

Abstract
Total parenteral nutrition (TPN) leads to a decline in phosphatidylinositol 3-kinase (PI3K)/phospho-Akt (p-Akt) activity, affecting downstream signaling, reducing epithelial cell (EC) proliferation, and contributing to intestinal mucosal atrophy. We hypothesized that promoting Akt activity would prevent these changes. We used a novel Akt-activating peptide, TCL1 (a head-to-tail dimer of the Akt-binding domain of T-cell lymphoma-1), or an inactive mutant sequence TCL1G conjugated to a transactivator of transcription peptide sequence to promote intracellular uptake. Four groups of mice were studied, enteral nutrition group (control), control mice given a functioning TCL1 (control + TCL1), TPN mice given TCL1G (control peptide, TPN + TCL1G); and TPN mice given TCL1. TPN mice given TCL1G showed a significant decrease in jejunal EC p-Akt (Ser473 and Thr308) abundance, whereas TPN + TCL1 mice showed increased p-Akt (Ser473) abundance. Phosphorylation of β-catenin and glycogen synthase kinase-3β (downstream targets of Akt signaling) were also decreased in the TPN + TCL1G group and completely prevented in the TPN + TCL1 group. Use of TCL1 nearly completely prevented the decline in EC proliferation seen in the TPN + TCL1G group, as well as partly returned EC apoptosis levels close to controls. The mammalian target of rapamycin pathway demonstrated a similar reduction in activity in the TPN + TCL1G group that was significantly prevented in the TPN + TCL1 group. These results support a significant loss of PI3K/p-Akt signaling upon replacing enteral nutrition with TPN, and prevention of this loss demonstrates the key importance of PI3K/p-Akt signaling in maintaining gut integrity including EC proliferation and reduction in apoptosis.
Keywords: intestinal epithelial cell, proliferation, apoptosis, mammalian target of rapamycin
translational highlights Removal of enteral nutrition in a mouse model of total parenteral nutrition leads to reduced epithelial cell proliferation, increased apoptosis, mucosal atrophy, and a decline in phosphatidylinositol 3-kinase/phospho-Akt activity. The authors used a novel Akt activating peptide, TCL1, conjugated to a transactivator of transcription peptide sequence to promote intracellular uptake in total parenteral nutrition-fed animals. Sustaining phosphatidylinositol 3-kinase/phospho-Akt signaling in this fashion prevented the decline in proliferation and increase in epithelial cell apoptosis and a decline in the mammalian target of rapamycin pathway. This study defines Akt as central to total parenteral nutrition-induced mucosal atrophy and demonstrates efficacy of a novel cell-permeant Akt activator.
Total parenteral nutrition (TPN), or the removal of all enteral nutrition, is critical for patients who cannot tolerate enteral feeding. Despite its clinical importance, TPN administration is associated with a decline in epithelial cell (EC) proliferation, increase in EC apoptosis, and mucosal atrophy, and these physiological findings may well contribute to an increase in septicemia and infectious complications (14, 16, 17). In normal gastrointestinal mucosa, intestinal epithelial proliferation is crucial for maintaining normal crypt-villus architecture (5, 29).
The mechanisms that contribute to the loss of normal crypt-villus structures with TPN administration are incompletely understood and are most certainly multifactorial. Our laboratory has recently shown that TPN administration led to a downregulation of phosphatidylinositol 3-kinase (PI3K)/phospho-Akt (p-Akt) activity (6). This observation may well be a strong contributing factor to the loss of EC proliferation and increases in EC apoptosis observed with TPN administration (33). The PI3K/p-Akt signaling pathway is a major signaling pathway that contributes to cell growth and survival (19, 20). PI3K/p-Akt signaling also contributes to the activation of mammalian target of rapamycin (mTOR), which integrates signals from nutrients and energy status into the regulation of a variety of processes, including cellular metabolism via activation of mTOR (4, 21). This latter function integrates signals from nutrients and energy status into the regulation of a variety of processes, including cellular metabolism (4, 21).
To test the importance of p-Akt signaling in the development of TPN-associated loss of EC proliferation, a recently described promoter of p-Akt production was used in this study (23). TCL1 (a head-to-tail dimer of the Akt-binding domain of T-cell lymphoma-1), which can promote the phosphorylation of p-Akt at two critical sites, was linked to a transactivator of transcription (TAT) sequence, which permitted the intracellular entrance of TCL1. We hypothesized that the use of this novel promoter would prevent many of the observed changes in the intestinal epithelium with the use of TPN.
