

Abstract
Cells in the Purkinje system (PS) are known to be more vulnerable than ventricular myocytes to secondary excitations during the action potential (AP) plateau or repolarization phases, known as early afterdepolarizations (EADs). Since myocytes have a lower intrinsic AP duration than the PS cells to which they are coupled, EADs occurring in distal branches of the PS are more likely to result in propagating ectopic beats. In this study, we use a computer model of the rabbit ventricles and PS to investigate the consequences of EADs occurring at different times and places in the cardiac conduction system. We quantify the role of tissue conductivity and excitability, as well as interaction with sinus excitation, in determining whether an EAD-induced ectopic beat will establish reentrant activity. We demonstrate how a single ectopic beat arising from an EAD in the distal PS can give rise to reentrant arrhythmia; in contrast, EADs in the proximal PS were unable to initiate reentry. Clinical studies have established the PS as a potential substrate for reentry, but the underlying mechanisms of these types of disorder are not well understood, nor are conditions leading to their development clearly defined; this work provides new insights into the role of the PS in such circumstances. Our findings indicate that simulated EADs in the distal PS can induce premature beats, which can lead to the tachycardias involving the conduction system due to interactions with sinus activity or impaired myocardial conduction velocity.
Keywords: computer modeling, bundle branch reentry, afterdepolarizations
early afterdepolarizations (EADs) are positive inflections in membrane potential (Vm) that occur during the late phases of the action potential (AP), before full repolarization. If sufficiently large, EADs can produce triggered activity and promote reentrant arrhythmias, including bundle branch reentrant ventricular tachycardia (BBRT) (1). At the ionic level, EADs result from decreased outward current, increased inward current, or combinations of the two. Electrophysiological studies have been performed using pharmacological agents and slow pacing modes to cause initial AP prolongation via rapidly activating K+ current reduction (cesium, quinidine) (53), slowed Na+ current (INa) inactivation (anthopleurin, veratradine) (10), and increased L-type Ca2+ current activity (BAY K 8644) (19). For a detailed description of ionic mechanisms of EADs, refer to the review by Volders et al. (48).
Ectopic beats arising from EADs are thought to be more common in the Purkinje system (PS) than in the ventricular myocardium for several reasons. EADs are easily induced in Purkinje cells by application of pharmacological agents that prolong the AP, which is already intrinsically longer than in typical ventricular myocytes (39, 40). In contrast, coupled ventricular cells have a shorter AP duration (APD) and become reexcitable, while the PS is still repolarizing and susceptible to EADs. Moreover, cells in the PS have a high membrane resistance, which allows small charge displacements to cause large changes in Vm (37). Finally, since Purkinje cells are only coupled in one dimension, there is reduced electrotonic suppression of myocardial EAD propagation.
Persistent bundle branch reentry (BBR) as a mechanism of sustained tachycardia has been observed in patients with significant conduction system impairment (6, 42, 50), which is often manifested as His-ventricle (H-V) interval prolongation, bundle branch block (BBB) QRS configuration, or both. Most BBRT studies have reported prolonged H-V interval during sinus rhythm, indicating underlying conduction abnormalities, but the pathology has also been observed in patients with idiopathic isolated conduction system disease and no apparent structural heart abnormalities (17, 38). Furthermore, BBRT has also been reported in patients with apparently normal His-PS conduction (23). These patients exhibited functional transient abnormalities in the conduction system that provided a substrate for reentry, such as temporary conduction block (CB) in the bundles. Overall, the exact mechanisms of BBRT initiation are not well understood.
Several experimental (14, 24) and computer modeling (2, 28, 37, 49) studies have documented how EADs generated in PS cells propagate to coupled myocytes and induce ectopic activity. However, the behavior of the PS changes significantly when it is coupled to a large mass of ventricular muscle due to electrotonic interactions and loading effects. Thus the EAD initiation and propagation mechanisms must be revisited in an anatomically realistic three-dimensional (3D) cardiac model.
Previously, our laboratory demonstrated that the PS is involved in initiation and maintenance of shock-induced arrhythmias, triggering postshock activations and providing alternative pathways for reentrant circuits (12), but it remains unclear whether triggered activity originating in Purkinje cells may induce reentry. Experimental difficulties in detecting and isolating Purkinje cells in subendocardial layers have restricted measurements of the intact PS. In this paper, we use a 3D computer model to investigate possible mechanisms of triggered activity and effects of conduction system disorders on arrhythmia initiation. We hypothesize that a single ectopic beat in the distal PS, if properly timed, can lead to arrhythmogenesis in ventricular tissue with impaired conduction or by interacting with sinus excitation. Furthermore, we hypothesize that a single EAD originating in the PS can establish BBRT in ventricles with reduced excitability and/or impaired conduction, whether or not functional BBB is present. We systematically examined conduction system abnormalities, such as temporary unidirectional BBB due to ectopic activity originating in the PS. We also studied how variations in tissue excitability and conductivity affect reentry initiation.
METHODS
Governing Equations and 3D Model
Cardiac electrical activity was described by the monodomain formulation (44):
| (1) |
where Im is membrane current, β is the tissue surface-to-volume ratio, and σ̄m is the monodomain conductivity tensor, which is the harmonic mean of the intracellular and extracellular conductivity tensors. The nominal extracellular conductivities in the longitudinal and transverse directions were 0.625 and 0.236 S/m, respectively, and the corresponding intracellular conductivities were 0.174 and 0.019 S/m [based on experiments by Clerc (9)].
Nonlinearities arising from the current-voltage relationship across the membrane were described by:
| (2) |
where Cm is the membrane capacitance per unit area, and Iion is the current density through ion channels, which depends on Vm and several other variables that are represented by ν, as described in the rabbit ventricular AP model developed by Mahajan et al. (26). Itrans is the transmembrane stimulus current density, as delivered by an intracellular electrode and removed by an adjacent extracellular electrode.
The system of equations was solved by the finite-element method using the CARP software package (46), with geometry and fiber oriention based on rabbit ventricular data (43). The system comprised ∼550,000 nodes with element edge lengths on the order of 300 μm. Equation 1 is a parabolic equation, which was decoupled from the set of ordinary differential equations (Eq. 2) describing ionic currents. Both sets of equations were solved explicitly, as detailed elsewhere (47). Simulations were carried out on four cores of an HP Opteron cluster (2.4 GHz) running Linux with 2 GB of physical memory per node.
Modeling the ventricular conduction system.
