

Abstract
Pulmonary arterial hypertension (PAH) is a life-threatening disease characterized by a sustained elevation in the pulmonary artery pressure and subsequent right heart failure. The activation of Rho/Rho-kinase activity and the beneficial effect of Rho-kinase inhibition have been demonstrated in several experimental models of pulmonary hypertension. However, it remains unclear whether Rho-kinase inhibitors can also be used against pulmonary hypertension associated with mutations in the type II bone morphogenetic protein receptor (BMPRII) gene. Transgenic mice expressing a dominant-negative BMPRII gene (with an arginine to termination mutation at amino acid 899) in smooth muscle by a tetracycline-gene switch system (SM22-tet-BMPR2R899X mice) were examined. They developed an elevated right ventricular systolic pressure (RVSP), right ventricular (RV) hypertrophy, muscularization of small pulmonary arteries, and an associated disturbed blood flow in their lungs. The Rho/Rho-kinase activity and Smad activity were determined by a Western blot analysis by detecting GTP-RhoA and the phosphorylation of myosin phosphatase target subunit 1, Smad1, and Smad2. In the lungs of SM22-tet-BMPR2R899X mice, the Rho/Rho-kinase activity was elevated significantly, whereas the Smad activity was almost unchanged. Fasudil, a Rho-kinase inhibitor, significantly decreased RVSP, alleviated RV hypertrophy and muscularization of small pulmonary arteries, and improved blood flow in SM22-tet-BMPR2R899X mice, although it did not alter Smad signaling. Our study demonstrates that Rho/Rho-kinase signaling is activated via a Smad-independent pathway in an animal model of pulmonary hypertension with a BMPRII mutation in the cytoplasmic tail domain. Rho-kinase inhibition is therefore a possible therapeutic approach for the treatment of PAH associated with genetic mutation.
Keywords: pulmonary circulation, Rho-kinase inhibitor, bone morphogenetic protein signaling
pulmonary arterial hypertension (PAH) is clinically characterized by a sustained elevation in the mean pulmonary artery pressure and subsequent right heart failure leading to significant morbidity and mortality. The histological features of PAH reveal a proliferation of endothelial and smooth muscle cells with vascular remodeling. PAH is usually fatal within three years if untreated. Whereas new therapeutic approaches have prolonged patient survival, the prognosis remains far from satisfactory.
At least 6% of all PAH cases are familial (FPAH) (26), and the causative gene for FPAH was identified as BMPR2, which encodes a type II bone morphogenetic protein receptor belonging to the transforming growth factor-β (TGF-β) superfamily, which plays a critical role in cell differentiation and cell growth (5, 9). Because only 10–20% of subjects with a BMPR2 mutation develop PAH, it is believed that other genes, genetic polymorphisms, or environmental factors are involved in the development of pulmonary hypertension (PH).
We previously reported that transgenic mice expressing a dominant-negative type II bone morphogenetic protein receptor (BMPRII) in smooth muscle show the phenotype of severe PH and right ventricular hypertrophy in the absence of other stimuli (31). We also demonstrated pulmonary vascular changes resembling the plexiform lesion and dysregulation of gene expression in mice expressing a dominant-negative BMPRII with a stop mutation in the cytoplasmic tail domain (BMPR2R899X) (32).
Rho-kinase is one of the downstream effectors of the small G protein Rho, which regulates gene transcription, cell differentiation, proliferation, migration, and smooth muscle contraction (11, 14, 18, 24). It is also known that Rho/Rho-kinase signaling is a major regulator of vascular tone (25); this is true in the pulmonary circulation as well (30). Rho-kinase inhibition has shown beneficial effects in treatment of hypoxia-induced or monocrotaline-induced PH (1, 2, 6, 21). We wanted to determine whether the administration of the Rho-kinase inhibitor fasudil may also be effective against PH in cases with a mutation in the BMPRII gene.
MATERIALS AND METHODS
All procedures in the present study were approved by the Chiba University Institutional Animal Care and Use Committee.
Transgenic mice.
The expression of a dominant-negative BMPRII gene was controlled by a tetracycline-gene switch system because of the necessity of bone morphogenetic protein (BMP) signaling in the early development of embryos. TetO7-BMPR2R899X transgenic mice were generated as previously described (32). R899X is an arginine to termination mutation at amino acid 899 in the BMPRII tail domain found in family US33 (9), thus leaving the Smad pathway intact. To induce the expression of BMPR2R899X in smooth muscle, we crossed TetO7-BMPR2R899X mice to SM22-reverse tetracycline transcriptional activator (rtTA) transgenic mice with smooth muscle-specific expression of the rtTA. Both transgenic mice were on the FVB/N background. Double-transgenic mice, SM22-tet-BMPR2R899X mice, were generated and used for all experiments.
Experimental design.