MATERIALS AND METHODS
Animals.
C57BL/6J male, specific pathogen-free mice (10–11 wk old; Jackson Laboratory, Bar Harbor, ME) were maintained under temperature-, humidity- and light-controlled conditions. Mice were initially fed ad libitum with standard mouse chow and water and allowed to acclimate for 1 wk before surgery. During the administration of intravenous solutions, mice were housed in metabolic cages to prevent coprophagia. The studies were approved by our University of Michigan Animal Use Committee (no. 7703).
Operative procedures and TPN.
Cannulation and administration of TPN was identical to that previously described (15). After 24 h, mice were randomized to four groups: 1) controls consisting of enterally fed mice plus intravenous saline, 2) control + TCL1 consisting of enterally fed mice receiving TAT-TCL1 in intravenous saline (see below), 3) TPN + TCL1G (control peptide), and 4) TPN + TCL1 (active peptide) group. Nitrogen delivery was controlled between each group (isonitrogenous). Caloric delivery was also kept isocaloric between groups (15). All animals were killed at 7 days using CO2.
Peptide-mediated activation of Akt.
A novel peptide was used to promote p-Akt activity (22, 23). This peptide links a polybasic TAT sequence, which allowed intracellular permeability to the Akt-binding domain of the endogenous Akt coactivator, TCL1 (14). This peptide was previously reported to stimulate two main Akt phosphorylation sites, which can result in maximal activity (308 in the kinase domain and Serine-473 in the regulatory domain). A control peptide TAT-TCL1G contained point mutation and had minimal activity. Acute in vivo use of this peptide led to increased Akt activity and phosphorylation of both proximal (glycogen synthase kinase, GSK-3β) and distal (p70S6k) as well as extracellular regulated kinase (ERK). The present study examined a chronic dosing regimen. Several doses (0.5 mg/day, 0.2 mg/day, and 0.1 mg/day) were examined for tolerance, as the use of TAT peptide may mediate a high cell membrane permeability, which could result in potential toxicity at high doses (22, 31, 34). No mortality (100% survival) was observed for the animals receiving 0.1 mg of peptide; therefore 0.1 mg of the respective peptide was added to each 4.8 ml TPN solution daily beginning on the second day after placement of the TPN cannulas. Additionally, 1 mg peptide was administered intraperitoneally 3 h before euthanasia. An intraperitoneal route was selected to avoid introduction of air or bacteria into the intravenous line.
Real-time PCR.
Mucosal scrapings were placed in TRIzol and homogenized, and RNA was extracted and purified as previously described. Oligomers were designed using an optimization program Primer Premier (www.premierbiosoft.com), and sequences of specific primers are described in Table 1. RT-PCR was performed using a Rotor-Gene 6000 (Corbett Life Science, Sydney, Australia), and β-actin was used as an internal control for normalization. Fold changes of target genes were calculated using comparative quantification to β-actin.
Table 1.
Primers used in study
| Gene | Genebank Accession | Forward | Reverse | Size |
|---|---|---|---|---|
| Cyclin D1 | NM_007631 | GCGTACCCTGACACCAATCTC | CTCCTCTTCGCACTTCTGCTC | 183 |
| Bax | NM_007527 | TGAAGACAGGGGCCTTTTTG | AATTCGCCGGAGACACTCG | 140 |
| Bad | NM_007522 | AAGTCCGATCCCGGAATCC | GCTCACTCGGCTCAAACTCT | 106 |
| Bcl-2 | NM_009741 | ATGCCTTTGTGGAACTATATGGC | GGTATGCACCCAGAGTGATGC | 120 |
| β-actin | NM_007614 | ATGGAGCCGGACAGAAAAGC | CTTGCCACTCAGGGAAGGA | 108 |
Western immunoblotting.