To model an anatomically realistic heart, a one-dimensional finite-element branching Purkinje network explained in Ref. 45 was implemented, as shown in Fig. 1A. This model enforced conservation of current at Purkinje-myocardial junctions (PMJs). At each PMJ, the Purkinje cell was coupled to n myocytes within a certain radius (rPMJ) by a fixed resistance RPMJ. The current load on each terminal Purkinje cell was calculated by summing the individual currents flowing into each junctional myocyte and scaling the total by a loading factor (KPMJ) to account for sink effects from surrounding tissue that was not directly coupled (4). Thus, while the current injected into a particular myocyte is derived from the straightforward Ohmic relationship, the load on a particular cable ending at xe is given by:
| (3) |
where IL is current load, ϕi(xe) is the intracellular potential of the terminal Purkinje cell, and ϕiM(xn) is the intracellular potential of the nth myocardial node inside the junctional radius. Model details can be found elsewhere (4, 12, 45).
Fig. 1.
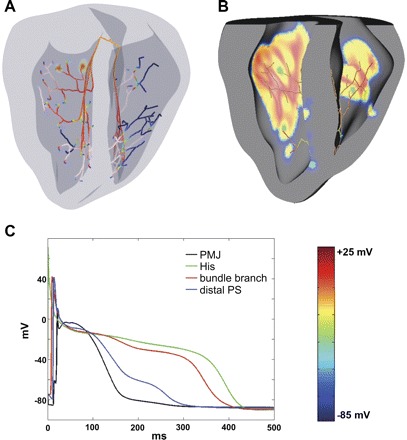
Computer model. A: three-dimensional model of rabbit ventricles with branching Purkinje system (PS). Colors indicate membrane potential (Vm; see the color bar). The same color scheme is used throughout the paper. B: excitation of the ventricles by the PS during normal sinus rhythm. Colors are as in A. C: action potentials (APs) in various parts of the conduction system. AP duration (APD) is longer in the bundle of His and bundle branches than in distal regions due to electrotonic interaction between terminal Purkinje cells and coupled myocytes. PMJ, Purkinje-myocardial junction.
PS potentials were calculated using, from the parabolic equation:
| (4) |
where s is a vector along the fiber (45), and σi is intracellular conductivity.
The DiFrancesco-Nobel equations (13) were used to govern membrane electrical activity in the PS. This allowed the model to reflect the distinct electrophysiological features between Purkinje cells and ventricular myocytes. Sodium conductance in the DiFrancesco-Nobel model was increased threefold (5) to increase maximum upstroke velocity (∂Vm/∂t) in uncoupled cells from 169 to 380 mV/ms, which is closer to the in vitro value reported by Lu et al. (25). Purkinje cell radius was set to 15 μm, and intracellular conductivity was set to 1.5 S/m to achieve the conduction velocity (CV) ratio of 4.4 with reference to the myocardium (4), which is within the experimental range (11).
Figure 1B shows ventricular excitation by the PS during normal sinus rhythm. Parameters for sodium conductance, tissue conductivity, and gap junction resistance were adjusted to reproduce normal ventricular activation patterns recorded during sinus rhythm (33). PMJ parameters were tuned to obtain realistic transmission characteristics (51), with retrograde propagation delays much shorter (≈0.96 ms) than anterograde delays (≈9.69 ms). Figure 1C shows APD variation between different parts of the system. Moving from the His bundle to the distal branches of the PS, APD was gradually abbreviated until the terminal cells at PMJs, which had APDs nearly as short as coupled myocytes. This feature, caused by electrotonic loading at the interface between tissues, made the PS an ideal substrate during reentry establishment.
Initiation of EADs in PS Cells
Ectopic activity in the distal PS was produced by injecting current in five contiguous PS nodes near the PMJs, as described in previous studies (28). In an alternative set of simulations, the PS was excited in the proximal region adjacent to the left (LBB) and right bundle branches (RBB). This approach, in contrast to altering the ionic model to produce genuine EADs, allowed for fine-grain control of the timing and location of simulated ectopic beats. Sinus beats were simulated by injecting current at the bundle of His at a fixed interval (tHis), which was varied between 600 and 1,000 ms in steps of 50 ms. The interval between the last sinus beat and the ectopic beat (tEB) was varied from 100 to 400 ms in steps of 10 ms. The time between an ectopic beat being triggered in one bundle branch and the same activation being picked up in the other was defined as tBB, which measures the ventricular conduction delay for either right bundle-ventricle-left bundle or left bundle-ventricle-right bundle pathways. To establish BBR, the effective path length of the reentrant circuit in our model was increased by reducing the conductivities and sodium channel conductance (gNa) in both the PS and myocardium. Myocardial (σVM) and PS conductivities (σPS) were varied between 20 and 50% of normal, while myocardial gNa was varied between 25 and 70%, and PS gNa was kept constant at 50%.
RESULTS
Ectopic Activity Triggered in the Distal PS
Ectopic activity was initiated in the left and right sides of the distal PS. The side of ectopic activity was chosen near PMJs, as observed in clinical studies (24). For the values of tEB between 100 and 400 ms, three different scenarios were observed.
Failed ectopic activity.
Ectopic beats induced when both the distal PS and ventricular myocytes were refractory due to the sinus rhythm failed to initiate ectopic activity (tEB < 190 ms). These stimuli elevated the potential of refractory tissue, but could not trigger new APs. This behavior is illustrated in Fig. 2B (indicated by “F”).
Fig. 2.
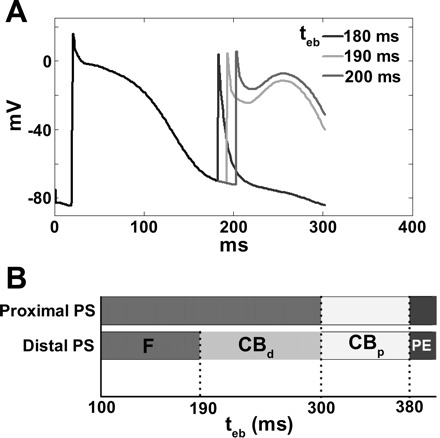
A: ectopic activity had varying consequences, depending on the interval between the last sinus beat and the ectopic beat (tEB): failure to elicit AP (tEB = 180 ms), triggered activity with conduction block (CB) in the distal PS (tEB = 190 ms), or triggered activity with CB in the bundle branch or proximal PS (tEB = 180 ms). B: outcome of the ectopic activity originating in the right proximal and distal PS at a range of tEB. F, failed ectopic activity; CBd, CB in the distal PS; CBp, CB in the proximal PS; PE, propagation of the ectopic activity without CB.
Premature excitation and CB.