Double-transgenic mice were divided into two groups. One group was fed doxycycline in their chow (1 g/kg) from 3 wk after birth. The other group was fed doxycycline in their chow from 3 wk and were treated with fasudil (Asahi Kasei Pharma, Tokyo, Japan), a Rho-kinase inhibitor (100 mg·kg−1·day−1), in drinking water from 10 wk of age for 14 days. Wild-type FVB/N mice fed doxycycline in their chow were prepared as controls. Phenotyping was performed when all mice were 12 wk of age. Some mice at 10 wk of age (n = 7) were challenged with a longer treatment with fasudil for 28 days, i.e., from 10 wk of age to 14 wk of age, to see whether the longer treatment led to different effects.
Heart catheterization.
Mice were initially anesthetized with an intraperitoneal injection of pentobarbital sodium (30 mg/kg) and placed on a heated table (ATB-1100; Nihon Kohden, Tokyo, Japan) to maintain their temperature during the procedure. To measure the right ventricular pressure, the right jugular vein was surgically exposed, and a 1.2-Fr pressure catheter (FT111B; Scisense, London, Ontario, Canada) connected to AP-621G (Nihon Kohden) was inserted in the right ventricle (RV) through the incision in the right jugular vein. To measure the left ventricular pressure, the left carotid artery was exposed, and a 1.2-Fr pressure catheter (Scisense) was inserted in the left ventricle (LV). The placement of the catheter tip was determined through observation of the pressure. The catheter was fixed with suture. The right ventricular systolic pressure (RVSP) and the left ventricular systolic pressure (LVSP) were recorded using the UAS-108S software package (Unique Medical, Tokyo, Japan).
Assessment of right ventricular hypertrophy.
After death, saline containing 5 U/ml heparin was flushed via the RV, and the heart was removed. The RV wall was separated from the LV wall and the ventricular septum (S). The degree of right ventricular hypertrophy was assessed as the ratio of the weight of the RV wall and that of the LV wall and ventricular septum (LV + S).
Morphometric analysis.
The right main bronchus was tied off and snap-frozen after removal of the heart. The left lung was inflated with 0.5% low-melting agarose in PBS via the trachea at a pressure of 25 cmH2O. The left lung was fixed in 10% buffered formalin for 48 h and then was embedded in paraffin wax. Serial 4-μm sections were stained with hematoxylin and eosin according to standard histological procedures. Immunohistochemistry was performed using the following primary antibodies: von Willebrand factor (1:200, goat polyclonal antibody; sc-8068; Santa Cruz Biotechnology, Santa Cruz, CA) and α-smooth muscle actin (1:100, rabbit monoclonal antibody; 1184–1; Epitomics). Pulmonary arteries ranging from 20 to 100 μm in diameter were classified into fully muscular (>75%), partially muscular (25–75%), or nonmuscular (<25%) to evaluate the muscularization of the pulmonary arteries using Eclipse E400 (Nikon) and Image-Pro plus instruments (Nihon Roper, Tokyo, Japan).
Fluorescent microangiography.
The mice were anesthetized with an intraperitoneal injection of a high dose of pentobarbital sodium (50 mg/kg), and the rib cage was cut away. A small incision was made at the left atrium, and saline containing 5 U/ml heparin was flushed via the RV. Next, a 45°C solution of 10% (vol/vol) fluorescent microspheres (FluoSpheres carboxylate-modified microspheres, 0.2 μm, yellow-green fluorescent; F8811; Molecular Probes, Eugene, OR) in 1% low-melting agarose in PBS was slowly flushed via the RV through the lungs until it flowed out of the left atrium. The lungs were immediately inflated with 0.8% low-melting agarose in PBS via the trachea at a pressure of 25 cmH2O and cooled with ice to congeal the gel. The lungs were fixed in 4% paraformaldehyde for 48 h and then soaked in a solution of 30% (wt/vol) sucrose in PBS. The lungs were frozen using optimum-cutting temperature compound (Sakura Finetek Japan, Tokyo, Japan). The sections were cut on a cryostat at 40 μm. The brightness of the green fluorescence was evaluated semiquantitatively using the Image J software program (National Institutes of Health, Bethesda, MD) according to the manufacturer's instructions to quantify the blood flow in the lungs. In brief, 10 pictures from 5 mice in each group were randomly chosen. The background brightness was subtracted from the pictures, they were divided into smaller regions (66 × 52 pixels), and 25 regions were randomly chosen for the analysis. In each region, the area of green fluorescence was traced, and the intensity of the green brightness was measured. The evaluation was done by two independent pulmonologists in a double-blind manner.
Western blot analysis.