Intestinal epithelial cells were prepared by using a modification of Grossmann's technique (13) and with immunoblotting techniques as previously described (26). The following antibodies (Ab) used included: mouse anti-β-catenin Ab (clone IgG1; BD Transduction Laboratories, BD Biosources, San Jose, CA), rabbit anti-phosphorylated Akt1/2/3 (Ser 473 and Thr308; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-phosphorylated GSK-3α/β (Ser21/9), mouse anti-cyclin D1, rabbit anti-phospho-p70 S6 kinase, rabbit anti-4E binding protein 1 (4EBP1), and mouse anti- proliferating cell nuclear antigen (PCNA) (Cell Signaling Technology, Beverly, MA). Secondary antibody was the horseradish peroxidase conjugate of either goat anti-mouse or goat anti-rabbit IgG (Cell Signaling).
Immunohistochemistry and immunofluorescent staining.
Zinc-formalin-fixed (10%) and paraffin-embedded tissue sections (5 μm) were stained using standard immunohistochemistry or immunofluorescent staining procedures, as previously described (8). The following primary antibodies and dilutions were used for immunohistochemistry: anti-Akt1/2/3 (Ser 473; at a 1:100 dilution, Santa Cruz), and anti-GSK-3β (Cell Signaling Technology, 1:50); biotinylated goat-anti-rabbit secondary antibody was used at a dilution of 1:200. The signal was amplified using streptavidin-horseradish peroxidase conjugate (Santa Cruz) and visualized with 3,3-diaminobenzidine substrate followed by hematoxylin. The following primary antibodies and dilutions were used for immunofluorescent staining: rabbit anti-Eukaryotic initiation factor 4EBP1 (1:200, Cell Signaling Technology), mouse anti PCNA (1:4,000, Cell Signaling Technology), mouse anti-β-catenin (clone IgG1; BD Transduction Laboratories), and goat anti-mouse-FITC IgG; goat anti-mouse-Cy3 IgG1 and goat anti-rabbit-FITC IgG were used as corresponding secondary antibody. Additional staining was performed for active caspase-3 as a marker of apoptosis (20), using anti-rabbit cleaved caspase-3 (Cell Signaling Technology). Immunofluorescent localization of p-p70 S6 was not done, as current available antibodies do not stain intestinal mucosal tissue adequately. Results are expressed as the percentage of active caspase-3-positive staining cell numbers in total EC numbers per villi; 10 villi were counted in each mouse.
EC proliferation was determined using 5-bromo-2-deoxyuridine (BrdU) (50 mg/kg; Roche Diagnostic, Indianapolis, IN) as previously described (26). An index of the crypt cell proliferation rate was calculated as the ratio of the number of crypt cells incorporating BrdU to the total number of crypt cells. The total number of proliferating cells per crypt was defined as the mean of proliferating cells in 10 crypts for each mouse.
Data analysis.
Data are expressed as the means ± SD. Statistical analyses employed Student's t-test for comparison of two means and a one-way ANOVA for comparison of multiple groups. For ANOVA a Bonferroni's post hoc analysis to assess statistical differences between groups was performed. The Chi-square test was used for categorical data (Prism software; GraphPad Software, San Diego, CA). Statistical significance was defined as P < 0.05.
RESULTS
TAT-TCL1 prevented TPN administration associate loss of p-Akt abundance.
TPN administration resulted in a significant reduction in the amount of activated Akt (Ser473) compared with the sham group (Fig. 1); the level of decline was similar to that previously described (8). Chronic administration of TAT-TCL1 (0.1 mg/day for 6 days) in the TPN solution led to a partial prevention in the decline of p-Akt (Ser473) abundance, whereas TPN mice treated with the Akt binding-deficient TAT-TCL1G showed a marked loss of pSer473 Akt abundance (Fig. 1B) compared with saline-treated controls. Enterally fed control mice administered the TAT-TCL1 peptide (0.1 mg/day for 6 days) had similar p-Akt abundance as saline-treated enteral control mice. The abundance of pThr308 Akt in TPN + TAT-TCL1G showed a marked decline compared with both control groups. There was no change in the abundance of pThr308 Akt in the TPN + TCL1 (Fig. 1C).
Fig. 1.