If the distal PS and the myocardium were excitable, but the proximal PS (His bundle and branches) remained refractory (190 ms ≤ tEB < 380 ms), the ectopic beat excited the myocardium and propagated retrogradely to nearby parts of the distal PS. However, activation was blocked at the refractory bundle branch, so retrograde excitation was not conducted to the other side of the PS. Figure 2A shows examples of ectopic activity triggering a premature AP in the PS.
In Fig. 3, an ectopic beat was induced in the right distal PS at tEB = 370 ms. Excitation propagated into the right ventricle (RV) (Fig. 3A) by anterograde propagation, but retrograde conduction into the RBB was blocked (Fig. 3B) due to refractoriness. Individual AP traces in the right PS (see Fig. 3E) clearly show the conduction failure in the RBB (shown by the dotted line). The asterisk indicates the location of CB in the RBB. Thus the left ventricle (LV) was excited after 45 ms by transmission across the septum, instead of by activity from within the PS. Shortly thereafter, LV activations propagated retrogradely into the left PS (Fig. 3C), but reentry into the right PS was prevented due to CB in the right BB (Fig. 3D). In other cases (190 ms ≤ tEB < 300 ms), retrograde activation did not occur as in Fig. 3C due to residual refractoriness in the distal PS (indicated by “CBd” in Fig. 2B).
Fig. 3.
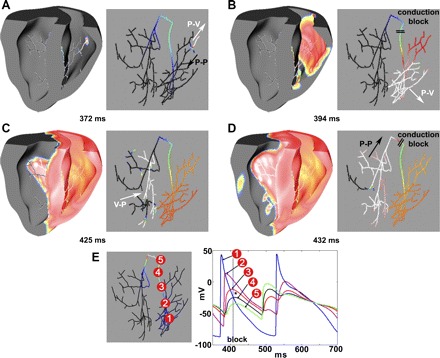
Temporary CB in the PS. A: 372 ms. An ectopic beat triggered an AP in the right distal PS. Excitation spread to right ventricle (RV; white arrow) and throughout the right distal PS (black arrow). B: 394 ms. The proximal right bundle branch (RBB) was still refractory, which caused temporary CB. C: 425 ms. Excitation spread through myocardium across the ventricular to reach the left ventricle (LV) and excited the left distal PS. D: 432 ms. The excitation spread through the left PS, and the left bundle branch (LBB), but the RBB was refractory, causing another temporary CB. E: individual AP traces (1–5) from five locations show retrograde conduction failure (dotted line) in the RBB. *The occurrence of CB at the corresponding location. P-V, anterograde conduction from PS to myocardium; V-P, retrograde conduction from myocardium to PS; P-P, retrograde conduction within the PS. Times are relative to the last His stimulation.
Premature excitation with no CB.
After 380 ms of recovery from sinus excitation, bundle branches were excitable, along with the distal PS network. Ectopic beats induced after this instance (tEB ≥ 380 ms) excited both the coupled ventricular tissue and the proximal PS. PS-enabled activation of the other ventricle occurred 40 ms after the ectopic beat. No CB was observed in the PS.
Ectopic Activity Triggered in the Proximal PS
APDs in the proximal PS are longer than the distal PS due to myocardial loading. Thus ectopic activity could not be initiated in the proximal PS branches for tEB ≤ 300 ms (see Fig. 2B). As above, functional CB was observed in the bundle branches (“CBp”) for 300 ms ≤ tEB < 380 ms, but CB did not occur in the distal PS for any ectopic beats originating in the proximal PS. Excitation patterns from premature stimuli with tEB ≥ 380 ms was similar to those observed for ectopic beats originating in the distal PS.
Induction of Reentry by an Ectopic Beat Interacting with Sinus Activity
When a sinus beat occurred while the tissue or part of the tissue was still recovering from a premature beat, the interactions were complex. The sinus beat was conducted by only one bundle branch due to refractoriness in the other, thus only one ventricle was excited. Excitation propagated to the unexcited ventricle through the myocardium, resulting in a delay. If the PS in that ventricle was excitable by the time this delay was elapsed, retrograde conduction occurred, forming a reentrant circuit.
One such scenario is summarized in Fig. 4. An ectopic beat induced in the right distal PS (tEB = 370 ms) produced a premature ventricular depolarization. During the next sinus activation at 700 ms (Fig. 4A), the left bundle was excitable and conducted the His activation (Fig. 4E, traces 2 and 3), but the right bundle was still refractory and blocked conduction into the right distal PS (Fig. 4B). AP traces 4 and 5 in Fig. 4E show CB in the RBB (indicated by asterisks). Consequently, the sinus beat only activated the LV (Fig. 4B, white arrow). By 800 ms, activation reached the RV by transseptal conduction, and retrograde propagation into the right distal PS occurred (Fig. 4C). At this point, the RBB was no longer refractory, and excitation continued through the bundles and left distal PS, establishing a reentrant circuit (Fig. 4D). Thus unidirectional block in the RBB due to an ectopic beat provided the necessary substrate for reentry for the next sinus activation.
Fig. 4.
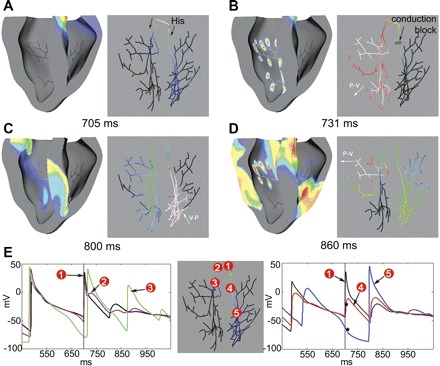
Reentry induced by an ectopic beat. A: 705 ms. A sinus beat originated at the His bundle at a fixed interval (tHis) = 700 ms (black arrow), following an ectopic activation in the right PS (tEB = 370 ms). B: 731 ms. Due to refractoriness in the RBB from the premature beat, sinus activation propagated into the left PS and ventricle but not into the right PS due to temporary CB. C: 800 ms. Activation spread across the septum from LV to RV, and the right distal PS, now fully excitable, was activated by retrograde propagation (white arrow). D: 860 ms. Reentry was initiated. E: individual APs at various locations in the PS. Traces 2 and 3 show conduction of the sinus beat in the LBB, while traces 4 and 5 show CB in the RBB (indicated by asterisks). Dotted line indicates the instance of sinus beat. Labels are as in Fig. 3.