The lung tissue specimens were homogenized in lysis buffer containing 50 mM Tris·HCl, pH 8.0, 0.5% Triton X-100, 4 M urea, 1 mM EDTA, 150 mM NaCl, proteinase inhibitor cocktail (Calbiochem), and phosphatase inhibitor cocktail (Nacalai tesque). Homogenates were centrifuged at 10,000 g for 10 min. The protein concentration of the supernatant was determined by the Bradford method (Bio-Rad Protein Assay; Nippon Bio-Rad Laboratories, Tokyo, Japan). Lysates were diluted with sample buffer (1:1) containing 100 mM Tris·HCl, pH 6.8, 4% SDS, 20% glycerol, and 12% β-mercaptoethanol and then boiled for 5 min. The samples were separated on 10% or 4–12% gradient Tris-glycine gels (Invitrogen Japan, Tokyo, Japan) and transferred to nitrocellulose membranes (Invitrogen Japan). Membranes were probed with the following primary antibodies: phospho-myosin phosphatase target subunit 1 on Thr696 (MYPT1Thr696, 1:2,000, rabbit polyclonal antibody; ABS45; Upstate Biotechnology), myosin phosphatase target subunit 1 (MYPT1) (1:1,000, rabbit polyclonal antibody; sc-25618; Santa Cruz Biotechnology), phosho-Smad1Ser463/465 (1:1,000, rabbit polyclonal antibody; no. 9511; Cell Signaling Technology), phospho-Smad2Ser465/467 (1:1,000, rabbit polyclonal antibody; no. 3101; Cell Signaling Technology), and β-actin (1:200, rabbit polyclonal antibody; sc-130656; Santa Cruz Biotechnology). The membranes were washed and incubated with a secondary antibody (1:100,000, horseradish peroxidase-conjugated anti-rabbit IgG; 111–035-003; Jackson ImmunoResearch Laboratories) and then the SuperSignal West Dura Extended Duration Substrate (Pierce, Rockford, IL). Chemiluminescence was detected by a LAS-1000 instrument (Fuji Film, Tokyo, Japan) and quantified by densitometry.
Measurement of RhoA activity.
RhoA activity was assessed using a RhoA activation assay kit (Cytoskeleton) according to the manufacturer's recommendations. The lung tissue specimens were homogenized in lysis buffer (50 mM Tris, pH 7.5, 10 mM MgCl2, 0.5 M NaCl, and 2% Igepal) including 1:100 diluted protease inhibitor cocktail (62 μg/ml leupeptin, 62 μg/ml pepstatin A, 14 mg/ml benzamidine, and 12 mg/ml tosyl arginine methyl ester). Homogenates were centrifuged at 10,000 g for 2 min. The protein concentration of the supernatant was determined by the BCA protein assay kit (Pierce). Lysates were incubated with rhotekin-RBD beads for 1 h. Samples were pelleted by centrifugation at 5,000 g for 1 min. The supernatant was removed, and the pellet was washed with wash buffer (25 mM Tris, pH 7.5, 30 mM MgCl2, and 40 mM NaCl). The pellet was then diluted with sample buffer (1:1) containing 100 mM Tris·HCl, pH 6.8, 4% SDS, 20% glycerol, and 12% β-mercaptoethanol and boiled for 2 min. The samples were separated on 12% Tris-glycine gels (Invitrogen Japan) and transferred to nitrocellulose membranes (Invitrogen Japan). Membranes were probed with an anti-RhoA monoclonal antibody (1:500, ARH03; Cytoskeleton). The membranes were washed and incubated with a secondary antibody (1:20,000, horseradish peroxidase-conjugated anti-mouse IgG; 115–035-003; Jackson ImmunoResearch Laboratories) and then the SuperSignal West Dura Extended Duration Substrate (Pierce). Chemiluminescence was detected by a LAS-1000 instrument (Fuji Film) and quantified by densitometry.
Data analysis.
All results are expressed as means ± SE. The data were analyzed using one-way ANOVA or the Kruskal-Wallis test. If a statistically significant effect was found, post hoc analysis was performed to detect the difference between the groups. A value of P < 0.05 was considered to be statistically significant.
RESULTS
The effects of fasudil on hemodynamics and right ventricular hypertrophy.