Phosphorylated-Akt (p-AKT). A: shown are representative immunoblot bands for p-Akt (Ser473 and Thr308, respectively) and total Akt expression for the following groups: control, control given active peptide (TCL1, a head-to-tail dimer of the Akt-binding domain of T-cell lymphoma-1), total parenteral nutrition (TPN) plus nonactive peptide (TCL1G), and TPN plus TCL1. B: results are expressed as the mean fold change in p-Akt at Ser473 mRNA relative to total Akt. C: results are expressed as the mean fold change in p-Akt at Thr308 mRNA relative to total Akt. Note that p-Akt significantly decreased with TPN administration as shown in the TPN + TAT-TCL1G group. Administration of TAT-TCL1 to control mice did not affect p-Akt abundance; however, TAT-TCL1 in TPN mice partially prevented the decline in p-Akt (Ser473) (B). However, TAT-TCL1 did not affect p-Akt abundance at the Thr308 phosphorylation site (C). Results of each group represent an n = 4–6. #P < 0.05; **P < 0.001.
TAT-TCL1 promotion of p-Akt abundance led to downstream signaling in the Wnt/β-catenin pathway.
The increased abundance of p-Akt does not necessarily reflect that Akt was able to transduce signals appropriately. The consequences of sustained p-Akt (Ser473) abundance in the TPN + TCL1 mice group were examined by determining whether proximal targets of p-Akt were phosphorylated. A classic p-Akt signaling pathway is the Wnt/β-catenin pathway. We began by examining GSK-3β and β-catenin expression.
Phospho-GSK-3β abundance was unaffected in the control + TCL1 group (Fig. 2; 0.62 ± 0.06 vs. 0.55 ± 0.13; control + saline vs. control + TCL1, respectively). P-GSK-3β was significantly (P < 0.01) decreased (∼2.5-fold) in the TPN + TCL1G group (0.23 ± 0.09). However, the loss of p-GSK-β was almost completely prevented (P < 0.01) in the group of TPN mice treated with TCL1 (0.58 ± 0.10; TPN + TCL1 group; Fig. 2B). Immunohistochemical staining showed that p-GSK-3β was primarily located in the jejunal crypts; a marked decline in p-GSK-3β was observed in the crypts of TPN + TCL1G mice, and a partial prevention of this loss was seen in the TPN + TCL1 group (data not shown).
Fig. 2.

Abundance of phospho-glycogen synthase-kinase (p-GSK)-3β. p-GSK-3β was decreased with TPN, and this was prevented with TCL1 administration. A: representative immunoblot bands of p-GSK-3β and β-actin are shown for each study group. Note that p-GSK-3β is shown as the lower of the two bands, and p-GSK-3α is the upper band. B: summarized results of Western immunoblotting as expressed as the ratio of p-GSK-3β expression to β-actin. Note the decline in p-GSK-3β in the TPN + TCL1G group and a significant prevention of this decline in the TPN + TCL1 group. Results of each group represent an n = 4–6. *P < 0.01.
A similar pattern of expression was seen for β-catenin (Fig. 3). A loss of β-catenin was observed in the TPN + TCL1G group, and a prevention of this loss of β-catenin was found in the TPN + TCL1 group. Immunofluorescent imaging demonstrated an almost complete loss of β-catenin in the TPN + TCL1G group, whereas the TPN + TCL1 cohort exhibited an intermediate level of fluorescence (Fig. 6, see below).
Fig. 3.

Intestinal epithelial cell β-catenin protein expression by Western immunoblots. A: representative gels for β-catenin. B: summarized results of β-catenin protein expression as normalized to β-actin. Intestinal epithelial cell β-catenin protein abundance was significantly decreased in the TPN + TCL1G group compared with controls. This decline was partially prevented in the TPN + TCL1 group. Results of each group represent an n = 5. **P < 0.001.
Fig. 6.
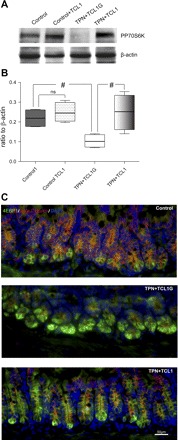
Alteration in mammalian target of rapamycin regulatory factors. A and B: representative immunoblot gels and summary of protein expression of phosphate P70S6K (PP70S6K) activity in study groups. Note the decline in PP70S6K in the TPN + TCL1G group and a partial prevention in the TPN + TCL1 group. Results of each group represent an n = 4–6. #P < 0.05. C: representative immunofluorescent images of 4E binding protein 1 (4EBP1) and β-catenin expression in control, TPN + TCL1G, and TPN + TCL1 groups. Note the marked increase in 4EBP1 (green fluorescence) in the TPN + TCL1G group and a return to lower levels of intensity in the TPN + TCL1 group. Also note the loss of β-catenin in the TPN + TCL1G group and a return to normal levels and cell membrane localization in the TPN + TCL1 group.
