

Abstract
Ischemic focal ventricular tachycardia (VT) occurs in animals and humans. Angiotensin-converting enzyme inhibitors and receptor blockers reduce sudden death in patients with ischemic heart disease. In our dog model of coronary artery occlusion (CAO), we tested the hypothesis that angiotensin II (AGII) will selectively promote focal VT and that the specific AT2 blocker PD-123319 (PD), or AT1 blocker losartan, will affect this VT. Anesthetized dogs (n = 90) underwent CAO, followed by three-dimensional activation mapping of inducible VT. Dogs without VT in 1–3 h after CAO received AGII, and those with VT received either PD or losartan. Focal endocardium excised from ischemic sites was studied in vitro with standard microelectrode. Of 33 dogs with no inducible VT, AGII infusion resulted in sustained VT of only focal Purkinje origin in 13 (39%) compared with 0 of 20 dogs with saline. Of 26 dogs with inducible VT at baseline, given PD, reinduction was blocked in 8 of 10 (P < 0.05) focal VT, but only 1 of 15 with reentry. In contrast, of 11 dogs given losartan, reinduction of either mechanism was not blocked. In vitro triggered activity in Purkinje was blocked by PD in 13 of 19 (P < 0.05), but not by losartan in 8. Also, triggered activity was promoted by AGII, losartan, or the combination in 9 of 12 tissues. AGII promotes only focal, mainly Purkinje ischemic VT. PD, but not losartan, preferentially blocked focal VT, which is likely due to triggered activity due to delayed afterdepolarizations in Purkinje.
Keywords: triggered activity, canine model, lorsartan
malignant ventricular arrhythmias are the principal cause of death during the acute phase of myocardial infarction. Understanding the mechanisms of ventricular tachycardia (VT) and ventricular fibrillation (VF) in the early hours after acute myocardial injury is hampered by the rapidly changing substrate, which is difficult to investigate clinically. Limited reports have implicated focal endocardial (6, 7), as well as reentrant mechanisms contributing to VT in humans with coronary artery disease and acute myocardial infarction. Mechanisms demonstrated in animal models may be useful to gain insight into new therapies to prevent VT in patients.
Animal experiments have shown significant contribution of the Purkinje system in the genesis of ischemic focal VT (1). Triggered activity (TA) due to delayed afterdepolarizations (DADs) appears to be a predominant mechanism underlying these arrhythmias (26). Previous studies have demonstrated that focal ischemic VT can be modulated by upstream therapies, such as adrenergic blockers (2), statins (27), and free radical scavengers (23).
The renin-angiotensin system is activated during myocardial ischemia and injury (17, 20–22). Angiotensin II (AGII) may have direct electrophysiological effects (13, 28) and could act as an endogenous arrhythmogenic agent, with mechanisms involving enhanced free radical formation, as well as release of sarcoplasmic calcium in cardiac myocytes via activation of a phosphatidylinositol response. Angiotensin-converting enzyme inhibitors and AGII receptor (AT) blockers reduce sudden cardiac death in patients with coronary artery disease and left ventricular dysfunction (11, 18). Furthermore, angiotensin-converting enzyme inhibitors have been shown to reduce spontaneous ventricular arrhythmias in patients with congestive heart failure (10). The acute effects of AGII and AGII receptor antagonists on ischemic VT in a large animal have not been studied. Most of the cardiovascular effects of AGII are thought to be mediated through the AT1 receptor. The exact role of AT2 receptor is still an open question (9). AT2 receptor blockade has been shown to be protective against ischemia-reperfusion injury-mediated VT in rats (22). Differential effects of AGII receptor modulation on induction of VT/VF during acute myocardial ischemia are not known.
The purpose of this study was to investigate the effects of AGII, AT1 receptor blocker losartan, and the selective, nonpeptide AT2 receptor blocker PD-123319 (PD) in modulation of mechanisms of VT induced during 1–3 h of acute myocardial ischemia in a well-studied dog model showing both focal and reentrant mechanisms.
METHODS
Ninety healthy dogs of either sex (mean weight: 19 ± 1 kg) were used for this study. The Animal Care and Use Review Board at the University of Iowa approved the protocol ACORP no. 0590902, conforming to the Guidelines of the American Physiological Society.
Surgical preparation.
Dogs were sedated with intramuscular injection of 10 mg/kg of ketamine and 0.5 mg/kg acepromazine maleate. Anesthesia was induced by intravenous administration of thiopental sodium (10 mg/kg), followed by bolus injection of α-chloralose (100–200 mg/kg). Maintenance of anesthesia was produced by constant infusion of α-chloralose (8 mg·kg−1·h−1 dissolved in saline and polyethylene glycol). The animals were intubated and ventilated using a volume-cycled respirator (Harvard) to maintain a Po2 of 80–110 Torr and a Pco2 of 35–45 Torr. The femoral vein and artery were cannulated using a cut-down technique, allowing continuous measurement of arterial blood pressure and for administration of fluid and drugs. Sodium bicarbonate was infused as necessary to maintain arterial pH in the 7.35–7.45 range.
The chest was opened through a midline sternotomy, and a pericardial “cradle” was fashioned to support the heart. A suture was passed under the left anterior descending coronary artery just distal to the first diagonal branch and was threaded in tubing, creating a snare for coronary artery occlusion (CAO). Epicardial temperature was maintained at ∼37°C by an infrared heating lamp, and a plastic sheet draped over the sternotomy. Warm saline was applied to the heart intermittently to prevent surface drying.
Electrophysiological preparation.
Surface electrocardiographic leads II and V5R were recorded continuously. The sinus node was clamped to control the heart rate. The atria were stimulated at 300-ms cycle length with a bipolar electrode. Constant current output was set at twice diastolic threshold, with a pulse width of 2 ms. As previously reported, twenty-three 16-pole plunge-needle electrodes (J. Kassell, Fayetteville, NC) inserted into the myocardium in and surrounding the risk zone of left anterior descending coronary artery recorded transmural signals (26). The interneedle distance was ∼1 cm, depending on coronary anatomy.
Bipolar electrograms were recorded from up to six different sites on each 16-pole electrode (1). Noise-free bipolar signals were chosen by sequential recording on a storage oscilloscope of each successive bipole, to maximize the capability of recording from Purkinje fibers. Purkinje, endocardial, midwall, and epicardial electrograms were chosen, as previously described (26).
Electrograms were recorded simultaneously on two computers: one for the three endocardial-most bipoles, and the other for the three epicardial-most bipoles. Data from both acquisition systems were incorporated, and three-dimensional (3D) activation maps were constructed, as previously described (1, 26, 27).
Experimental protocol.