For ectopic activity in the right distal PS (tEB = 370 ms, σPS and σVM at 33.33% of base values), early sinus beats (tHis < 680 ms) failed to propagate at all beyond the bundle branches. For intermediate cases (680 ms ≤ tHis ≤ 1,000 ms), refractoriness in the RBB led to tachycardias with the RBB block morphology, as illustrated in Fig. 4. For later sinus beats (tHis > 1,000 ms), if interactions resulted in an arrhythmia, the morphology was a tachycardia without BBB. Similar behavior was observed when the ectopic beat originated in left PS (not shown). Premature excitation spread through the LV and septum to reach the RV, producing asymmetric refractoriness in the bundles with the LBB nonexcitable. The sinus beat coming down the His bundle within 300–500 ms after the ectopic beat was thus conducted only through the RBB, resulting in left BBB morphology.
In contrast, simulations with ectopic activity in the proximal PS did not result in reentry. Since the values of tEB associated with triggered activity for such stimuli were significantly higher (≥300 ms), the distal PS and both ventricles were excitable and conducted the premature beats without CB. Thus, when the following sinus beats interacted with these activation patterns, there was no asymmetry between the LV and RV, and reentry was avoided.
Reentry Induction by Ectopic Beat in Presence of Slow Conduction
Initiation of BBRT requires unidirectional block in one bundle branch and slow conduction in the other. A subset of simulations was performed with impaired conduction in the tissue to reproduce conditions in which the BBB morphology can occur. Slower conduction in the ventricles and in the PS increased the effective path length for establishing reentrant circuits with ectopic beats. Figure 5 shows the outcome of ectopic activity (tEB = 370 ms) for several tissue conductivities (both σPS and σVM) and myocardial gNa. PS gNa was kept constant at 50%. For lower values of myocardial gNa, APs could not be elicited due to insufficient depolarizing current; in contrast, for higher values, a temporary CB was observed in the LBB. In the latter cases, no reentry through the bundles was observed, as the RBB was refractory when activation reached the RV. BBRT was observed for σPS = σVM = 35% with gNa = 50%, σPS = σVM = 30% with gNa = 58%, and σPS = σVM = 25% with gNa = 67%. Other combinations, indicated by “T”, produced tachycardia involving the bundle branches and PS, but not BBRT. The shaded region (between the two lines) represents the tachycardia-prone parameter range. Reentrant activations were considered tachycardias if they were sustained for at least 500 ms.
Fig. 5.
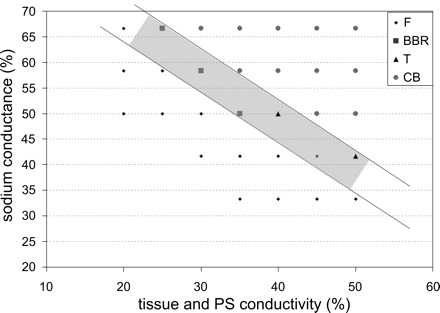
Outcome of ectopic activity with varying tissue conductivities [PS (σPS) and myocardial (σVM)] and myocardial sodium channel conductance (gNa). BBR, bundle branch reentry; F, failure to trigger propagated premature activations; CB, CB in the bundle branch (no reentry); T, tachycardia. The shaded rectangle indicates the vulnerable region.
If either of σPS or σVM was slightly increased, BBRT was converted into normal tachycardia; if any of σPS, σVM, or gNa was increased substantially, the BBRT degraded into temporary CB, and the reentry died down.
BBRT
Figure 6 shows a typical activation sequence during successful BBRT induction by ectopic activity (tEB = 370 ms) in the right distal PS. Excitation spread through the right distal PS (traces 1 and 2 in Fig. 6E) and the RV (white arrow, Fig. 6A), but retrograde CB occurred in the RBB (asterisk in trace 3 in Fig. 6E). Myocardial excitation reached the LV by transseptal conduction (black arrow, Fig. 6B). The left PS was activated by retrograde propagation 165 ms after the ectopic beat (trace 4 in Fig. 6E). Due to slow conduction through the PS and myocardium, activations reached the RBB, now excitable, 185 ms after the ectopic beat (Fig. 6C, and trace 5 in Fig. 6E), and the BBRT circuit was established by propagation into the right PS and RV (denoted by a two-way arrow between traces 1 and 3 in Fig. 6E). Thus the anterograde and retrograde limbs of the BBRT circuit are formed by the RBB and LBB, respectively, as shown schematically in Fig. 7.
Fig. 6.
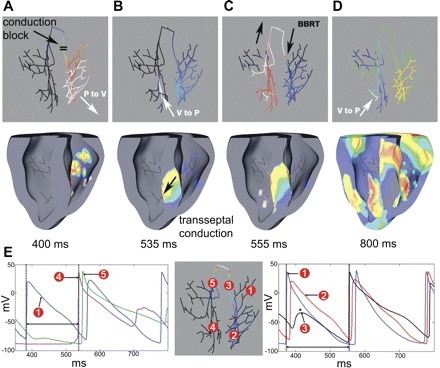
Bundle branch reentrant tachycardia (BBRT). A: ectopic activity spread through the right distal PS and into the RV (white arrow). CB was observed in the RBB. B: activations reached the LV by transseptal conduction (black arrow) and were picked up by the left PS 165 ms after the ectopic beat (white arrow). C: activations were conducted retrogradely through the LBB and bundle of His to reach the RBB after 185 ms. D: excitation spread through the right distal PS and RV. E: individual AP traces at various locations in the PS. Ectopic beat was originated near trace 1. *Retrograde CB in the RBB. The two-way arrow between dotted lines in the lefthand traces (between trace 1 and 4) indicate the time required by the ectopic beat to travel from right PS to the left PS (through septum). The two-way arrow between the dotted lines in the righthand traces (between trace 1 and 3) indicate the time taken by the ectopic activation to reach back to the RBB via LBB and His bundle.
Fig. 7.
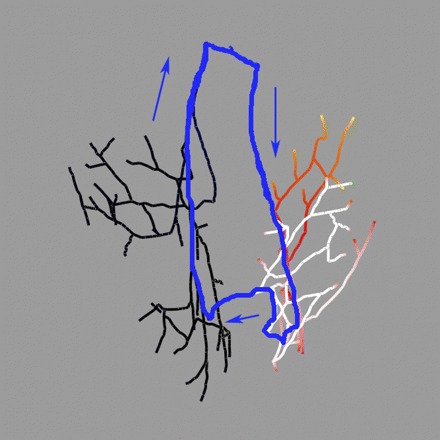
BBRT reentrant circuit as described in text. The retrograde (LBB) and anterograde (RBB) limbs are easily identified.
Interestingly, BBRT was not observed for ectopic activity originating in the left or right proximal PS. In this case, the ectopic activity spread rapidly through the entire distal PS, leading to near-simultaneous excitation of the LV and RV. This reduced the path length of the reentrant circuit, making the establishment of BBRT impossible.