TAT-TCL1 promoted EC proliferation through activation of the T cell factor/lymphoid enhancer factor transcription factor cyclin D1.
The distal end of the β-catenin pathway is T cell factor (TCF)-4 mediated transcriptional activation. As a marked loss of β-catenin (nuclear and cytoplasmic) was detected in TPN + TCL1G mice, it was next determined whether this resulted in a loss of TCF-4 transcriptional activation by measuring the RNA abundance of two known TCF-4 target genes, c-myc and cyclin D1. Compared with both control groups after 7 days of TPN, there was a nearly threefold decline in mRNA and protein expression for cyclin D1 in the TPN + TCL1G group, and this decline was prevented in the TPN + TCL1 group (Fig. 4). c-Myc expression was also found to be decreased in the TPN + TCL1G group; however, no significant difference was observed with the use of TCL1 (data was not shown).
Fig. 4.

Cyclin D1 expression in intestinal epithelial cells. A: mRNA expression of cyclin D1. B: representative immunoblot gels for cyclin D1. C: summarized results of cyclin D1 protein expression. Cyclin D1 decreased significantly in the TPN + TCL1G group compared with controls. This decline was partially prevented in the TPN + TCL1 group. Results of each group represent an n = 5. **P < 0.001, *P < 0.01.
Cyclin D1 and c-myc are two major regulators of the progression of cells into the proliferative stage of the cell cycle (13, 31, 34). Therefore, the tissue-level functional consequences of TCL1-mediated sustained p-Akt abundance and activity were assessed. BrdU incorporation was assessed as a marker of proliferation. TPN + TCL1G mice showed a marked decline in EC proliferation compared with both control groups (0.23 ± 0.06 vs. 0.49 ± 0.06 and 0.53 ± 0.12, TPN + TCL1G vs. control + saline and control + TCL1, respectively). TPN + TCL1 mice had a preservation of proliferation to nearly the same level as the control group (0.41 ± 0.07 TPN + TCL1), and the level was not significantly different than controls (Fig. 5F).
Fig. 5.

Intestinal epithelial cell proliferation. A–E: proliferating cell nuclear antigen (PCNA) abundance. A, B, and C are representative immunofluorescent images of PCNA-positive crypt cells in control, TPN + TCL1G, and TPN + TCL1 groups. D and E are representative immunoblot gels and summaries of immunoblot data. F: intestinal epithelial cell proliferation. Proliferation is determined by 5-bromo-2-deoxyuridine (BrdU) crypt cell incorporation, as represented by a proliferation index (ratio of BrdU-positive cells to total number of crypt cells). Note that for PCNA and BrdU data, proliferation was significantly decreased in the TPN + TCL1G group compared with controls and that this decline was partially prevented in the TPN + TCL1 group. Results of each group represent an n = 5. **P < 0.001, *P < 0.01.
The BrdU incorporation data were supported by measuring PCNA expression by Western blot and immunohistochemistry. TAT-TCL1 did not affect PCNA expression in the control group (0.51 ± 0.07 vs. 0.53 ± 0.07 control + saline vs. control + TCL1, respectively), whereas PCNA expression was approximately fivefold lower in the TPN + TCL1G group (0.1 ± 0.05). A prevention in this loss of PCNA was noted in the TPN + TCL1 group (0.39 ± 0.08; Fig. 5, D and E). Immunofluorescent staining showed the number of PCNA-positive staining crypt cells (Fig. 5). Immunofluorescent intensity was markedly lost in the TPN + TCL1G group (Fig. 5B) compared with the control + TCL1 group (Fig. 5A), and this loss was partially prevented in the TPN + TCL1G group (Fig. 5C). No difference was noted between control + saline vs. control + TCL1 groups (data not show).
TAT-TCL1 administration prevented TPN-associated changes in the mTOR pathway.
The scope of the activity of p-Akt was assessed by determining the effects on the mTOR pathway, p70 S6 kinase (pp70S6K; or PP70), and expression/localization of 4EBP1, proteins that mediate mTOR signaling and mRNA translation (26). Administration of TAT-TCL1 did not affect the phosphorylation of p70S6K in the control group (0.22 ± 0.05 vs. 0.25 ± 0.05; control + saline vs. control + TCL1, respectively). A significant (P < 0.05) loss of pp70S6K expression was detected in TPN + TCL1G group (0.10 ± 0.03), whereas a retention of normal pp70S6K expression was measured in TPN mice treated with TCL1 (0.25 ± 0.09; P < 0.05 compared with the TPN + TCL1G group, Fig. 6).