Left anterior descending CAO was performed after confirming target blood-gas measurements and adequate anesthesia. One hour after CAO, the effective refractory period (ERP) was determined by delivering single extrastimuli after eight ventricular-paced complexes, with a drive cycle length of 300 ms at constant current output set at four times diastolic threshold. The ERP was defined as the longest interval between the last drive pacing stimulus (S1) and the first extrastimulus (S2) that failed to capture the ventricle. Extrastimulus testing was employed to induce VT from normal endocardial sites (apical, septal, lateral) bordering the ischemic zone using up to four premature stimuli (S2–S5).
Pacing protocols were performed in the 1- to 3-h period after CAO; previous studies using this model have shown that VT can be reproducibly induced over this period (2, 23, 24 25, 27). Specifically, when the same morphology of VT was induced twice from one pacing site, continuing induction of the same VT morphology and mechanism over the first 3 h of coronary occlusion is predicted, providing a basis for comparison of antiarrhythmic interventions. Sustained VTs were terminated by pacing or cardioverted with 10- to 20-J epicardial shocks. Induction was done from apical, septal, or lateral left ventricular pacing sites, repeated every 20 min, and continued until two episodes of VT with similar surface lead morphology were induced.
In dogs with reproducibly inducible VT of similar morphology over this time period, PD (200 μg/kg, Sigma-Aldrich) or losartan (200 μg/kg, Sigma-Aldrich) was given randomly by IV infusion. Ten minutes into each infusion, extrastimulus testing was performed from the same pacing site(s), which induced VT during the control period. Failure to induce previously reproducible VT with the same or a more aggressive protocol was considered as block of VT.
In dogs in which no VT was induced by three rounds of extrastimulus testing from three normal pacing sites, in the 2 h after CAO, the occurrence of induced VT in the third hour is negligible (3). AGII (0.1 μg·kg−1·min−1, Sigma-Aldrich) was given by IV infusion over 10 min. Blood pressure recordings were obtained at baseline and at 5 and 10 min. During the last minutes of infusion, extrastimulus testing was repeated from the apical, septal, and lateral ventricular pacing sites. Coronary sinus blood was collected in 10 dogs for measurement of plasma AGII levels to correlate with inducibility of VTs. This analysis was performed at the Analytical Core Laboratory at the University of Iowa using radioimmunoassay. Blood samples were prepared with 1 mg of ethylenediamine tetraacetic acid and 1,000 units of the kallikrein inhibitor aprotinin. These samples were rapidly frozen and stored at −80°C. AGII level in the blood was then measured using a kit (Peninsula Laboratories, Belmont, CA).
In vitro techniques.
Once the in vivo experiment was complete, the heart was excised and placed in cold Tyrode solution (composition in mM: 125 NaCl, 24 NaHCO3, 4.5 KCl, 1.8 CaCl2, 0.5 MgCl2, 0.25 NaHPO4, and 5.5 dextrose; pH 7.4), with the mapping electrode recording the focal site of origin of VT left in situ. Endocardial tissue (measuring 5 × 5 × 2 mm) from the site identified by the mapping electrode or an ischemic site, if a focal mechanism was not identified, was then excised and placed in a 3-ml tissue bath (Warner Instrument). The bath was superfused with Tyrode solution (37°C) at 9 ml/min, and the tissue was then stimulated with a bipolar electrode using 2-ms pulse width at twice diastolic threshold. The most superficial cells were then impaled with a 3 M KCl filled glass capillary microelectrode with tip resistance ranging from 6 to 10 MΩ. The microelectrode was connected to a high-input impedance preamplifier (Axoclamp-2A, Axon Instruments, Foster City, CA). The bath was grounded with a Ag/AgCl pellet.
Action potential measurements were performed during constant pacing at 1.5 Hz for at least 5 min in control and after addition of superfused agents. Assessment for DADs and TA was performed during pacing at 1–4 Hz for at least 15 stimuli at a new frequency before the stimulator was turned off to assess for DADs and TA. Potentials were recorded and stored on a computer with the use of commercial software (Axon Instruments) with data filtered at 1 kHz and sampled at 2 kHz. Zero offsets were recorded to correct for drift. Impalements of Purkinje tissue were identified by spontaneous phase 4 depolarization or automaticity at cycle lengths >1 s, while myocardial cells do not have these findings (26). Depolarization occurring after phase 3 of paced action potentials was defined as a DAD. Nonpaced action potentials occurring at the peak of a DAD showing overdrive stimulation were defined as TA.
Tissues were first assessed for the presence or absence of DADs and TA during pacing alone, and, if not induced, pacing was repeated during the 5th minute of superfusion with 5 × 10−7 M isoproterenol. Reproducibility of induction of TA due to DADs was defined as occurring as a result of at least two of five rounds of pacing for at least 15 stimuli. Once DADs and TA were induced, either PD (10−6 M) or losartan (10−6 M) were added to the tissue bath, and pacing (at similar cycle length that induced DAD/TA) was repeated at 5 and 10 min to determine effects on action potentials recorded at constant pacing of 1.5 Hz, as well as induced DAD/TA. If DAD/TA was blocked, drug washout for 10–20 min was then performed to validate reproducibility of the DADs and TA with the original pre-AT-blocker milieu.
When DADs or TA were not inducible de novo or with isoproterenol, AGII in increasing concentrations (10−6 M and 10−5 M) was added to the solution to assess for inducibility of DAD/TA. If no DAD/TA were inducible with AGII, losartan was then added to the AGII to assess whether AT1 receptor blockade had a proarrhythmic effect. Tissues that had inducible DAD/TA with a combination of AGII and losartan (with or without isoproterenol) were treated with PD superfusion to assess for suppressibility of DAD/TA. Washout of the drugs and isoproterenol was also attempted if the impalement was maintained.
Definitions and analysis.
3D mapping of electrograms was done offline, as previously described (26). The following definitions were used.
VT was defined as at least five consecutive nonstimulated ventricular complexes, and sustained VT was defined as VT that did not self-terminate within 10 s; most episodes of VT were pace terminated, and cardioversion was required for the rest because of hemodynamic collapse. The cycle length of each VT episode was measured by averaging the first 10 cycles.
VF was defined as a rapid, irregular, multimorphic ventricular rhythm that resulted in hemodynamic collapse and required a shock to terminate. Usually the first eight ventricular complexes of VT were mappable by the system before acceleration to VF.
Focal VT occurred when the earliest electrical activity was recorded on one electrogram, proceeding sequentially to all surrounding electrograms, and when no electrical activity was recorded on all adjacent sites in three dimensions between the latest activation of one QRS complex and the earliest of the next QRS. Moreover, adjacent electrodes could not manifest conduction delay, accounting for the majority of cycle length of the VT. Focal origin thus implies that the VT has a single site of origin, with activation spreading out from that site in all directions (5).