In a previous report, we demonstrated that approximately one-third of SM22-tet-BMPR2R899X mice that were fed doxycycline from 4 wk of age until 13 wk of age showed elevated RVSP, accompanied with smaller pulmonary arterial occlusion (32). Hence, we first confirmed this using the present experimental design (i.e., in mice fed doxycycline from 3 wk of age for 9 wk). We also examined whether the mice treated with doxycycline until 10 wk of age, when the fasudil treatment was started, developed PH. Hemodynamic measurements and evaluation of RV hypertrophy were performed to confirm the impact of dominant-negative BMPRII on pulmonary circulation and to investigate the effects of fasudil (Fig. 1). In general, the majority of SM22-tet-BMPR2R899X mice in the present study developed PH by the time they were 10 wk of age (RVSP = 39.0 ± 1.7 mmHg, n = 5). The high incidence of PH in the present study was probably due to the fact that the mice started receiving doxycycline at a young age. Starting doxycycline at weaning likely led to an impact on late lung development (31). The RVSP of mice fed doxycycline from 3 wk of age to 12 wk of age further increased and was significantly higher compared with that in the controls (SM22-tet-BMPR2R899X: 43.3 ± 1.0 mmHg, n = 20; controls: 26.5 ± 0.8 mmHg, n = 10). Fasudil significantly decreased the RVSP in SM22-tet-BMPR2R899X mice (37.3 ± 0.9 mmHg, n = 14), although the RVSP in SM22-tet-BMPR2R899X mice treated with fasudil was still significantly higher than that in the controls. RV hypertrophy developed in SM22-tet-BMPR2R899X mice as a consequence of PH. The ratio of RV and LV + ventricular septum (RV/LV + S) was significantly elevated in SM22-tet-BMPR2R899X mice (0.34 ± 0.01) compared with that in the controls (0.22 ± 0.01). The RV/LV + S in SM22-tet-BMPR2R899X mice treated with fasudil significantly decreased (0.28 ± 0.01) as a consequence of an improvement in PH. We also used a longer treatment with fasudil (for 4 wk) but found no further decrease in the RVSP (36.7 ± 1.0 mmHg, n = 7) or the RV/LV + S (0.28 ± 0.01, n = 7) was achieved compared with the mice treated for 2 wk. No differences in the LVSP were found between the groups (controls, SM22-tet-BMPR2R899X, and SM22-tet-BMPR2R899X with fasudil = 118.5 ± 4.9, 118.1 ± 4.2, and 116.6 ± 5.3 mmHg, respectively, n = 5).
Fig. 1.
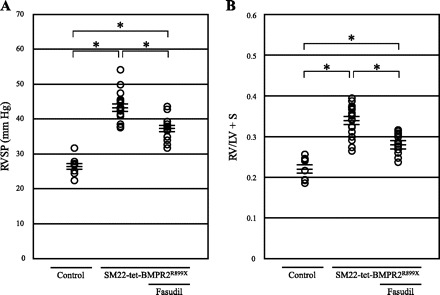
Treatment with fasudil alleviates pulmonary hypertension and right ventricular (RV) hypertrophy in transgenic mice expressing a dominant-negative type II bone morphogenetic protein receptor gene (with an arginine to termination mutation at amino acid 899) in smooth muscle by a tetracycline-gene switch system (SM22-tet-BMPR2R899X). A: right ventricular systolic pressure (RVSP) in SM22-tet-BMPR2R899X mice was elevated significantly (43.3 ± 1.0 mmHg, n = 20) compared with that in the controls (26.5 ± 0.8 mmHg, n = 10). Fasudil decreased RVSP in SM22-tet-BMPR2R899X mice (37.3 ± 0.9 mmHg, n = 14). B: the ratio of RV and left ventricle + ventricular septum (RV/LV + S) in SM22-tet-BMPR2R899X mice was elevated significantly (0.34 ± 0.01) compared with that in the controls (0.22 ± 0.01). Fasudil decreased RV/LV + S in SM22-tet-BMPR2R899X mice (0.28 ± 0.01). *P < 0.01.
The effects of fasudil on the muscularization of pulmonary arteries.
We assessed the degree of muscularization of pulmonary arteries ranging from 20 to 100 μm in diameter by immunohistological evaluation. The proportions of partially and fully muscularized pulmonary arteries were significantly increased in SM22-tet-BMPR2R899X mice (26.5 and 4.3%, respectively) compared with those in the controls (7.8 and 0.5%, respectively) (Fig. 2). Treatment with fasudil decreased the proportions of partially and fully muscularized pulmonary arteries (19.5 and 1.8%, respectively), although these values were still higher than those in the controls.
Fig. 2.
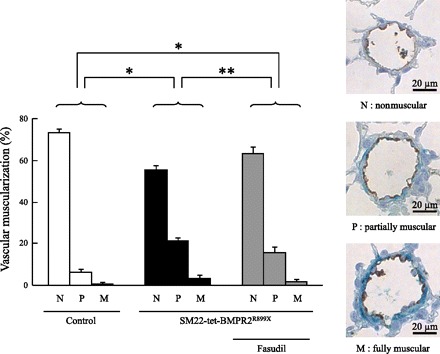
The effects of fasudil on the degree of muscularization. The proportions of partially and fully muscularized pulmonary arteries significantly increased in SM22-tet-BMPR2R899X mice (26.5 and 4.3%, respectively) compared with those in the controls (7.8 and 0.5%, respectively). Fasudil decreased the proportions of partially and fully muscularized pulmonary arteries (19.5 and 1.8%, respectively). N, nonmuscular; P, partially muscular; M, fully muscular. Brown, von Willebrand factor; green, α-smooth muscle actin. *P < 0.0001 and **P < 0.01.