Loss of PI3K/p-Akt signaling in intestinal EC was found to increase 4EBP1 expression and was associated with the dissociation of the β-catenin/4EBP1 complex. Immunofluorescent detection of 4EBP1 in histological sections revealed that most of the 4EBP1-positive cells were located at the bottom of the crypts in the TPN + TCL1G group, and this intensity was markedly increased compared with the control groups (control + TCL1, Fig. 7A). As a control, administration of TAT-TCL1G did not affect the 4EBP1 intensity in the control group (data not shown). Additionally, histological sections from control mice showed colocalization of 4EBP1 and β-catenin, which was lost in the TPN + TCL1G cohort. 4EBP1 signal intensity was decreased in the TPN + TCL1 group compared with TPN + TCL1G mice although still slightly higher than the control group. Treatment of TPN mice with TCL1 also preserved the membrane localization of β-catenin.
Fig. 7.

Alteration in intestinal epithelial cell apoptosis. A–C: summarized results of Bad, Bax, and Bcl-2 mRNA abundance with real-time PCR in 3-day mucosal samples. Note the increase in Bad and Bax expression in the TPN + TCL1G group, and a partial prevention of these changes in the TPN + TCL1 group. No significant difference was noted in Bcl-2 expression in all groups. Results of each group represent the mean ± SD of n = 5–6, #P < 0.05; *P < 0.01; **P < 0.001. D–G: active caspase-3 immunofluorescent staining. Note the marked increase in the number and density of caspase-3-positive staining cells in the TPN + TCL1G group (E) compared with the control group (D), with most of the positive staining epithelial cells located at the base of the villi. The TPN + TCL1 group was associated with a reduction in the number of caspase-3-positive cells (F). Summation of caspase-3 activity is expressed as the percentage of active caspase-3-positive epithelial cells (G).
TAT-TCL1 administration partially prevented TPN-associated epithelial cell apoptosis.
Another major physiological role of the PI3K/Akt signaling pathway is to inhibit apoptosis. Gut epithelial cell apoptosis is a relatively early event following the transition from enteral to parenteral nutrition; therefore epithelial cell apoptosis was determined in gut epithelial cells after 3 days. First, mRNA expression was measured for three members of the Bcl-2 family, including Bad, Bax, and Bcl-2. Both Bad and Bax expression increased significantly in the TPN + TCL1G group compared with controls (Fig. 7, A and B). There was no significant difference for Bcl-2 expression across the four groups (Fig. 7C).
Immunofluorescent staining for active caspase-3 was then performed on histological sections of small bowel. The result showed very low levels of active caspase-3 staining cells in the control + saline group (Fig. 7, D and G). Although not shown, this low caspase-3 activity was also observed in control + TCL1 mice (0.013 ± 0.005; caspase-3 EC per villi). The percentage of active caspase-3-positive staining cells was 10-fold higher (0.12 ± 0.04) in the TPN + TCL1G group, with most of the positive staining EC cells being located at the base of the villi (Fig. 7E). The percentage of active caspase-3-positive staining cells significantly decreased to 0.06 ± 0.02 in TPN + TCL1 group, a twofold decline compared with the TPN + TCL1G cohort (Fig. 7, F and G).
DISCUSSION
Our laboratory has previously showed that the administration of TPN resulted in a loss of intestinal EC p-Akt phosphorylation (19). In this former study, glutamine was used to prevent the loss of p-Akt expression in our TPN model, and glutamine partially prevented the observed loss of EC proliferation with TPN. Glutamine is known to stimulate epithelial proliferation (30, 35). However, although glutamine has been shown to modulate the PI3K/Akt pathway (18), several reports have shown that glutamine can also affect many other intracellular signaling pathways in addition to PI3K/Akt (3, 6, 7). To determine the relevance of this TPN-associated loss in p-Akt, the present study used a novel promoter of phosphorylation, TAT-TCL1 (23). This peptide prevented a significant portion of the TPN-induced loss of Akt phosphorylation, which resulted in 1) the partial prevention in TPN-induced loss of EC proliferation, 2) the attenuation of TPN-induced increase in EC apoptosis, and 3) the maintenance of mTOR activation. Taken together these data suggest that the PI3K/p-Akt signaling pathway is critical for maintaining gut integrity, and many of the GI histopathological findings with TPN administration can be explained by loss of PI3K/p-Akt activity.
