills. 2004;99:679–687. doi: 10.2466/pms.99.2.679-687. [DOI] [PubMed] [Google Scholar]
- Edwards RR, Smith MT, Stonerock G, Haythornthwaite JA. Pain-related catastrophizing in healthy women is associated with greater temporal summation of and reduced habituation to thermal pain. Clinical Journal of Pain. 2006;22:730–737. doi: 10.1097/01.ajp.0000210914.72794.bc. [DOI] [PubMed] [Google Scholar]
- France CR, al'Absi M, Ring C, France JL, Harju A, Wittmers LE. Nociceptive flexion reflex and pain rating responses during endogenous opiate blockade with naltrexone in healthy young adults. Biological Psychology. 2007;75:95–100. doi: 10.1016/j.biopsycho.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frid M, Singer G, Rana C. Interactions between personal expectations and naloxone: Effects on tolerance to ischemic pain. Psychopharmacology (Berlin) 1979;65:225–231. doi: 10.1007/BF00492208. [DOI] [PubMed] [Google Scholar]
- Gjerstad J, Tjolsen A, Svendsen F, Hole K. Inhibition of spinal nociceptive responses after intramuscular injection of capsaicin involves activation of noradrenergic and opioid systems. Brain Research. 2000;859:132–136. doi: 10.1016/s0006-8993(00)01970-3. [DOI] [PubMed] [Google Scholar]
- Goffaux P, de Souza JB, Potvin S, Marchand S. Pain relief through expectation supersedes descending inhibitory deficits in fibromyalgia patients. Pain. 2009;145:18–23. doi: 10.1016/j.pain.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Goffaux P, Redmond WJ, Rainville P, Marchand S. Descending analgesia—when the spine echoes what the brain expects. Pain. 2007;130:137–143. doi: 10.1016/j.pain.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Gonzalez JP, Brogden RN. Naltrexone. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the management of opioid dependence. Drugs. 1988;35:192–213. doi: 10.2165/00003495-198835030-00002. [DOI] [PubMed] [Google Scholar]
- Goodin BR, McGuire LM, Stapleton LM, Quinn NB, Fabian LA, Haythornthwaite JA, et al. Pain catastrophizing mediates the relationship between self-reported strenuous exercise involvement and pain ratings: Moderating role of anxiety sensitivity. Psychosomatic Medicine. 2009;71:1018–1025. doi: 10.1097/PSY.0b013e3181bc62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracely RH, Geisser ME, Giesecke T, Grant MA, Petzke F, Williams DA, et al. Pain catastrophizing and neural responses to pain among persons with fibromyalgia. Brain. 2004;127:835–843. doi: 10.1093/brain/awh098. [DOI] [PubMed] [Google Scholar]
- Granot M, Granovsky Y, Sprecher E, Nir RR, Yarnitsky D. Contact heat-evoked temporal summation: Tonic versus repetitive-phasic stimulation. Pain. 2006;122:295–305. doi: 10.1016/j.pain.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Granot M, Weissman-Fogel I, Crispel Y, Pud D, Granovsky Y, Sprecher E, et al. Determinants of endogenous analgesia magnitude in a diffuse noxious inhibitory control (DNIC) paradigm: Do conditioning stimulus painfulness, gender and personality variables matter? Pain. 2008;136:142–149. doi: 10.1016/j.pain.2007.06.029. [DOI] [PubMed] [Google Scholar]
- Heymen S, Maixner W, Whitehead WE, Klatzkin RR, Mechlin B, Light KC. Central processing of noxious somatic stimuli in patients with irritable bowel syndrome compared with healthy controls. Clinical Journal of Pain. 2010;26:104–109. doi: 10.1097/AJP.0b013e3181bff800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JB, Hanson RN, Portoghese PS, Takemori AE. Stereochemical studies on medicinal agents. 23. Synthesis and biological evaluation of 6-amino derivatives of naloxone and naltrexone. Journal of Medicinal Chemistry. 1977;20:1100–1102. doi: 10.1021/jm00218a023. [DOI] [PubMed] [Google Scholar]
- Kelly SJ, Franklin KB. Role of peripheral and central opioid activity in analgesia induced by restraint stress. Life Sciences. 1987;41:789–794. doi: 10.1016/0024-3205(87)90460-7. [DOI] [PubMed] [Google Scholar]
- King CD, Devine DP, Vierck CJ, Mauderli A, Yezierski RP. Opioid modulation of reflex versus operant responses following stress in the rat. Neuroscience. 2007;147:174–182. doi: 10.1016/j.neuroscience.2007.04.012. [DOI] [PubMed] [Google Scholar]