Purkinje origin of VT was defined as a focal endocardial mechanism, with recording of a Purkinje potential before the QRS on an endocardial electrogram recording the earliest activity. Purkinje potentials had to be identified on electrograms during atrial pacing before and after CAO and during VT to merit consideration for mechanistic involvement (1, 27).
Reentrant VT occurred when the electrode recording the earliest activity was immediately adjacent to the site of the latest activation from the previous complex, and continuous diastolic activity was recorded between complexes. Reentrant mechanisms also demonstrated unidirectional and functional block to the subsequent earliest site of activation (1, 24, 27).
If the electrode recording the earliest activity for a VT complex was not surrounded by electrodes recording subsequent activation, the possibility could not be excluded that either a focal mechanism or reentry from a remote site outside the electrode array could have been responsible for initiating the complex. In such cases, the mechanism could not be defined (1, 27).
Ischemic zone was defined by voltage reduction resulting from CAO as in a prior report (26).
Longitudinal conduction velocity (in m/s) was measured during regular ventricular pacing from the 3D activation maps (24) before and after administration of AGII, PD, and losartan. The surface distance was measured between two plunge electrodes in the longitudinal direction. Time interval between intrinsicoid deflections of local electrograms inscribed at the two plunge electrode sites was measured from the activation map. Longitudinal conduction velocity was calculated using the surface distance divided by time interval.
Western blot assay.
To analyze angiotensin and its receptor protein expression, Purkinje and myocardial tissues were collected from three experiments and put in the cold normal Tyrode solution. Samples were homogenized in RIPA buffer (1% Nonidet P-40, 0.5% sodium deoxycholic acid, 0.1% SDS in PBS, pH 7.4) containing protease inhibitors (29). The protein extracts (100 μg) were loaded to SDS-PAGE system under reducing conditions and were transferred to nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ). The membranes were incubated with AGI/AGII, its receptor AT2 polyclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) were diluted 1:100–1:200, and anti-GAPDH monoclonal antibody (Chemicon, Temecula, CA) was diluted 1:8,000 in 3% nonfat dry milk at 4°C overnight. The primary antibodies were visualized with the corresponding IRDye-800-conjugated secondary antibodies by using the Odyssey Infrared Imaging System (LI-COR, Lincoln, NE). The intensity of protein expression was assessed by means of quantitative densitometry analysis and then normalized to corresponding GAPDH expression in each sample. The level of individual protein expression is expressed as fold change from the level in normal Purkinje fiber.
Statistics.
The effect of AGII, PD, and losartan on the incidence of VT, DADs, and TA was analyzed by a two-tailed Fishers exact test at each dose level. The effect of these agents on ventricular ERP, systolic, diastolic, and mean arterial pressure, pacing threshold, longitudinal conduction velocity, and infarct size was analyzed using a two-way ANOVA with repeated measurements. T-tests were performed to assess the effects of drug-induced changes in continuous variables. P < 0.05 was considered statistically significant. All values are reported as means ± SE.
RESULTS
Effects of intravenous AGII infusion.
Fifty-three dogs had no inducible VT after 1–2 h of CAO, and 33 were given intravenous AGII infusion. Repeat induction during AGII infusion resulted in sustained VT in 13 (39%) compared with none of 20 animals that received only saline during the same time after coronary occlusion. Induction of VT required three extrastimuli in seven dogs and four extrastimuli in six dogs. All of the induced VTs were of focal origin, with 11 having an endocardial focus (6 with Purkinje focus) and 2 having an epicardial focus. The characteristics of VT induced with AGII administration are shown in Table 1. In 6 of 13, VT degenerated into VF, requiring defibrillation. There was no significant difference in plasma AGII levels between inducible vs. noninducible dogs (127 ± 26 vs. 136 ± 24 pg/ml; P = nonsignificant).
Table 1.
Characteristics of ventricular tachycardia induced with angiotensin II
| Dog No. | VT Type | S3-5 | Mechanism | Location |
|---|---|---|---|---|
| 1 | SVT-VF | S4 | Focal | Endo |
| 2 | SVT-VF | S4 | Focal | Endo |
| 3 | SVT-VF | S5 | Focal | Purkinje |
| 4 | SVT | S5 | Focal | Endo |
| 5 | SVT-VF | S4 | Focal | Endo |
| 6 | SVT | S5 | Focal | Epi |
| 7 | SVT | S4 | Focal | Purkinje |
| 8 | SVT-VF | S4 | Focal | Endo |
| 9 | SVT | S4 | Focal | Purkinje |
| 10 | SVT | S5 | Focal | Purkinje |
| 11 | SVT-VF | S5 | Focal | Purkinje |
| 12 | SVT | S4 | Focal | Purkinje |
| 13 | SVT | S5 | Focal | Epi |
VT, ventricular tachycardia; SVT, sustained ventricular tachycardia; VF, ventricular fibrillation; Endo, endocardial origin; Epi, epicardial origin; SVT-VF, sustained VT degenerating to VF; S3-5, extrastimuli following drive train (S1) resulting in VT induction.
An example of induction of focal Purkinje VT during AGII administration is shown in Fig. 1. The first downward arrow denotes the last pacing complex of the eight-beat pacing drive train (S1). The next three downward arrows show ventricular extrastimuli at progressively shorter coupling intervals. The cycle length for the drive train (S1S1) is 300 ms. The first extrastimulus is delivered at a coupling interval of 170 ms (S1S2). The coupling interval progressively reduced for the second (S2S3 = 140 ms) and third (S3S4 = 130 ms), resulting in VT, of which seven complexes are shown. Vertical lines indicate onset of surface QRS of the first, third, and fifth VT complexes. For S1 through S4, Purkinje potentials [upward arrows on PF (Purkinje focus) and EN (east north)] preceded muscle activity with greater delay on each successive premature. Starting with the first VT complex, Purkinje potentials on PF (upward arrows) were seen earlier than the surface QRS. Site PF (Purkinje focus of origin) persists as the site of origin for all subsequent VT complexes, showing Purkinje spikes (upward arrows) preceding all surrounding muscle activity. There was also a sequential delay from PF to the adjacent Purkinje potentials on EN. All surrounding activity suggests focal activation with no late activation that would be expected with reentrant excitation.
Fig. 1.