Classification Based on Conduction Time
To quantify the effects of slowed ventricular conduction, we looked at the time between an ectopic beat in one bundle branch and the associated wave front exciting the other bundle branch. Figure 8A classifies events based on tBB for ectopic activity in the right PS (tEB = 370 ms). In the scenario of just the CB in RBB, the average tBB was 109 ± 15 ms (n = 10). To produce tachycardia involving the conduction system, considerable slow conduction was required in the tissue with an average tBB = 138 ± 0.7 ms (n = 2), whereas BBRT was produced at extremely slow conduction velocities in myocardial mass as well as PS. The average tBB recorded in the cases with BBRT was 165 ± 4 ms (n = 3). These trends are in agreement with the longer H-V intervals observed in most of the patients with BBRT and tachycardia involving His-PS (6, 27).
Fig. 8.
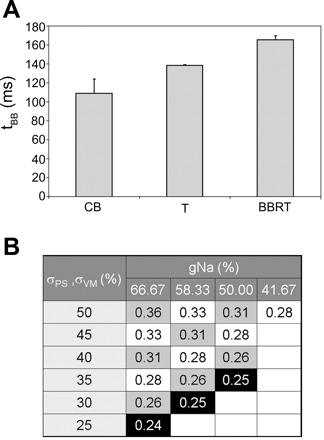
A: classification of events based on average time between an ectopic beat being triggered in one bundle branch and the same activation being picked up in the other (tBB) values during CB, tachycardia involving His-PS (T), and BBRT. B: variation in longitudinal conduction velocity (m/s) in ventricular myocytes with varying σPS and σVM and myocardial gNa (numbers given as percentage of nominal values). Combinations yielding similar conduction velocity are grouped diagonally; combinations leading to BBRT are highlighted as white on black.
Figure 8B shows longitudinal CV for each combination of reduced conductivities (σPS and σVM) and myocardial gNa. CV during normal excitation was 0.5 m/s. Reducing the conductivities to 50% lowered CV to 0.36 m/s. Further reductions had the same effect as reducing gNa with higher conductivity. In Fig. 8, combinations yielding the same CV are grouped diagonally. Reducing conductivities to 25% roughly halved CV. Note that white-on-black highlighted combinations correspond to states resulting in BBRT, with CV in the range of 0.24–0.25 m/s. CV in the PS, which was ∼1.6 m/s in normal conditions, was reduced to 1.35 m/s with σPS = σVM = 50%, and 0.8 m/s for σPS = σVM = 30%.
DISCUSSION
This paper investigates the effects of conduction system abnormalities on reentry induction. Specifically, we examined how ectopic foci in the PS might induce arrhythmias during pathological states conducive to BBRT. The main findings of this research are as follows: 1) EADs originating in the distal PS may initiate reentry in the ventricles due to temporary CB; 2) EADs originating in the proximal PS could not initiate similar reentry despite producing CB; 3) a single, critically timed ectopic beat was sufficient to produce reentry involving the His-PS; and 4) in the presence of extremely slow or impaired ventricular conduction, a single ectopic beat could initiate BBRT. We reproduced common features of patients suffering from BBRT, such as slow conduction, transient CB in the PS, and reduced myocardial excitability. The results presented help explain the mechanisms involved at the Purkinje-myocardial interface and the conditions necessary for the initiation of arrhythmias due to EADs originating in the PS.
Ectopic Activity Triggered by the PS
Purkinje cells are susceptible to EAD formation because of their higher membrane resistance and longer APD (37), which provides sufficient time for L-type calcium channel reactivation, leading to EAD formation. Coupled myocardial cells have a shorter APD and become reexcitable while PS cells are still repolarizing. Consequently, the incidence of phase II or II EADs in Purkinje fibers is increased, which can give rise to propagated ectopic beats. Triggered activity depends on differences in Vm between connected cells and also on the excitability threshold of already-repolarized cells (48). Several experimental studies have documented how EADs generated in the PS propagate to ventricular muscle cells (24, 14). Computer modeling studies have shown that suppression or facilitation of triggered activity depends on the connection between conductive and myocardial tissue (28, 37). Very high resistance at PMJs and certain physiological circumstances resulting in poor coupling between the two cell types facilitate triggered activity (49). EADs in the PS cause reactivation of INa in nearby groups of coupled cells, generating triggered APs that propagate to the ventricular myocardium as ectopic activity (37).
Although we have constantly referred to EADs, delayed afterdepolarizations (DADs) are also calcium-mediated events leading to premature depolarizations, but, after the cell has fully repolarized, DADs can also lead to ectopic beats in the PS (52, 16). Thus propagated DADs may trigger reentry as well, and the arguments we put forth apply to the timing of the ectopic beat, whether it be an EAD or DAD, and do not distinguish between the two.
Previously, simulations have studied the possibility of EAD propagation based on two-cell models (15, 49) or one-dimensional models (30, 36) by analyzing the effects of different factors, such as electrical coupling, EAD conditions, and stimulation frequency. A more recent study by Monserrat et al. (28) was based on a two-dimensional computer model of Purkinje fibers connected to a thin sheet of myocardium. Our study differs from previous work, because it used an anatomically realistic 3D ventricular model, complete with a PS, to analyze organ level behavior. PS junctional parameters were tuned to match experimental observations under normal conditions. The timing of EAD induction from phases II and III of repolarization was systematically varied to study propagation or suppression of premature beats under these conditions.
Arrhythmia Induction by Triggered Activity
We observed that triggered activity from diseased PS (slow conduction) led to tachycardia induction when facilitated by temporary CB and/or confronted with sinus activation. Electrotonic interactions at PMJs shortened APD in the distal PS, as has been observed experimentally. Thus a considerable APD gradient developed within the PS, facilitating functional CB in the bundles.
Myerburg et al. (29) reported a progressive increase in Purkinje APD, reaching a maximum in the distal PS before decreasing again at the insertion sites. This distal shortening of APD can be explained by electrotonic loading from the ventricular mass, as reproduced in our model. Based on their observations, they proposed a “gating mechanism” at distal sites, which collectively acted as “limiting segments” for the passage of premature impulses between the two tissues. This was in agreement with Hoffman et al. (18), who suggested that peripheral Purkinje fibers might be especially prone to CB during premature impulses. However, Lazzara et al. (21) later pointed out that in vitro preparations studied by Myerburg et al. (29) were dissected so that a false tendon was the sole bridge between islands of tissue barring conduction through shorter and quicker pathways to the myocardium through interior fibers of the LBB and septal fibers of the RBB. In fact, Lazzara et al. (21) showed that, in the intact PS, the bundle branches are the preferential sites for CB during premature activations. Our findings are in agreement with this observation.