Fluorescent microangiography.
The specific vascular structural changes in SM22-tet-BMPR2R899X mice include the pulmonary artery filling with CD45+ cells, although this is not frequently detected (32). In the present study, these lesions were rarely observed, probably because of the differences in the experimental design (data not shown). However, the more common feature of the vascular lesions seen in this mouse model, namely the occlusion of smaller vessels, was reproducibly observed in this study. Fluorescent microangiography with 0.2 μm fluorescent microspheres was performed to visualize the blood flow in the lungs. A considerable decrease in the blood perfusion in the lungs of SM22-tet-BMPR2R899X mice was observed compared with the control mice (Fig. 3, A and B). The treatment with fasudil apparently improved the blood flow. However, complete recovery of microcirculation throughout the lung was not achieved, and some disturbance of the blood flow was still observed.
Fig. 3.
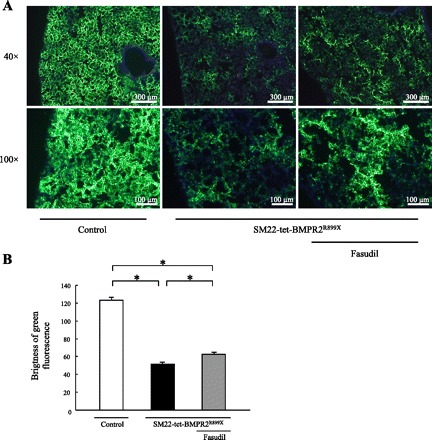
Fluorescent microangiography with 0.2 μm yellow-green fluorescent microsphere. A: the blood flow of SM22-tet-BMPR2R899X mice decreased considerably. Treatment with fasudil apparently improved the blood flow. B: a semiquantitative analysis was performed by evaluating the brightness of the green fluorescence. *P < 0.01.
Rho-kinase activity and the effect of fasudil on Rho-kinase activity.
Rho-kinase inhibits myosin light-chain phosphatase, resulting in phosphorylation of MYPT1Thr696 (12, 28). To determine the Rho-kinase activity, the level of phosphorylated MYPT1Thr696 was measured by a Western blot analysis (Fig. 4A). The level of MYPT1 phosphorylation was upregulated in SM22-tet-BMPR2R899X mice, indicating that Rho-kinase activity is elevated in these mice. In contrast, treatment with fasudil apparently decreased the level of MYPT1 phosphorylation.
Fig. 4.
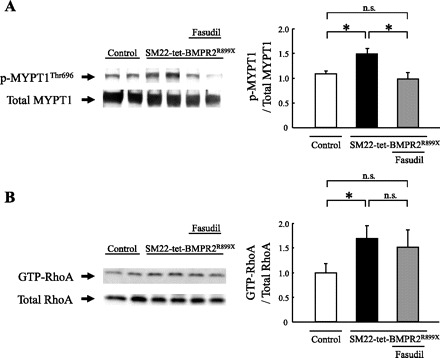
Rho-kinase activity and RhoA activity. A: to determine the Rho-kinase activity, the level of phosphorylated myosin phosphatase target subunit 1 (MYPT1) on Thr696 (MYPT1Thr696) was measured by a Western blot analysis. The bands for phospho-MYPT1Thr696 were normalized to those of total MYPT1. The level of MYPT1 phosphorylation was upregulated in SM22-tet-BMPR2R899X mice. Treatment with fasudil apparently decreased the level of MYPT1 phosphorylation. B: to determine the RhoA activity, the level of GTP-RhoA was measured. The bands for GTP-RhoA were normalized to those of total RhoA. The level of GTP-RhoA was upregulated in SM22-tet-BMPR2R899X mice. Treatment with fasudil did not affect the level of GTP-RhoA. *P < 0.05. ns, Not significant.
RhoA activity and the effect of fasudil on RhoA activity.
After confirming that Rho-kinase was overactive in SM22-tet-BMPR2R899X mice, we next wanted to determine whether Rho, the upstream regulator of Rho-kinase, was elevated in these mice. To determine the RhoA activity, the level of GTP-RhoA, which activates Rho-kinase, was measured by using rhotekin-RBD beads. The level of GTP-RhoA was found to be upregulated in SM22-tet-BMPR2R899X mice, indicating that RhoA activity is elevated in these mice (Fig. 4B). However, treatment with fasudil did not significantly alter the level of GTP-RhoA.
Smad signaling.
BMPRII signaling transfers extracellular signals through the Smad pathway, leading to gene transcription (19, 23). The phosphorylation of Smad1, which is a downstream effector of BMPRII, and the phosphorylation of Smad2, which is an effector of the Smad pathway mainly activated by TGF-β stimulation, were detected by a Western blot analysis. The phospho-Smad1Ser463/465 and phospho-Smad2Ser465/467 levels were normalized to that of β-actin. No differences were observed between the controls and SM22-tet-BMPR2R899X mice in the phosphorylation of Smad1 and Smad2. In addition, treatment with fasudil did not affect the Smad phosphorylation (Fig. 5, A and B).