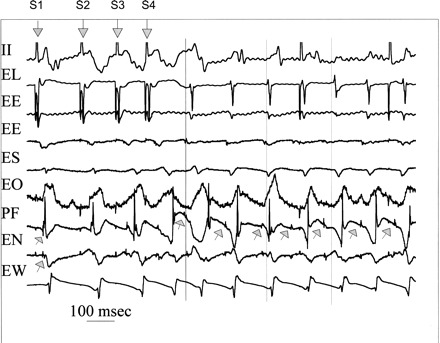
Induction of focal Purkinje ventricular tachycardia (VT) during angiotensin II (AGII) administration. Shown is the last pacing complex of the drive train and three extrastimuli (downward arrows labeled S1 through S4), and seven complexes of the induced VT. Recordings are surface ECG lead II and bipolar endocardial (E) electrograms from the lateral (L) site of pacing in the normal zone and ischemic zone recording sites east (E), south (S), overlying (O), north (N), and west (W) immediately surrounding (within 10 mm) the Purkinje (P) focus (F) of origin of VT. During the stimulated complexes, the upward arrows indicate Purkinje spikes (upward arrows), which are progressively delayed, along with the local muscle potentials. Site PF is the site of origin for the first and all subsequent VT complexes preceding the adjacent muscle activity. The vertical lines indicate surface QRS onsets for first, third, and fifth VT complexes to assist the reader. All activity surrounding PF occurs afterward and suggests focal activation with no very late activation that would be expected with reentrant excitation. See text for details.
Figure 2 shows a 3D activation map of the third (last) extrastimulus, labeled S4, in Fig. 1. Each rectangle represents a layer in the anterior wall of the heart extending from epicardium (EPI), subepicardial (S-EPI), MIDWALL, subendocardial (S-ENDO), endocardial (ENDO) to Purkinje (PURK) layers. The left side of each layer (west) represents left ventricle adjacent to the anterior descending coronary artery, and the north side, the area of CAO. The east side of each layer represents the lateral aspect of the risk zone of the occluded artery, while the south side represents the apex of the anterior wall of the left ventricle. Activation times are given in milliseconds (ms) for each electrode site. Maps are drawn in 20-ms isochrones, indicated in bold numbers and different colors (with earliest shown in orange) after the stimulus. Earliest ventricular activation is located in the S-ENDO layer (6 ms after S4), with activation proceeding outward to EPI and inward to the ENDO and PURK, and with no significant conduction delay or block in the latter layers adjacent to where the first activation of the induced VT occurs (Fig. 3).
Fig. 2.
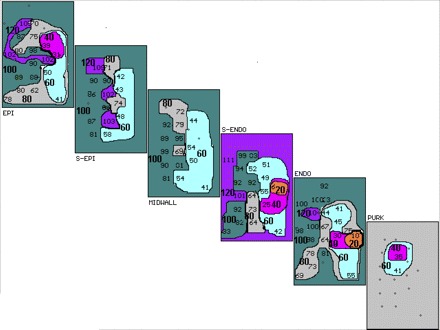
Three-dimensional activation map of the last premature stimulated complex (S4) in Fig. 1. Activation maps of the epicardial (EPI), subepicardial (S-EPI), midwall, subendocardial (S-ENDO), endocardial (ENDO), and Purkinje (PURK) planes are shown. Activation times are in milliseconds (ms) for each electrode site. Maps are drawn in 20-ms isochrones (different colors, marked by bold numbers with earliest shown in orange) after the stimulus. Earliest intracardiac activity is seen in S-ENDO layer at 6 ms. Activation proceeds to the PURK layer and out to the EPI layers, with no conduction block and no significant conduction delay in the PURK or ENDO layers adjacent to where the first activation of VT occurs (Fig. 3).
Fig. 3.
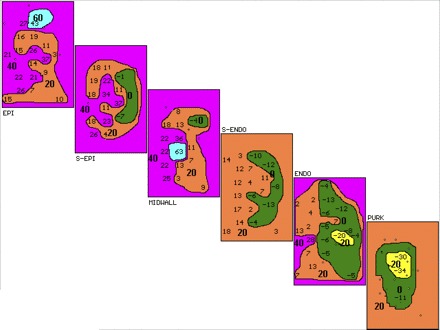
Three-dimensional activation map of third VT complex in Fig. 1. Activation maps of the planes through the ventricular wall labeled as in Fig. 2 are shown. Isochrones (20 ms shown in bold) start with the earliest activity shown in yellow before (−) or after the onset of the surface QRS. Transmural activation starts in PURK and proceeds to EPI. Earliest activation is seen in the PURK, 34 ms before the onset of surface QRS, with activation proceeding to surrounding PURK and transmurally to midwall, where the latest activity is seen 63 ms after QRS. There is not enough conduction delay or retrograde conduction from EPI to PURK to suggest reentry.
Figure 3 shows the 3D activation map of the third VT complex shown in Fig. 1. The setup of the map layers is identical to that in Fig. 2. Earliest activation is seen in the PURK layer 34 ms before the onset of surface QRS (denoted as −34 in yellow), with activation proceeding to surrounding PURK and ENDO and then transmurally to MIDWALL, where the latest activity is seen (63 ms after QRS onset). There is not enough conduction delay shown, nor is there retrograde EPI to PURK activation to support reentry. These figures demonstrate AGII-mediated induction of typical focal VT originating from the PURK layer. This experiment is exemplary of the majority of VTs facilitated during angiotensin infusion.
AGII infusion significantly increased systolic, diastolic, and mean arterial pressure (P < 0.01). AGII infusion did not result in any significant change in ventricular ERP, pacing threshold, longitudinal conduction velocity, or infarct size (Table 2). AGII-induced changes in systolic, diastolic, and mean arterial pressure did not predict VT induction following AGII infusion (Table 3).
Table 2.
Effects of angiotensin II, PD-123319, and losartan on hemodynamic and electrophysiological parameters
| Baseline | AGII | P Value | Baseline | PD | P Value | Baseline | Losartan | P Value | |
|---|---|---|---|---|---|---|---|---|---|
| SBP, mmHg | 116±3 | 181±6 | <0.01 | 120±8 | 114±9 | NS | 132±5 | 116±7 | NS |
| DBP, mmHg | 79±2 | 125±5 | <0.01 | 82±6 | 77±6 | NS | 87±3 | 77±6 | NS |
| MAP, mmHg | 92±2 | 144±5 | <0.01 | 98±3 | 95±3 | <0.01 | 101±3 | 90±5 | NS |
| ERP, ms | 155±2 | 153±2 | NS | 150±3 | 151±3 | NS | 152±4 | 154±5 | NS |
| PT, mV | 41±3 | 44±3 | NS | 49±4 | 50±3 | NS | 52±7 | 54±8 | NS |
| IS, % | 51±3 | 54±3 | NS | 59±4 | 61±4 | NS | 55±1 | 55±1 | NS |
| CV, m/s | 1.04±0.1 | 0.94±0.1 | NS | 0.75±0.1 | 0.78±0.1 | NS | 0.75±0.1 | 0.78±0.1 | NS |
Values are means ± SE.
SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure; ERP, effective refractory period; PT, pacing threshold; IS, infarct size; CV, longitudinal conduction velocity; AGII, angiotensin II; PD, PD-123319; NS, nonsignificant.
Table 3.
Effects of angiotensin II on hemodynamic and electrophysiological parameters separated by inducibility
| VT Inducible | VT Noninducible | P Value | |
|---|---|---|---|
| n | 13 | 20 | |
| ΔSBP, mmHg | 65±9 | 64±7 | NS |
| ΔDBP, mmHg | 48±10 | 45±5 | NS |
| ΔMAP, mmHg | 53±9 | 51±5 | NS |
| ΔERP, ms | −6±3 | −2±2 | NS |
| ΔPT, mV | 4±2 | 2±3 | NS |
| ΔIS, % | 2±3 | 5±2 | NS |
| ΔCV, m/s | −0.1±0.1 | −0.1±0.2 | NS |
Values are means ± SE;
n, no. of animals. Δ, Mean change with AGII.
Effects of PD.
In 26 dogs with reproducibly inducible VT after CAO, 15 were pace terminated, and the rest were defibrillated. Induction of VT required 2 extrastimuli in 4 dogs, 3 extrastimuli in 8, and 4 extrastimuli in 14 dogs. Of the VTs induced, 15 had a reentrant mechanism, which was predominantly epicardial, 10 had a focal endocardial or Purkinje origin, and the mechanism was indeterminate in 1. Of the 15 dogs with reentrant VT, PD blocked 1 (7%), whereas, in the 10 dogs with focal VT, 8 (80%) were blocked by PD infusion (P = 0.01). In the one dog where the VT mechanism was indeterminate, PD infusion did not block reinduction.
PD infusion resulted in a statistically significant reduction in mean arterial pressure (Table 2). However, the effect of PD on ischemic VT was independent of PD-induced change in mean arterial pressure, i.e., the reduction in blood pressure did not predict block of VT. PD infusion did not result in significant changes in ventricular ERP, pacing threshold, longitudinal conduction velocity, or infarct size.
Effects of losartan.
In 11 dogs with reproducibly inducible VT after CAO, 5 were pace terminated, and 6 were defibrillated. Induction of VT required two extrastimuli in two dogs, three extrastimuli in four dogs, and four extrastimuli in five dogs. Of the inducible VTs, five were of focal origin, with three having an endocardial focus, one having a Purkinje focus, and one having a focus in the midwall. Four of the inducible VTs had epicardial reentry. In the other two dogs, the VT mechanism remained indeterminate. One out of five focal and one out of four reentrant VTs were blocked by losartan infusion. Losartan infusion did not result in significant changes in systolic, diastolic, and mean arterial pressure, ERP, pacing threshold, longitudinal conduction velocity, or infarct size (Table 2).
In vitro experiments.
AGII, PD, or losartan had no significant effects on action potential characteristics of ischemic tissues, as shown in Table 4, taken from sites of origin of VT or other ischemic sites confirmed by reduction in voltage. Nineteen tissues, 16 Purkinje and 3 muscle, had inducible TA due to DADs. As previously described (26), TA was reproducibly induced with higher pacing frequencies, especially with isoproterenol. PD superfusion at 10−6 M blocked TA in 13 out of 19 (68%). Of the 19 tissues, 4 had DAD/TA with pacing alone, and PD blocked DAD/TA in 3. Eight tissues had DAD/TA with isoproterenol superfusion with PD blocking DAD/TA in five (Fig. 4). The remaining seven tissues had DAD/TA inducible only with a combination of isoproterenol and either AGII (10−6 M), losartan (10−6 M), or both. In this last group, DAD/TA was blocked by PD in five out of seven tissues (Fig. 5). With PD superfusion, block was complete in seven tissues (Figs. 4 and 5) and partial, reducing the number of TA complexes by one-half, in six; the average number of triggered beats was reduced to 3.5 ± 2.0 complexes from a baseline of 8 ± 2.2 complexes (P = 0.01). PD superfusion did not result in a significant change in DAD amplitude (5.53 ± 1.1 mV) compared with baseline (5.92 ± 1.1 mV; P = nonsignificant). TA was re-inducible in tissues following PD washout with normal Tyrode solution (Fig. 4).
Table 4.
Effects of angiotensin II, PD-123319, and losartan on action potential characteristics of pooled ischemic endocardium and Purkinje tissues
| AGII (n = 21) |
PD (n = 12) |
Losartan (n = 10) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline | AGII | P Value | Baseline | PD | P Value | Baseline | Losartan | P Value | |
| RMP, mV | −81±3 | −81±3 | NS | −77±3 | −78±5 | NS | −92±4 | −88±2 | NS |
| APA, mV | 87±3 | 84±4 | NS | 83±5 | 82±6 | NS | 94±5 | 90±5 | NS |
| APD50, ms | 172±9 | 172±11 | NS | 188±13 | 192±13 | NS | 188±14 | 179±13 | NS |
| APD90, ms | 258±11 | 247±11 | NS | 256±15 | 258±14 | NS | 247±16 | 233±14 | NS |
Values are means ± SE;
n, no. of animals. RMP, resting membrane potential; APA, action potential amplitude; APD50, action potential duration at 50% amplitude; APD90, action potential duration at 90% amplitude.
Fig. 4.
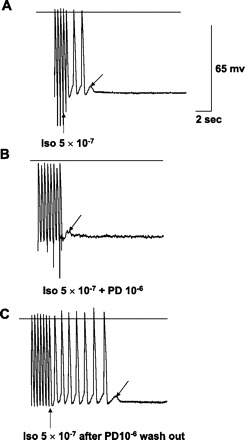
In vitro recordings from an excised endocardial site of origin of focal VT. Horizontal lines mark the zero voltage. Voltage (65 mV) and time (2 s) calibrations are shown. Upward arrow points to the last paced action potential. Downward arrow points to delayed afterdepolarizations (DADs). A: with isoproterenol (Iso), pacing at 2.5 Hz produces three DADs, two of which reach threshold and result in triggered activity (TA). B: TA is blocked by addition of PD-123319 (PD), but a DAD remains, not reaching threshold. C: TA is again induced after PD washout, stopping spontaneously with a single DAD. Note that none of the tracings shows spontaneous phase 4 depolarization, so this cell is endocardial muscle.
Fig. 5.