Previous experimental studies have used pharmalogical intervention to test whether tachyarrhythmias with left BBB and right BBB patterns arise from enhanced automaticity or triggered behavior (20). The effects of ATP, nicorandil, and verapamil were evaluated in 17 patients with bundle block ventricular tachycardia (VT), which was found to be EADs and DADs. VT sensitive to ATP but not verapamil was caused by triggered activity from the PS. If a Purkinje fiber has abnormal automaticity in a deep resting potential, an L-type calcium blocker such as verapamil might not affect it, but ATP suppresses such triggered activity (20, 22). Nicorandil was found to abolish VT arising from afterpolarizations in long-QT syndrome. In another case study, BBRT with no structural heart disease was reproducibly terminated by intravenous adenosine (35). During an electrophysiology study, BBRT was repeatedly initiated by isoproterenol infusion, suggesting that it originated from triggered activity (35). Schafferhofer-Steltzer et al. (37) have also shown how to induce EADs in Purkinje fibers by locally superfusing them with isoproterenol solution. In a study of RyR2/RyR2R4496C mutated mice hearts, both calcium overload and adrenergic stimulation produced catecholaminergic polymorphic VT (7). Epicardial breakthroughs and endocardial mapping in these cases revealed that all episodes of catecholaminergic polymorphic VT originated from focal sources in the His-PS, which were EAD/DAD-induced triggered activities.
In their study of unidirectional CB in a one-dimensional cable of modeled cardiomyocytes, Qu et al. (32) systematically demonstrate that the critical refractoriness gradient is larger in cases where the blocked wave front is travelling in the opposite direction relative to the preceding wave. In configurations where BBRT occurred in this study, the spatial dispersion of refractoriness that gave rise to reentry was analogous, since the wave front from the ectopic beat blocked in the bundle branch following a sinus beat. Here, the incidence of BBRT suggests that the APD gradient due to myocardial loading effects in the distal PS, also observed experimentally (29), is a sufficiently large barrier to produce CB.
Favorable Conditions for Reentry
We reduced the excitability (sodium channel conductance) and tissue conductivities in both the PS and myocytes to induce reentry involving the conduction system. It is evident from the results that reentry involving the His-PS cannot occur under healthy conditions, since the effective reentrant path length in such cases is too short. Path length can be increased in the scenario where conduction is hampered in both conductive tissue and the ventricular mass. This is in agreement with clinical observations that sustained BBRT occurs in patients with delays in Purkinje-ventricle activation time and interventricular conductance (6, 27). Anti-arrhythmic drug-induced slowing of CV has been shown to favor the incidence of macro-reentrant VT involving the His-PS (8). In another clinical report, amiodarone-induced slowing of CV within the His-PS led to the occurrence of recurrent BBRT or interfascicular tachycardias in patients (34).
Two types of reentry were observed. In the first, triggered activity conducted through diseased PS and myocardium was confronted by sinus activation, inducing reentry. This could also occur due to interaction with excitation from some other source coming down the bundle or the proximal PS. In the second, the triggered activity itself induced BBRT due to slow conduction and temporary retrograde CB in the distal PS. This case depended on the extent of CV reduction in the ventricles and the PS. This is in accord with the model work of Ten Tusscher and Panfilov (41), who also had to increase effective path length of reentry in their human ventricular model by decreasing PS conductance to 12.5% and lowering INa in both PS and myocardium to 40% to produce BBRT. Likewise, in their early simulation analysis of arrhythmias involving the PS, Berenfeld and Jalife (3) employed a FitzHugh-Nagumo ionic kinetics, which results in reduced excitability due to slower upstroke. Clinical reports have also documented prolonged H-V intervals in most BBRT patients (6, 27), indicating slow conduction through the PS.
In most clinical studies, though, the cause of BBRT initiation has not been clear. Typically, a diseased PS with either of the branches blocked and slower conduction through the other branch are considered as precursors to reentry (27). We observed that ectopic activity originating in the distal PS could result in temporary functional CB in either bundle branch, which provided a substrate for reentry. Structural impairment of the conduction system was not needed, so no H-V prolongation was necessary. Our observations agree with the clinical findings of Li et al. (23), who observed BBRT in patients with normal H-V intervals in the presence of functional conduction impairment. Moreover, our results show that a single ectopic beat, occurring during the specific time window when the distal PS and myocardium are reexcitable, but the proximal PS is still refractory, was sufficient to induce reentry in tissue with reduced CV, which approximate diseased ventricles.
Limitations
While we used a rabbit model and not human, rabbits have similar PS penetration depths as humans, as well as a similar effective electrical size (31). A homogeneous APD distribution was used in the ventricles. This would have altered specific timings for reentry occurrence, but would not alter the basic phenomenon of one-sided BBB. Similarly, variations in the physical structure of the PS and the parameters responsible for PMJ coupling would surely result in the formation of different reentrant circuits, and the size and timing of window of vulnerability might also be affected. Nonetheless, as long as the critical PS features emphasized in this study, rapid propagation, retrograde and anterograde transmission, electrical isolation from the ventricles except at endpoints, and a pronounced gradient in refractoriness from distal to proximal sites due to electrotonic loading, the key phenomena demonstrated, such as arrhythmogenesis from a single well-timed ectopic beat in the PS, would continue to be observed.
Conclusions
We have presented a computer simulation study of arrhythmogenesis due to conduction system disorders in the PS. Ectopic excitation in the distal PS led to triggered activity in the ventricles, which induced reentrant VT when facilitated by functional CB within the proximal PS and/or interaction with sinus activity. A single, well-timed ectopic beat in the distal PS could initiate reentry involving the His-PS, whereas ectopic activity in the proximal PS could not. Initiation of BBRT required extremely slow conduction in the ventricles and temporary CB in one of the bundle branches. This paper describes possible mechanisms of arrhythmia initiation involving the specialized ventricular conduction system. Our findings may help clarify the elements underlying clinical entities, such as BBRT and fascicular tachycardias.
GRANTS
This research was supported by the Natural Sciences and Engineering Research Council of Canada, the Alberta Ingenuity Fund, and the Mathematics of Information Technology and Complex Systems NCE.
DISCLOSURES
E. J. Vigmond holds equity in Cardiosolv LLC.
ACKNOWLEDGMENTS
The authors acknowledge the provision of computational resources by WestGrid.