- King CD, Wong F, Currie T, Mauderli AP, Fillingim RB, Riley JL. Deficiency in endogenous modulation of prolonged heat pain in patients with irritable bowel syndrome and temporomandibular disorder. Pain. 2009;143:172–178. doi: 10.1016/j.pain.2008.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlyar M, al'Absi M, Brauer LH, Grant JE, Fong E, Kim SW. Naltrexone effect on physiological and subjective response to a cold pressor task. Biological Psychology. 2008;77:233–236. doi: 10.1016/j.biopsycho.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus E, Besson JM, Lebars D. Behavioral model for diffuse noxious inhibitory controls (DNIC)—potentation by 5-hydroxytryptophan. Brain Research. 1982;231:461–465. doi: 10.1016/0006-8993(82)90384-5. [DOI] [PubMed] [Google Scholar]
- Kraus E, Le Bars D, Besson JM. Behavioral confirmation of “diffuse noxious inhibitory controls” (DNIC) and evidence for a role of endogenous opiates. Brain Research. 1981;206:495–499. doi: 10.1016/0006-8993(81)90554-0. [DOI] [PubMed] [Google Scholar]
- Lautenbacher S, Prager M, Rollman GB. Pain additivity, diffuse noxious inhibitory controls, and attention: A functional measurement analysis. Somatosensory and Motor Research. 2007;24:189–201. doi: 10.1080/08990220701637638. [DOI] [PubMed] [Google Scholar]
- Le Bars D, Chitour D, Kraus E, Dickenson AH, Besson JM. Effect of naloxone upon diffuse noxious inhibitory controls (DNIC) in the rat. Brain Research. 1981;204:387–402. doi: 10.1016/0006-8993(81)90597-7. [DOI] [PubMed] [Google Scholar]
- Lee MC, Wagner HN, Jr, Tanada S, Frost JJ, Bice AN, Dannals RF. Duration of occupancy of opiate receptors by naltrexone. Journal of Nuclear Medicine. 1988;29:1207–1211. [PubMed] [Google Scholar]
- Maixner W, Fillingim R, Booker D, Sigurdsson A. Sensitivity of patients with painful temporomandibular disorders to experimentally evoked pain. Pain. 1995;63:341–351. doi: 10.1016/0304-3959(95)00068-2. [DOI] [PubMed] [Google Scholar]
- Meyer MC, Straughn AB, Lo MW, Schary WL, Whitney CC. Bioequivalence, dose-proportionality, and phar-macokinetics of naltrexone after oral administration. Journal of Clinical Psychiatry. 1984;45:15–19. [PubMed] [Google Scholar]
- Millan MJ. Descending control of pain. Progress in Neurobiology. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Moont R, Pud D, Sprecher E, Sharvit G, Yarnitsky D. ‘Pain inhibits pain’ mechanisms: Is pain modulation simply due to distraction? Pain. 2010;150:113–120. doi: 10.1016/j.pain.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Naert AL, Kehlet H, Kupers R. Characterization of a novel model of tonic heat pain stimulation in healthy volunteers. Pain. 2008;138:163–171. doi: 10.1016/j.pain.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Pert CB, Pasternak G, Snyder SH. Opiate agonists and antagonists discriminated by receptor binding in brain. Science. 1973;182:1359–1361. doi: 10.1126/science.182.4119.1359. [DOI] [PubMed] [Google Scholar]
- Pertovaara A, Kemppainen P, Johansson G, Karonen SL. Ischemic pain non-segmentally produces a predominant reduction of pain and thermal sensitivity in man—a selective role for endogenous opioids. Brain Research. 1982;251:83–92. doi: 10.1016/0006-8993(82)91276-8. [DOI] [PubMed] [Google Scholar]
- Peters ML, Schmidt AJ, Van den Hout MA, Koopmans R, Sluijter ME. Chronic back pain, acute postoperative pain and the activation of diffuse noxious inhibitory controls (DNIC) Pain. 1992;50:177–187. doi: 10.1016/0304-3959(92)90159-9. [DOI] [PubMed] [Google Scholar]
- Picavet HS, Vlaeyen JW, Schouten JS. Pain catastrophizing and kinesiophobia: Predictors of chronic low back pain. American Journal of Epidemiology. 2002;156:1028–1034. doi: 10.1093/aje/kwf136. [DOI] [PubMed] [Google Scholar]
- Piche M, Arsenault M, Rainville P. Cerebral and cerebrospinal processes underlying counterirritation analgesia. Journal of Neuroscience. 2009;29:14236–14246. doi: 10.1523/JNEUROSCI.2341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielsticker A, Haag G, Zaudig M, Lautenbacher S. Impairment of pain inhibition in chronic tension-type headache. Pain. 2005;118:215–223. doi: 10.1016/j.pain.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Popescu A, LeResche L, Truelove EL, Drangsholt MT. Gender differences in pain modulation by diffuse noxious inhibitory controls: A systematic review. Pain. 2010;150:309–318. doi: 10.1016/j.pain.2010.05.013. [DOI] [PubMed] [Google Scholar]