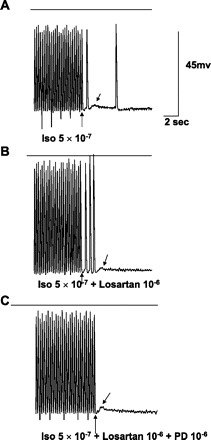
In vitro recordings from an excised Purkinje site of origin of focal VT labeled as Fig. 4, including calibrations. A: with Iso, pacing at 4 Hz produces two DADs, with one TA. Note the spontaneous action potential occurring ∼4 s after the last DAD, indicating slow Purkinje automaticity. B: addition of losartan to Iso allows three TA complexes at the same frequency of pacing. C: adding PD to Iso and losartan blocks TA, leaving a small DAD.
Another eight tissues, of which seven were Purkinje, had inducible DAD-mediated TA; losartan superfusion at 10−6 M blocked DAD/TA in none of them.
AGII (10−6 M and 10−5 M, n = 23) and/or losartan (10−6 M, n = 3) superfusion alone themselves or in combination (n = 4) did not induce DAD/TA in any of the ischemic tissues. However, in 12 ischemic tissues without inducible DAD/TA with isoproterenol alone, addition of losartan (Fig. 5), or the combination with AGII (Fig. 6) and isoproterenol resulted in TA in nine; all but one was Purkinje tissue.
Fig. 6.
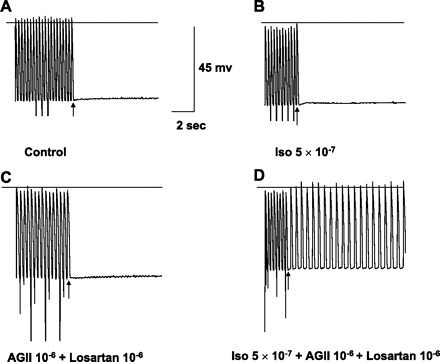
In vitro recordings from an excised Purkinje site of origin of focal VT labeled as in Fig. 4, including calibrations. A and B: no DAD/TA with pacing at 3 Hz in control or with addition of Iso, although spontaneous phase 4 depolarization is present, indicating Purkinje origin. C: after washout of Iso, addition of AGII and losartan superfusion did not result in DAD/TA, but phase 4 depolarization persists. D: with Iso, AGII, and losartan, pacing at the same frequency as A, B, and C now results in sustained TA.
To assess receptors in Purkinje and muscle as modulated by ischemia, we measured AGII and AT2 receptors, which showed higher or unchanged numbers in ischemic muscle (Fig. 7). We could not measure ischemic Purkinje, but our data document AGII and AT2 receptor exist on Purkinje.
Fig. 7.
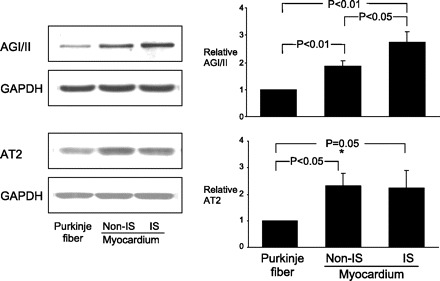
Western blot analysis of AGI/II and AGII receptor type 2 (AT2) protein expression in Purkinje fiber, and myocardium with and without ischemia (IS). Representative Western blots (left) correspond to graphs showing means ± SE (right). AGI/II and AT2 are expressed in both Purkinje fiber and cardiomyocytes. AGI/II and AT2 expression were higher in myocardium than those in Purkinje fibers. Ischemia significantly increased AGI/II expression, but not AT2, in cardiomyocytes. Expression levels of protein were normalized to those of GAPDH and are shown as fold increase over Purkinje fibers.
DISCUSSION
In a clinically relevant canine model of acute ischemic VT/VF, we have demonstrated that AGII infusion promotes only focal Purkinje, and not reentrant, VT. VT was strongly influenced by the AT2 receptor, since focal Purkinje VT was blocked by PD and not by losartan. In endocardial tissue excised from the very sites of ischemic origin of VT, PD, but not losartan, superfusion markedly reduced DAD-mediated TA. In fact, our in vitro experiments suggest that losartan may have proarrhythmic properties in conjunction with AGII and isoproterenol superfusion.
Renin-angiotensin system is activated during myocardial ischemia and injury (17, 20–22). AGII, acting through both AT1 and AT2 receptors, have been reported to have both direct and indirect electrophysiological effects (13). Effects on L-type Ca2+ current, transient outward K+ current, and slow component of delayed rectifier K+ current might predict changes in action potential duration and the plateau, but we saw no evidence for this on our in vitro ischemic tissues, and we saw no effects on refractory periods in vivo. We certainly cannot exclude important intracellular calcium loading via the L-type Ca2+ current; however, the amplitude of DADs before and after effects of drugs makes this possibility an unlikely major effect. Significant interactions between the renin-angiotensin system and the adrenergic system have also been demonstrated (13), but we found effects of PD in vitro, with and without adrenergic stimulation with isoproterenol. Although the AT2 receptor is abundant in human myocardium (8), its pathophysiological role has not been well defined.
Although the exact mechanism(s) by which AGII triggers VT/VF remain unclear, the following potential mechanisms are possible. AGII, in the presence of catecholaminergic stress, could result in calcium release from the sarcoplasmic reticulum, resulting in DADs and TA. This may be mediated through the ventricular AT2 receptor. In vivo block of focal VT by PD with in vitro suppression of DAD-mediated TA could be consistent with action on sarcoplasmic calcium release. The fact that neither AGII nor PD had any significant effects on ventricular ERP, as well as action potential parameters, suggests minimal ion channel involvement in mediating these effects. AGII has been shown to provoke cesium-induced VT/VF by increasing calcium release from sarcoplasmic reticulum through activation of a phosphatidylinositol response (14). Other potential mechanisms include effects on the Na+/Ca2+ exchanger controlling cytosolic calcium (16), promotion of myocyte apoptosis (12, 15, 28), and involvement of reactive oxygen species (23).
In our model, PD blocks focal VT in ischemic Purkinje and probably myocardium. This effect was independent of any hemodynamic changes attributable to PD. It also blocks DAD and TA without or with isoproterenol, either alone or when added to AGII and/or losartan. Thus TA is probably the underlying mechanism of focal VT. This suggests a significant role for myocardial AT2 receptor in the immediate post-CAO period. No change in action potential parameters appears to be responsible for this effect. To assess effects of receptor changes, we measured tissue receptors in three experiments, showing that normal Purkinje had only one-fifth to one-half of AGII or AT2 receptors measured in muscle, respectively, which either did not change or increased with ischemia. Unfortunately, there was inadequate Purkinje tissue from ischemic sites to assess the effect of receptor changes, preventing us from further understanding our major electrophysiological results.