REFERENCES
- 1. Akhtar M, Damato AN, Batsford WP, Ruskin JN, Ogunkelu JB, Vargas G. Demonstration of re-entry within the his-Purkinje system in man. Circulation 50: 1150–1162, 1974 [DOI] [PubMed] [Google Scholar]
- 2. Aslanidi OV, Stewart P, Boyett MR, Zhang H. Optimal velocity and safety of discontinuous conduction through the heterogeneous purkinje-ventricular junction. Biophys J 97: 20–39, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berenfeld O, Jalife J. Purkinje-muscle reentry as a mechanism of polymorphic ventricular arrhythmias in a 3-dimensional model of the ventricles. Circ Res 82: 1063–1077, 1998 [DOI] [PubMed] [Google Scholar]
- 4. Boyle P, Deo M, Plank G, Vigmond E. The role of the Purkinje system in the response of the quiescent ventricles to defibrillation-strength shocks: a computer modeling study. Ann Biomed Eng 38: 456–468, 2010 [DOI] [PubMed] [Google Scholar]
- 5. Cabo C, Barr RC. Propagation model using the DiFrancesco-Noble equations: comparison to reported experimental results. Med Biol Eng Comput 30: 292–302, 1992 [DOI] [PubMed] [Google Scholar]
- 6. Caceres J, Jazayeri M, McKinnie J, Avitall B, Denker ST, Tchou P, Akhtar M. Sustained bundle branch reentry as a mechanism of clinical tachycardia. Circulation 79: 256–270, 1989 [DOI] [PubMed] [Google Scholar]
- 7. Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O'Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, Priori SG, Jalife J. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res 101: 1039–1048, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chalvidan T, Cellarier G, Deharo JC, Colin R, Savon N, Barra N, Peyre JP, Djiane P. His-Purkinje system reentry as a proarrhythmic effect of flecainide. Pacing Clin Electrophysiol 23: 530–533, 2000 [DOI] [PubMed] [Google Scholar]
- 9. Clerc L. Directional differences of impulse spread in trabecular muscle from mammalian heart. J Physiol 255: 335–346, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clusin WT. Calcium and cardiac arrhythmias: DADs, EADs, and alternans. Crit Rev Clin Lab Sci 40: 337–375, 2003 [DOI] [PubMed] [Google Scholar]
- 11. Coghlan HC, Coghlan AR, Buckberg GD, Cox JL. “The electrical spiral of the heart”: its role in the helical continuum, the hypothesis of the anisotropic conducting matrix. Eur J Cardiothorac Surg 29: S178–S187, 2006 [DOI] [PubMed] [Google Scholar]
- 12. Deo M, Boyle P, Plank G, Vigmond E. Arrhythmogenic mechanisms of the purkinje system during electric shocks: a modeling study. Heart Rhythm 6: 1782–1789, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DiFrancesco D, Nobel D. A model of cardiac electrical activity incorporating ionic pumps and concentration changes. Philos Trans R Soc Lond B Biol Sci 307: 353–398, 1985 [DOI] [PubMed] [Google Scholar]
- 14. el Sherif N, Zeiler RH, Craelius W, Gough WB, Henkin R. QTU prolongation and polymorphic ventricular tachyarrhythmias due to bradycardia-dependent early afterdepolarizations. Afterdepolarizations and ventricular arrhythmias. Circ Res 63: 286–305, 1988 [DOI] [PubMed] [Google Scholar]
- 15. Gibb WJ, Wagner MB, Lesh MD. Modeling triggered cardiac activity: an analysis of the interactions between potassium blockade, rhythm pauses, and cellular coupling. Math Biosci 137: 101–133, 1996 [DOI] [PubMed] [Google Scholar]
- 16. Gough WB, el Sherif N. Dependence of delayed afterdepolarizations on diastolic potentials in ischemic purkinje fibers. Am J Physiol Heart Circ Physiol 257: H770–H777, 1989 [DOI] [PubMed] [Google Scholar]
- 17. Gupta AK, Vajifdar BU, Vora AM. Bundle branch re-entrant ventricular tachycardia in a patient with structurally normal heart. Indian Heart J 51: 80–82, 1999 [PubMed] [Google Scholar]
- 18. Hoffman BF, Moore EN, Stuckey JH, Cranefield PF. Functional properties of the atrioventricular conduction system. Circ Res 13: 308–328, 1963 [DOI] [PubMed] [Google Scholar]
- 19. January C, Riddle J, Salata J. A model for early afterdepolarizations with the calcium channel agonist bayk8644. Circ Res 62: 563–571, 1988 [DOI] [PubMed] [Google Scholar]
- 20. Kobayashi Y, Yazawa T, Adachi T, Kawamura M, Ryu S, Asano T, Obara C, Katagiri T. Ventricular arrhythmias with left bundle branch block pattern and inferior axis: assessment of their mechanisms on the basis of response to ATP, nicorandil and verapamil. Jpn Circ J 64: 835–841, 2000 [DOI] [PubMed] [Google Scholar]
- 21. Lazzara R, El-Sherif N, Befeler B, Scherlag B. Regional refractoriness within the ventricular conduction system. An evaluation of the “gate” hypothesis. Circ Res 39: 254–262, 1976 [DOI] [PubMed] [Google Scholar]
- 22. Lerman B, Belardinelli L, West G, Berne R, DiMarco J. Adenosine-sensitive ventricular tachycardia: evidence suggesting cyclic AMP-mediated triggered activity. Circulation 74: 270–280, 1986 [DOI] [PubMed] [Google Scholar]
- 23. Li YG, Grnefeld G, Israel C, Bogun F, Hohnloser SH. Bundle branch reentrant tachycardia in patients with apparent normal his-Purkinje conduction: the role of functional conduction impairment. J Cardiovasc Electrophysiol 13: 1233–1239, 2002 [DOI] [PubMed] [Google Scholar]
- 24. Li ZY, Maldonado C, Zee-Cheng C, Hiromasa S, Kupersmith J. Purkinje fibre-papillary muscle interaction in the genesis of triggered activity in a guinea pig model. Cardiovasc Res 26: 543–548, 1992 [DOI] [PubMed] [Google Scholar]
- 25. Lu HR, Marin R, Saels A, Clerck FD. Species plays an important role in drug-induced prolongation of action potential duration and early afterdepolarizations in isolated purkinje fibers. J Cardiovasc Electrophysiol 12: 93–102, 2001 [DOI] [PubMed] [Google Scholar]