Fig. 5.
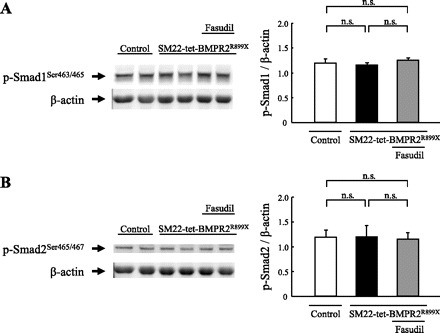
Smad phosphorylation. A: no differences in the Smad1 phosphorylation levels were observed between the controls and SM22-tet-BMPR2R899X mice. Treatment with fasudil did not affect the level of Smad1 phosphorylation. B: no differences in the Smad2 phosphorylation levels were observed between the controls and SM22-tet-BMPR2R899X mice. Treatment with fasudil did not affect the level of Smad2 phosphorylation. The bands for phosho-Smad1Ser463/465 and phospho-Smad2Ser465/467 were normalized to those of β-actin.
DISCUSSION
The present study demonstrated that the Rho-kinase inhibitor fasudil alleviates PH, RV hypertrophy, and muscularization of pulmonary arteries induced by a BMPRII mutation in the cytoplasmic tail domain in a transgenic animal model. Atkinson et al. (3) reported the expression of BMPRII to be markedly reduced in the lungs of PAH patients regardless of the presence of BMPRII mutations. Yang et al. (34) demonstrated that the growth-suppressive response to BMP4 was attenuated in pulmonary artery smooth muscle cells isolated from PAH patients both with and without BMPRII mutation (34). Moreover, disruption of BMP/TGF-β signaling was demonstrated in two rat models of PH (15). The important role of Rho-kinase and the beneficial effects of Rho-kinase inhibition for PAH have attracted increasing attention (24); increased Rho-kinase activity and the beneficial effects of Rho-kinase inhibition have been reported in some rodent models of PH (1, 2, 6, 21); and an acute vasodilatory effect of a Rho-kinase inhibition has also been demonstrated in PAH patients (8, 10). Because it is supposed that BMPRII mutation or reduced BMPRII expression plays a key role in the development of PAH, not only in FPAH patients but also in idiopathic pulmonary arterial hypertension patients, we wanted to elucidate whether a Rho-kinase inhibition could be effective against PAH caused by a dominant-negative BMPRII.
In the present study, we observed elevated RVSP and RV hypertrophy in SM22-tet-BMPR2R899X mice with activation of Rho/Rho-kinase signaling. However, the effects of the Rho-kinase inhibition on decreasing RVSP and improving RV hypertrophy were limited, as it was shown that RVSP and the degree of RV hypertrophy in SM22-tet-BMPR2R899X mice treated with fasudil were still higher than those of the controls. These limitations were also observed in the assessment of muscularization of the pulmonary arteries and the blood flow in the lungs. We believe that this is because BMPRII mutation works through both increased tone (29, 35) and through structural alterations, including vascular stiffening, dropout, and occlusion (17, 32). Our data suggest that chronic fasudil is doing an excellent job of reversing tone-related defects (Figs. 1 and 4) but has little impact on the progressive loss of vessels (Fig. 3). We are unclear as to the mechanism of vascular dropout; complex vascular lesions were not seen in these mice, possibly because they are younger and at lower altitude than in previous studies.
It remains unclear why Rho-kinase is upregulated in SM22-tet-BMPR2R899X mice. We hypothesize the following mechanism (Fig. 6): competing for common-partner Smad (co-Smad; Smad4), which receptor-mediated Smads (R-Smads) must complex with to translocate to the nucleus, BMP signaling via Smad1/5/8 antagonizes TGF-β signaling via Smad2/3. This interaction and a reduction in BMP signaling, which induces an increase in TGF-β signaling, are considered to be a possible mechanism underlying the development of PAH in FPAH patients (20). However, BMPRII mutations with truncation of the cytoplasmic tail domain have been reported to retain the transcriptional activity of BMPRII, although the BMPRII mutations within the extracellular and kinase domains reduced this activity (22). The mutation used in our model, R899X, is located in the BMPRII cytoplasmic tail domain. Indeed, we did not find any significant alterations of either Smad1 phosphorylation or Smad2 phosphorylation in the present study. Although TGF-β signaling via the Smad pathway induces the expression of Rho guanine nucleotide exchange factor, which mediates Rho activation (13), this mechanism of alteration of the Smad pathway to activate Rho-kinase does not apply to our model. It is also known that BMPs and TGF-β regulate MAP kinases, including ERK, JNK, and p38 in certain cell types (23, 36) and that TGF-β activate RhoA (4) via a Smad-independent pathway. We reported the activation of p38 in the whole lungs of SM22-tet-BMPR2R899X mice (32). In addition, Rudarakanchana et al. (27) reported the activation of p38 and p38-dependent proliferation of cells by transfection of a R899X mutant to NMuMG cells. Rho/Rho-kinase signaling may also be activated by a dysfunction of the BMPRII cytoplasmic tail domain via the Smad-independent pathway. Alternatively, BMPRII directly interacts with and regulates LIMK, Tctex, and c-Src, all of which have been reported to regulate Rho activity (7, 16, 33); LIMK in particular also directly interacts with Rho-kinase. Because the functions of the BMPRII cytoplasmic tail domain are not well-understood, further studies are therefore required.