- Porter SJ, Somogyi AA, White JM. In vivo and in vitro potency studies of 6beta-naltrexol, the major human metabolite of naltrexone. Addiction Biology. 2002;7:219–225. doi: 10.1080/135562102200120442. [DOI] [PubMed] [Google Scholar]
- Price DD, McHaffie JG. Effects of heterotopic conditioning stimuli on first and second pain: A psychophysical evaluation in humans. Pain. 1988;34:245–252. doi: 10.1016/0304-3959(88)90119-4. [DOI] [PubMed] [Google Scholar]
- Pruessner JC, Kirschbaum C, Meinlschmid G, Hellhammer DH. Two formulas for computation of the area under the curve represent measures of total hormone concentration versus time-dependent change. Psychoneuroendocrinology. 2003;28:916–931. doi: 10.1016/s0306-4530(02)00108-7. [DOI] [PubMed] [Google Scholar]
- Pud D, Granovsky Y, Yarnitsky D. The methodology of experimentally induced diffuse noxious inhibitory control (DNIC)-like effect in humans. Pain. 2009;144:16–19. doi: 10.1016/j.pain.2009.02.015. [DOI] [PubMed] [Google Scholar]
- Quiton RL, Greenspan JD. Across- and within-session variability of ratings of painful contact heat stimuli. Pain. 2008;137:245–256. doi: 10.1016/j.pain.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley JL, Hastie BA, Glover TL, Fillingim RB, Staud R, Campbell CM. Cognitive-affective and somatic side effects of morphine and pentazocine: Side-effect profiles in healthy adults. Pain Medicine. 2010a;11:195–206. doi: 10.1111/j.1526-4637.2009.00680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley J, King C, Wong F, Fillingim RB, Mauderli AP. Lack of endogenous modulation and reduced decay of prolonged heat pain in older adults. Pain. 2010b;150:153–160. doi: 10.1016/j.pain.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring C, France CR, al'Absi M, Beesley L, Edwards L, McIntyre D, et al. Effects of opioid blockade with naltrexone and distraction on cold and ischemic pain in hypertension. Journal of Behavioral Medicine. 2007;30:59–68. doi: 10.1007/s10865-006-9084-1. [DOI] [PubMed] [Google Scholar]
- Rosa C, Ghione S, Mezzasalma L, Pellegrini M, Basile Fasolo C, Giaconi S, et al. Relationship between pain sensitivity, cardiovascular reactivity to cold pressor test and indexes of activity of the adrenergic and opioid system. Clin Exp Hypertens A. 1988;10:383–390. doi: 10.3109/10641968809075994. [DOI] [PubMed] [Google Scholar]
- Song GH, Venkatraman V, Ho KY, Chee MW, Yeoh KG, Wilder-Smith CH. Cortical effects of anticipation and endogenous modulation of visceral pain assessed by functional brain MRI in irritable bowel syndrome patients and healthy controls. Pain. 2006;126:79–90. doi: 10.1016/j.pain.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Sprenger C, Bingel U, Buchel C. Treating pain with pain: Supraspinal mechanisms of endogenous analgesia elicited by heterotopic noxious conditioning stimulation. Pain. 2011;152:428–439. doi: 10.1016/j.pain.2010.11.018. [DOI] [PubMed] [Google Scholar]
- Staud R, Robinson ME, Vierck CJ, Jr, Price DD. Diffuse noxious inhibitory controls (DNIC) attenuate temporal summation of second pain in normal males but not in normal females or fibromyalgia patients. Pain. 2003;101:167–174. doi: 10.1016/s0304-3959(02)00325-1. [DOI] [PubMed] [Google Scholar]
- Sullivan MJ, Bishop SR, Pivik J. The pain catastrophizing scale: Development and validation. Psychological Assessment. 1995;7:524–532. [Google Scholar]
- Suzuki K, Maekawa K, Minakuchi H, Yatani H, Clark GT, Matsuka Y, et al. Responses of the hypothalamic-pituitary-adrenal axis and pain threshold changes in the orofacial region upon cold pressor stimulation in normal volunteers. Archives of Oral Biology. 2007;52:797–802. doi: 10.1016/j.archoralbio.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Takemori AE, Portoghese PS. Comparative antagonism by naltrexone and naloxone of mu, kappa, and delta agonists. European Journal of Pharmacology. 1984;104:101–104. doi: 10.1016/0014-2999(84)90374-1. [DOI] [PubMed] [Google Scholar]