The specific AT1 blocker losartan did not show significant anti-arrhythmic effects on any mechanism of VT in vivo. Consistent with this includes in vitro data that losartan did not block de novo or isoproterenol-induced DAD/TA (Fig. 5). Moreover, when combined with isoproterenol, losartan by itself and in combination with AGII seemed to facilitate DAD/TA (Fig. 6). This indicates that AGII mediated arrhythmogenecity in ischemic myocardium requires catecholaminergic stress and is enhanced by AT1 receptor blockade. Taken together with the PD data, our results suggest a predominant electrophysiological role for AT2 and not the AT1 receptor in the immediate peri-infarct period. There is some additional experimental data (rat model) to support this. Pretreatment with AT1 receptor blockers did not reduce 24-h postmyocardial infarction ventricular arrhythmias or survival, and high doses increased mortality by causing excessive hypotension (19).
In this study, we investigated inducible VT in the dog in the immediate 1–3 h following left anterior descending CAO. Previous data show that transmural infarct extension is almost complete at 1 h after CAO and forms the basis of our induction protocols. Our laboratory's previous studies have shown that VT induced by this protocol is stable, reproducible, and, therefore, forms a basis for interpreting interventions (2, 23, 24, 25, 27). Thus α2-adrenergic receptor agonists and lovastatin (27) have been shown to prevent inducible VT correlated with suppression of DADs and TA in vitro (27). Conversely, ZP123, an agent that phosphorylates gap junctions inactivated by ischemia, prevents ischemic reentry in this model (24), with no effect on DADs and TA (25). Thus we have a valid, reliably reproducible, and clinically relevant animal model to study and understand these arrhythmias that are difficult to observe and study in humans.
Focal VT is an increasingly recognized VT type, in humans, in the immediate period after CAO. This type, when recognized, can be therapeutically targeted by radio-frequency ablation (6, 7). In this study, we demonstrate for the first time the role of AGII in triggering focal VT in the immediate peri-infarct period and the preferential suppression of this VT type by the specific AT2 receptor blocker PD. In vitro, we further demonstrate that DAD-mediated TA can be blocked by AT2 receptor blockade and can be initiated by AGII and, surprisingly, by losartan in the presence of isoproterenol. This in vitro-in vivo correlation strengthens our observations.
Translating our findings into clinical practice, our data support use of angiotensin-converting enzyme inhibitors in the first few hours after an acute myocardial infarction. Our data in vitro suggest that AT1 blockers may be proarrhythmic. Further studies should be undertaken to explore the latter, as well as the potential role of AT2 receptor blockade in acute myocardial infarction and ischemic heart disease.
Study limitations.
Our laboratory's prior studies have been performed with placebo controls and reported in detail both for inducibility (2, 23, 24, 25, 27) and noninduciblilty (3). We have demonstrated that, over the same time course, both reentry and focal mechanisms are dramatically reproducible when VT was reproducible in the baseline state; this is the nature of the model after a wait of 1 h of coronary occlusion. So reproducible induction of the same morphology of VT from the same site of induction does predict continuing inducibility over the subsequent 2 h with only saline infusion (2, 23–25, 27). In this paper, we again report that inducibility of reentrant VT was persistent given all agents, and inducibility of focal VT was persistent given losartan. Given these findings in preliminary studies, we did not plan a parallel control group of animals that received only saline. Our in vitro studies back up the in vivo data, showing that TA was blocked by PD similar to the focal VT. Finally when AGII caused VT in hearts with prior noninducibility of VT (24), focal VT was the consistent finding. We reviewed our experiments over the same year when AGII was given and found 20 dogs that received only saline over the 3 h after occlusion, comparable to the time when AGII was given. We believe that these data, taken together, support our conclusion that the ATII receptor plays the dominant role in modulation of focal VT due to TA, given our prior reports.
Even with using 23 multipolar plunge electrodes in and surrounding the region of left anterior descending coronary artery, as well as employing 3D activation mapping with well-defined criteria, micro-reentry as a mechanism cannot be completely excluded. The high correlation demonstrated between focal VT in vivo and TA in vitro, however, argues for a focal mechanism and against a reentrant mechanism. Drug dosages used were based on standard practice. This may have caused an enhanced or attenuated response in certain cases, given the dog weight was only one-fourth that of an adult human. Also, only single weight-based doses were used for in vivo studies. It is possible that different doses could have differing effects on focal vs. reentrant mechanisms.
Conclusions.
AGII promotes only focal VT in the setting of myocardial ischemia by a mechanism independent of mean arterial pressure, plasma AGII levels, and infarct size. PD, but not losartan, preferentially blocked focal VT in this canine model. TA due to DADs is the likely mechanism that is blocked by PD. Our data support angiotensin-converting enzyme inhibition in the setting of acute myocardial ischemia, and myocardial AT2 blockade may be a further potential anti-arrhythmic target for prevention of sudden cardiac death in patients with myocardial ischemia.
GRANTS
This study was supported by funding from the University of Iowa Hospitals and Clinics and the Veterans Affairs Medical Center.
ACKNOWLEDGMENTS
We thank Linda Bang for excellent secretarial assistance.