Fig. 6.
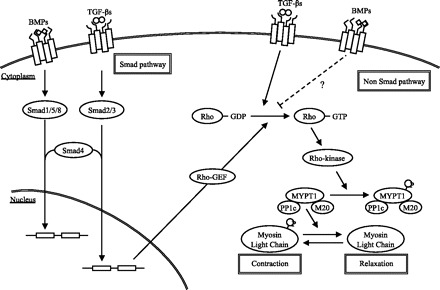
Schematic representation of the interactions between Rho/Rho-kinase signaling and bone morphogenetic protein (BMP)/transforming growth factor (TGF)-β signaling. In SM22-tet-BMPR2R899X mice, the levels of GTP-RhoA and MYPT1 phosphorylation were elevated, thus indicating that Rho/Rho-kinase signaling was activated. Two possible pathways, namely the Smad-dependent and Smad-independent pathways, in which BMP/TGF-β signaling activated Rho/Rho-kinase signaling are known. We confirmed that the Smad-dependent pathway was not altered. We therefore speculate that Rho/Rho-kinase signaling was activated via the Smad-independent pathway. Rho-GEF, Rho guanine nucleotide exchange factor; PP1c, catalytic subunit; M20, a 20-kDa subunit of unknown function; P, inorganic phosphate.
In summary, we demonstrated that the upregulation of Rho/Rho-kinase in mice expressing a BMPRII mutant with truncation of the cytoplasmic tail domain results in PH, which also shows the Rho-kinase inhibitor fasudil to be effective against the PH caused by this BMPRII mutant.
GRANTS
This research was partially supported by the Ministry of Education, Science, Sports and Culture, Grant-in-Aid for Scientific Research (C) 19590884.
DISCLOSURES
No conflicts of interest are declared by the authors.
REFERENCES
- 1. Abe K, Shimokawa H, Morikawa K, Uwatoku T, Oi K, Matsumoto Y, Hattori T, Nakashima Y, Kaibuchi K, Sueishi K, Takeshita A. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ Res 94: 385–393, 2004 [DOI] [PubMed] [Google Scholar]
- 2. Abe K, Tawara S, Oi K, Hizume T, Uwatoku T, Fukumoto Y, Kaibuchi K, Shimokawa H. Long-term inhibition of Rho-kinase ameliorates hypoxia-induced pulmonary hypertension in mice. J Cardiovasc Pharmacol 48: 280–285, 2006 [DOI] [PubMed] [Google Scholar]
- 3. Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 105: 1672–1678, 2002 [DOI] [PubMed] [Google Scholar]
- 4. Deaton RA, Su C, Valencia TG, Grant SR. Transforming growth factor-beta1-induced expression of smooth muscle marker genes involves activation of PKN and p38 MAPK. J Biol Chem 280: 31172–31181, 2005 [DOI] [PubMed] [Google Scholar]
- 5. Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 67: 737–744, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, McMurtry IF. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol 287: L656–L664, 2004 [DOI] [PubMed] [Google Scholar]
- 7. Foletta VC, Lim MA, Soosairajah J, Kelly AP, Stanley EG, Shannon M, He W, Das S, Massague J, Bernard O. Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J Cell Biol 162: 1089–1098, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fukumoto Y, Matoba T, Ito A, Tanaka H, Kishi T, Hayashidani S, Abe K, Takeshita A, Shimokawa H. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart 91: 391–392, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. International Consortium PPH Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips 3rd JA, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat Genet 26: 81–84, 2000 [DOI] [PubMed] [Google Scholar]
- 10. Ishikura K, Yamada N, Ito M, Ota S, Nakamura M, Isaka N, Nakano T. Beneficial acute effects of rho-kinase inhibitor in patients with pulmonary arterial hypertension. Circ J 70: 174–178, 2006 [DOI] [PubMed] [Google Scholar]
- 11. Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, Watanabe N, Saito Y, Kakizuka A, Morii N, Narumiya S. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J 15: 1885–1893, 1996 [PMC free article] [PubMed] [Google Scholar]
- 12. Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 295: L515–L529, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kardassis D, Murphy C, Fotsis T, Moustakas A, Stournaras C. Control of transforming growth factor beta signal transduction by small GTPases. FEBS J 276: 2947–2965, 2009 [DOI] [PubMed] [Google Scholar]
- 14. Leung T, Manser E, Tan L, Lim L. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J Biol Chem 270: 29051–29054, 1995 [DOI] [PubMed] [Google Scholar]
- 15. Long L, Crosby A, Yang X, Southwood M, Upton PD, Kim DK, Morrell NW. Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation 119: 566–576, 2009 [DOI] [PubMed] [Google Scholar]