- Tepperman FS, Hirst M, Smith P. Brain and serum levels of naloxone following peripheral administration. Life Sciences. 1983;33:1091–1096. doi: 10.1016/0024-3205(83)90665-3. [DOI] [PubMed] [Google Scholar]
- Tousignant-Laflamme Y, Page S, Goffaux P, Marchand S. An experimental model to measure excitatory and inhibitory pain mechanisms in humans. Brain Research. 2008;1230:73–79. doi: 10.1016/j.brainres.2008.06.120. [DOI] [PubMed] [Google Scholar]
- Tricklebank MD, Hutson PH, Curzon G. Analgesia induced by brief or more prolonged stress differs in its dependency on naloxone, 5-hydroxytryptamine and previous testing of analgesia. Neuropharmacology. 1984;23:417–421. doi: 10.1016/0028-3908(84)90249-1. [DOI] [PubMed] [Google Scholar]
- van Wijk G, Veldhuijzen DS. Perspective on diffuse noxious inhibitory controls as a model of endogenous pain modulation in clinical pain syndromes. J Pain. 2010;11:408–419. doi: 10.1016/j.jpain.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Verebey K. The clinical pharmacology of naltrexone: Pharmacology and pharmacodynamics. NIDA Research Monograph. 1981;28:147–158. [PubMed] [Google Scholar]
- Villanueva L. Diffuse noxious inhibitory control (DNIC) as a tool for exploring dysfunction of endogenous pain modulatory systems. Pain. 2009;143:161–162. doi: 10.1016/j.pain.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Walsh SL, Sullivan JT, Preston KL, Garner JE, Bigelow GE. Effects of naltrexone on response to intravenous cocaine, hydromorphone and their combination in humans. Journal of Pharmacology and Experimental Therapeutics. 1996;279:524–538. [PubMed] [Google Scholar]
- Washington LL, Gibson SJ, Helme RD. Age-related differences in the endogenous analgesic response to repeated cold water immersion in human volunteers. Pain. 2000;89:89–96. doi: 10.1016/S0304-3959(00)00352-3. [DOI] [PubMed] [Google Scholar]
- Weissman-Fogel I, Sprecher E, Pud D. Effects of catastrophizing on pain perception and pain modulation. Experimental Brain Research. 2008;186:79–85. doi: 10.1007/s00221-007-1206-7. [DOI] [PubMed] [Google Scholar]
- Wen YR, Wang CC, Yeh GC, Hsu SF, Huang YJ, Li YL, et al. DNIC-mediated analgesia produced by a supramaximal electrical or a high-dose formalin conditioning stimulus: Roles of opioid and alpha2-adrenergic receptors. Journal of Biomedical Science. 2010;17:19. doi: 10.1186/1423-0127-17-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder-Smith CH, Schindler D, Lovblad K, Redmond SM, Nirkko A. Brain functional magnetic resonance imaging of rectal pain and activation of endogenous inhibitory mechanisms in irritable bowel syndrome patient subgroups and healthy controls. Gut. 2004;53:1595–1601. doi: 10.1136/gut.2003.028514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder-Smith CH, Song G, Yeoh KG, Ho KY. Activating endogenous visceral pain modulation: A comparison of heterotopic stimulation methods in healthy controls. European Journal of Pain. 2009;13:836–842. doi: 10.1016/j.ejpain.2008.09.015. [DOI] [PubMed] [Google Scholar]
- Willer JC, Le Bars D, De Broucker T. Diffuse noxious inhibitory controls in man: Involvement of an opioidergic link. European Journal of Pharmacology. 1990;182:347–355. doi: 10.1016/0014-2999(90)90293-f. [DOI] [PubMed] [Google Scholar]
- Willer JC, Roby A, Le Bars D. Psychophysical and electrophysiological approaches to the pain-relieving effects of heterotopic nociceptive stimuli. Brain. 1984;107:1095–1112. doi: 10.1093/brain/107.4.1095. [DOI] [PubMed] [Google Scholar]
- Wong F, Vierck CJ, Riley JL, King C, Mauderli AP. A new thermal stimulation method for human psycho-physical studies: Pain intensity clamping. Journal of Neuroscience Methods. 2010;188:83–88. doi: 10.1016/j.jneumeth.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarnitsky D, Arendt-Nielsen L, Bouhassira D, Edwards RR, Fillingim RB, Granot M, et al. Recommendations on terminology and practice of psychophysical DNIC testing. European Journal of Pain. 2010;14:339. doi: 10.1016/j.ejpain.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Zimmerman DM, Leander JD. Opioid antagonists: Structure activity relationships. NIDA Research Monograph. 1990;96:50–60. [PubMed] [Google Scholar]