- 26. Mahajan A, Shiferaw Y, Sato D, Baher A, Olcese R, Xie LH, Yang MJ, Chen PS, Restrepo JG, Karma A, Grafinkel A, Qu Z, Weiss J. A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys J 94: 392–410, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mazur A, Kusniec J, Strasberg B. Bundle branch reentrant ventricular tachycardia. Indian Pacing Electrophysiol J 5: 86–95, 2005 [PMC free article] [PubMed] [Google Scholar]
- 28. Monserrat M, Saiz J, Ferrero JMJ, Ferrero JM, Thakor NV. Ectopic activity in ventricular cells induced by early afterdepolarizations developed in Purkinje cells. Ann Biomed Eng 28: 1343–1351, 2000 [DOI] [PubMed] [Google Scholar]
- 29. Myerburg RJ, Stewart JW, Hoffman BF. Electrophysiological properties of the canine peripheral A-V conducting system. Circ Res 26: 361–378, 1970 [DOI] [PubMed] [Google Scholar]
- 30. Nordin C. Computer model of electrophysiological instability in very small heterogeneous ventricular syncytia. Am J Physiol Heart Circ Physiol 272: H1838–H1856, 1997 [DOI] [PubMed] [Google Scholar]
- 31. Panfilov AV. Is heart size a factor in ventricular fibrillation? Or how close are rabbit and human hearts? Heart Rhythm 3: 862–864, 2006 [DOI] [PubMed] [Google Scholar]
- 32. Qu Z, Garfinkel A, Weiss JN. Vulnerable window for conduction block in a one-dimensional cable of cardiac cells. 1. Single extrasystoles. Biophys J 91: 793–804, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ramanathan C, Jia P, Ghanem R, Ruy K, Rudy Y. Activation and repolarization of the normal human heart under complete physiological conditions. Proc Natl Acad Sci USA 103: 6309–6314, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reithmann C, Hahnefeld A, Oversohl N, Ulbrich M, Remp T, Steinbeck G. Reinitiation of ventricular macroreentry within the his-Purkinje system by back-up ventricular pacing-a mechanism of ventricular tachycardia storm. Pacing Clin Electrophysiol 30: 225–235, 2007 [DOI] [PubMed] [Google Scholar]
- 35. Rubenstein DS, Burke MC, Kall JG, Kinder CA, Kopp DE, Wilber DJ. Adenosine-sensitive bundle branch reentry. J Cardiovasc Electrophysiol 8: 80–88, 1997 [DOI] [PubMed] [Google Scholar]
- 36. Saiz J, Ferrero JJM, Monserrat M, Ferrero JM, Thakor NV. Influence of electrical coupling on early afterdepolarizations in ventricular myocytes. IEEE Trans Biomed Eng 46: 138–147, 1999 [DOI] [PubMed] [Google Scholar]
- 37. Schafferhofer-Steltzer I, Hofer E, Huelsing DJ, Bishop S, Pollard A. Contributions of Purkinje-myocardial coupling to suppression and facilitation of early afterdepolarization-induced triggered activity. IEEE Trans Biomed Eng 52: 1522–1531, 2005 [DOI] [PubMed] [Google Scholar]
- 38. Simons GR, Sorrentino RA, Zimerman LI, Wharton JM, Natale A. Bundle branch reentry tachycardia and possible sustained interfascicular reentry tachycardia with a shared unusual induction pattern. J Cardiovasc Electrophysiol 7: 44–50, 1996 [DOI] [PubMed] [Google Scholar]
- 39. Szabo B, Kovacs T, Lazzara R. Role of calcium loading in early afterdepolarizations generated by Cs+ in canine and guinea pig Purkinje fibers. J Cardiovasc Electrophysiol 6: 796–812, 1995 [DOI] [PubMed] [Google Scholar]
- 40. Szabo B, Sweidan R, Rajagopalan CV, Lazzara R. Role of Na+:Ca2+ exchange current in Cs+-induced early afterdepolarizations in Purkinje fibers. J Cardiovasc Electrophysiol 5: 933–944, 1994 [DOI] [PubMed] [Google Scholar]
- 41. Ten Tusscher K, Panfilov AV. Modelling of the ventricular conduction system. Prog Biophys Mol Biol 96: 152–170, 2008 [DOI] [PubMed] [Google Scholar]
- 42. Touboul P, Kirkorian G, Atallah G, Moleur P. Bundle branch reentry: a possible mechanism of ventricular tachycardia. Circulation 67: 674–680, 1983 [DOI] [PubMed] [Google Scholar]
- 43. Vetter F, McCulloch A. Three-dimensional analysis of regional cardiac function: a model of rabbit ventricular anatomy. Prog Biophys Mol Biol 69: 157–183, 1998 [DOI] [PubMed] [Google Scholar]
- 44. Vigmond E, Aguel F, Trayanova N. Computational techniques for solving the bidomain equations in three dimensions. IEEE Trans Biomed Eng 49: 1260–1269, 2002 [DOI] [PubMed] [Google Scholar]
- 45. Vigmond EJ, Clements C. Construction of a computer model to investigate sawtooth effects in the Purkinje system. IEEE Trans Biomed Eng 54: 389–399, 2007 [DOI] [PubMed] [Google Scholar]
- 46. Vigmond EJ, Hughes M, Plank G, Leon LJ. Computational tools for modeling electrical activity in cardiac tissue. J Electrocardiol 36, Suppl: 69–74, 2003 [DOI] [PubMed] [Google Scholar]
- 47. Vigmond EJ, Weber Dos Santos R, Prassl AJ, Deo M, Plank G. Solvers for the cardiac bidomain equations. Prog Biophys Mol Biol 96: 3–18, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Volders P, Vos M, Szabo B, Sipido K, Groot S, Gorgels A, Wellens H, Lazzara R. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: time to revise current concepts. Cardiovasc Res 42: 376–392, 2000 [DOI] [PubMed] [Google Scholar]
- 49. Wagner MB, Gibb WJ, Lesh MD. A model study of propagation of early afterdepolarizations. IEEE Trans Biomed Eng 42: 991–997, 1995 [DOI] [PubMed] [Google Scholar]
- 50. Welch WJ, Strasberg B, Coelho A, Rosen KM. Sustained macroreentrant ventricular tachycardia. Am Heart J 104: 166–169, 1982 [DOI] [PubMed] [Google Scholar]
- 51. Wiedmann RT, Tan RC, Joyner RW. Discontinuous conduction at Purkinje-ventricular muscle junction. Am J Physiol Heart Circ Physiol 271: H1507–H1516, 1996 [DOI] [PubMed] [Google Scholar]
- 52. Wit AL, Rosen MR. Pathophysiologic mechanisms of cardiac arrhythmias. Am Heart J 106: 798–811, 1983 [DOI] [PubMed] [Google Scholar]
- 53. Zeng J, Rudy Y. Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys J 68: 949–964, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]