- 16. Machado RD, Rudarakanchana N, Atkinson C, Flanagan JA, Harrison R, Morrell NW, Trembath RC. Functional interaction between BMPR-II and Tctex-1, a light chain of Dynein, is isoform-specific and disrupted by mutations underlying primary pulmonary hypertension. Hum Mol Genet 12: 3277–3286, 2003 [DOI] [PubMed] [Google Scholar]
- 17. Majka S, Hagen M, Blackwell T, Harral J, Johnson JA, Gendron R, Paradis H, Crona D, Loyd JE, Nozik-Grayck E, Stenmark KR, West J. Physiologic and molecular consequences of endothelial Bmpr2 mutation (Abstract). Respir Res 12: 84, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J 15: 2208–2216, 1996 [PMC free article] [PubMed] [Google Scholar]
- 19. Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem 147: 35–51, 2010 [DOI] [PubMed] [Google Scholar]
- 20. Morrell NW. Pulmonary hypertension due to BMPR2 mutation: a new paradigm for tissue remodeling? Proc Am Thorac Soc 3: 680–686, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, McMurtry IF, Oka M. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med 171: 494–499, 2005 [DOI] [PubMed] [Google Scholar]
- 22. Nishihara A, Watabe T, Imamura T, Miyazono K. Functional heterogeneity of bone morphogenetic protein receptor-II mutants found in patients with primary pulmonary hypertension. Mol Biol Cell 13: 3055–3063, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nohe A, Keating E, Knaus P, Petersen NO. Signal transduction of bone morphogenetic protein receptors. Cell Signal 16: 291–299, 2004 [DOI] [PubMed] [Google Scholar]
- 24. Oka M, Fagan KA, Jones PL, McMurtry IF. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. Br J Pharmacol 155: 444–454, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ratz PH, Berg KM, Urban NH, Miner AS. Regulation of smooth muscle calcium sensitivity: KCl as a calcium-sensitizing stimulus. Am J Physiol Cell Physiol 288: C769–C783, 2005 [DOI] [PubMed] [Google Scholar]
- 26. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Koerner SK, Levy PC, Reid LM, Vreim CE, Williams GW. Primary pulmonary hypertension. A national prospective study. Ann Intern Med 107: 216–223, 1987 [DOI] [PubMed] [Google Scholar]
- 27. Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, Trembath RC, Morrell NW. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet 11: 1517–1525, 2002 [DOI] [PubMed] [Google Scholar]
- 28. Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 83: 1325–1358, 2003 [DOI] [PubMed] [Google Scholar]
- 29. Tada Y, Majka S, Carr M, Harral J, Crona D, Kuriyama T, West J. Molecular effects of loss of BMPR2 signaling in smooth muscle in a transgenic mouse model of PAH. Am J Physiol Lung Cell Mol Physiol 292: L1556–L1563, 2007 [DOI] [PubMed] [Google Scholar]
- 30. Wang Z, Jin N, Ganguli S, Swartz DR, Li L, Rhoades RA. Rho-kinase activation is involved in hypoxia-induced pulmonary vasoconstriction. Am J Respir Cell Mol Biol 25: 628–635, 2001 [DOI] [PubMed] [Google Scholar]
- 31. West J, Fagan K, Steudel W, Fouty B, Lane K, Harral J, Hoedt-Miller M, Tada Y, Ozimek J, Tuder R, Rodman DM. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res 94: 1109–1114, 2004 [DOI] [PubMed] [Google Scholar]
- 32. West J, Harral J, Lane K, Deng Y, Ickes B, Crona D, Albu S, Stewart D, Fagan K. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am J Physiol Lung Cell Mol Physiol 295: L744–L755, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wong WK, Knowles JA, Morse JH. Bone morphogenetic protein receptor type II C-terminus interacts with c-Src: implication for a role in pulmonary arterial hypertension. Am J Respir Cell Mol Biol 33: 438–446, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C, Chen H, Trembath RC, Morrell NW. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res 96: 1053–1063, 2005 [DOI] [PubMed] [Google Scholar]
- 35. Young KA, Ivester C, West J, Carr M, Rodman DM. BMP signaling controls PASMC KV channel expression in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol 290: L841–L848, 2006 [DOI] [PubMed] [Google Scholar]
- 36. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res 19: 128–139, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]