REFERENCES
- 1. Arnar DO, Bullinga JR, Martins JB. Role of the Purkinje system in spontaneous ventricular tachycardia during acute ischemia in a canine model. Circulation 96: 2421–2429, 1997 [DOI] [PubMed] [Google Scholar]
- 2. Arnar DO, Xing D, Lee H, Martins JB. Prevention of ischemic ventricular tachycardia of Purkinje origin: role for α2-adrenoceptors in Purkinje? Am J Physiol Heart Circ Physiol 280: H1182–H1190, 2001 [DOI] [PubMed] [Google Scholar]
- 3. Arnar DO, Xing D, Martins JB. Alpha-2 adrenergic antagonism enhances risk of ventricular tachycardia during acute ischemia. Scand Cardiovasc J 41: 378–385, 2007 [DOI] [PubMed] [Google Scholar]
- 4. Arnar DO, Xing D, Martins JB. Overdrive pacing of early ischemic ventricular tachycardia: evidence for both reentry and triggered activity. Am J Physiol Heart Circ Physiol 288: H1124–H1130, 2005 [DOI] [PubMed] [Google Scholar]
- 5. Arnar DO, Cai JJ, Lee HC, Martins JB. Electrophysiologic effects of civamide (zucapsaicin) on canine cardiac tissue in vivo, and in vitro. J Cardiovasc Pharmacol 32: 875–883, 1998 [DOI] [PubMed] [Google Scholar]
- 6. Ashwath ML, Sogade FO. Focal origin of ventricular fibrillation in a patient with ischemic cardiomyopathy. J Natl Med Assoc 96: 1228–1231, 2004 [PMC free article] [PubMed] [Google Scholar]
- 7. Bansch D, Oyang F, Antz M, Arentz T, Weber R, Val-Mejias JE, Ernst S, Kuck KH. Successful catheter ablation of electrical storm after myocardial infarction. Circulation 108: 3011–3016, 2003 [DOI] [PubMed] [Google Scholar]
- 8. Brink M, Erne P, de Gasparo M, Rogg H, Schmid A, Stulz P, Bullock G. Localization of the angiotensin II receptor subtypes in the human atrium. J Mol Cell Cardiol 28: 1789–1799, 1996 [DOI] [PubMed] [Google Scholar]
- 9. Chorvatova A, Gallo-Payet N, Casanova C, Payet MD. Modulation of membrane potential and ionic currents by the AT1 and AT2 receptors of angiotensin II. Cell Signal 8: 525–532, 1996 [DOI] [PubMed] [Google Scholar]
- 10. Cleland JG, Dargie HJ, Robertson JI. Angiotensin converting enzyme inhibition in heart failure. Br J Clin Pharmacol 18, Suppl 2: 157S–160S, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cohn JN, Johnson G, Ziesche S, Cobb F, Francis G, Tristani F, Smith R, Dunkman WB, Loeb H, Wong M. A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med 325: 303–310, 1991 [DOI] [PubMed] [Google Scholar]
- 12. Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133: 462–474, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goette A, Lendeckel U. Electrophysiological effects of angiotensin II. I. Signal transduction and basic electrophysiological mechanisms. Europace 10: 238–241, 2008 [DOI] [PubMed] [Google Scholar]
- 14. Gondo N, Kumagai K, Nakashima H, Saku K. Angiotensin II provokes cesium-induced ventricular tachyarrhythmias. Cardiovasc Res 49: 381–390, 2001 [DOI] [PubMed] [Google Scholar]
- 15. Haendeler J, Ishida M, Hunyady L, Berk BC. The third cytoplasmic loop of the angiotensin II type 1 receptor exerts differential effects on extracellular signal-regulated kinase (ERK1/ERK2) and apoptosis via Ras- and Rap1-dependent pathways. Circ Res 86: 729–736, 2000 [DOI] [PubMed] [Google Scholar]
- 16. Homma N, Amran MS, Nagasawa Y, Hashimoto K. Topics on the Na+/Ca2+ exchanger: involvement of Na+/Ca2+ exchange system in cardiac triggered activity. J Pharm Sci 102: 17–21, 2006 [DOI] [PubMed] [Google Scholar]
- 17. Noda K, Sasaguri M, Ideishi M, Ikeda M, Arakawa K. Role of locally formed angiotensin II and bradykinin in the reduction of myocardial infarct size in dogs. Cardiovasc Res 27: 334–340, 1993 [DOI] [PubMed] [Google Scholar]
- 18. Pfeffer MA, McMurray JJ, Velazquez EJ, Rouleau JL, Kober L, Maggioni AP, Solomon SD, Swedberg K, Van de Werf F, White H, Leimberger JD, Henis M, Edwards S, Zelenkofske S, Sellers MA, Califf RM. Valsartan in Acute Myocardial Infarction Trial Investigators Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 20: 1893– 1906, 2003 [DOI] [PubMed] [Google Scholar]
- 19. Pourdjabbar A, Parker TG, Nguyen QT, Desjardins JF, Lapointe N, Tsoporis JN, Rouleau JL. Effects of pre-, peri-, and postmyocardial infarction treatment with losartan in rats: effect of dose on survival, ventricular arrhythmias, function, and remodeling. Am J Physiol Heart Circ Physiol 288: H1997–H2005, 2005 [DOI] [PubMed] [Google Scholar]
- 20. Reiss K, Capasso JM, Huang HE, Meggs LG, Li P, Anversa P. ANG II receptors, c-myc, and c-jun in myocytes after myocardial infarction and ventricular failure. Am J Physiol Heart Circ Physiol 264: H760–H769, 1993 [DOI] [PubMed] [Google Scholar]
- 21. Santos RA, Brum JM, Brosnihan KB, Ferrario CM. The renin-angiotensin system during acute myocardial ischemia in dogs. Hypertension 15, Suppl: I121–I127, 1990 [DOI] [PubMed] [Google Scholar]
- 22. Wang LX, Ideishi M, Yahiro E, Urata H, Arakawa K, Saku K. Mechanism of the cardioprotective effect of inhibition of the renin-angiotensin system on ischemia/reperfusion-induced myocardial injury. Hypertens Res 24: 179–187, 2001 [DOI] [PubMed] [Google Scholar]
- 23. Xing D, Chaudhary AK, Miller FM, Martins JB. Free radical scavenger specifically prevents ischemic focal ventricular tachycardia. Heart Rhythm 6: 530–536, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xing D, Kjølbye AL, Nielsen MS, Petersen JS, Harlow KW, Holstein-Rathlow NH, Martins JB. ZP123 increases gap junctional conductance, and prevents reentrant ventricular tachycardia during myocardial ischemia in open chest dogs. J Cardiovasc Electrophysiol 14: 510–520, 2003 [DOI] [PubMed] [Google Scholar]
- 25. Xing D, Kjolbye AL, Petersen JS, Martins JB. Pharmacological stimulation of cardiac gap junction coupling does not affect ischemia-induced focal ventricular tachycardia or triggered activity in dogs. Am J Physiol Heart Circ Physiol 288: H511–H516, 2005 [DOI] [PubMed] [Google Scholar]
- 26. Xing D, Martins JB. Triggered activity due to delayed afterdepolarizations in sites of focal origin of ischemic ventricular tachycardia. Am J Physiol Heart Circ Physiol 287: H2078–H2084, 2004 [DOI] [PubMed] [Google Scholar]
- 27. Xing D, Murry DJ, Schmidt MS, Hohl RJ, Martins JB. Lovastatin specifically prevents focal ischemic ventricular tachycardia due to triggered activity. Heart Rhythm 4: 629–637, 2007 [DOI] [PubMed] [Google Scholar]
- 28. Yamada T, Horiuchi M, Dzau VJ. Angiotensin II type 2 receptor mediates programmed cell death. Proc Natl Acad Sci USA 93: 156–160, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zheng W, Weiss RM, Wang X, Zhou R, Arlen AM, Lei L, Lazartigues E, Tomanek RJ. DITPA stimulates arteriolar growth and modifies myocardial postinfarction remodeling. Am J Physiol Heart Circ Physiol 287: H2739–H2745, 2004 [DOI] [PubMed] [Google Scholar]