Akt is known to be a key regulatory pathway in several disease states, including inflammatory bowel disease and in other acute injuries such as myocardial and cerebral ischemia/reperfusion. Thus there has been an intense interest in understanding how to modulate this critical signaling pathway (9). The use of TCL1 in our work is one such approach. The βA strand of TCL1 forms a homodimer, each face of which then binds one molecule of Akt (1, 23). TCL1 may then aid in recruiting Akt to the membrane where the activating kinases PDK1 and PDK2 are located. Alternative mechanisms include bringing two molecules of Akt into proximity to facilitate transactivation, or that the complex provides steric shielding for the phosphorylation sites, rendering them less accessible to the phosphatases that inactivate Akt (for example, protein phosphatase 2A). This approach, and linking TCL1 to the transfer domain of the HIV-1 protein TAT (27), has shown that Akt can be activated in vivo in a specific, receptor-independent fashion in a nontransgenic acute septic mouse model (23). However, this previous work used this TAT-TCL1 peptide for a relatively short period of time. In our present study, TAT-TCL1 and its nonactive peptide were used in vivo in a much more chronic process (1-wk period). As high doses of TAT may be toxic, we carefully titrated the dosing to 0.1 mg/day, which led to excellent mouse survival during TPN administration. Clearly, future adjustment of this dosing may be needed to better understand this complex signaling pathway in our TPN model.
In our present study, both p-Akt at Ser473 and at Thr308 were downregulated with TPN administration using the control peptide, TAT-TCL1G, whereas, in the TPN + TCL1 group, there was significantly increased abundance of p-Akt, as detected by Western immunoblotting and immunohistochemistry staining at Ser473. It was interesting to note that there was no significant change in the abundance at the Thr308 phosphorylation site. This differential expression may not be entirely surprising. In McDunn's initial work (23), it also appeared that use of TCL1 on serum-starved Jurkat cells led to a far greater abundance of Ser473 phosphorylation compared with the Thr308 site. Furthermore, due to the chronic nature of the present study, it is also possible that we may have missed an earlier peak in the abundance of p-Akt at the Thr308 site. Our present work emphasizes that relatively small levels of sustained p-Akt abundance have tremendous potential to sustain cell viability and function. The mechanisms by which TCL1 differentially affected each study group is not fully known. It was interesting that TCL1 exerted minimal action in control mice and marked effect in the TPN group. It is possible that the action of TCL1 is to prevent p-Akt activity from declining below a certain level. It is also possible that TCL1 exerts a differential effect on normal proliferating epithelial cells (minimal action) compared with those epithelial cells with low levels of p-Akt activity (proliferative effect).
TPN-associated loss of p-Akt has been linked to a downregulation in p-GSK-3β and β-catenin (8). In our present study a decline in p-GSK-3β and β-catenin was observed in the TPN + TCL1G group, and this was prevented in the TPN + TCL1 group. Additionally, the use of this active compound led to a prevention in the decline in EC proliferation observed in TPN mice receiving TAT-TCL1G. Additionally, this strongly supports the fact that TPN-associated intestinal mucosal atrophy is due to a loss of PI3K/Akt activity. This appears to be supported by a previous study that showed that loss of p-Akt has been observed in mice given an elemental diet (e.g., glucose, electrolytes, and water), with p-Akt expression rapidly rebounding with administration of a solid diet (28). Additionally, such actions could clearly influence downstream factors including the fate of intracellular β-catenin and cell proliferation.
In addition, Akt has been reported to be involved in the phosphorylation and activation of mTOR at Ser2448, and then affecting downstream signals PP70s6K and 4EBP1. Therefore, to further determine the functional affects of sustaining p-Akt abundance in our TPN model, this pathway was also studied. TPN administration in mice given TAT-TCL1G led to a decline in PP70S6K expression and activated 4EBP1 expression. The administration of TAT-TCL1 to TPN mice prevented both of these changes. The mTOR pathway is tightly related to cell growth and the G1 cell cycle and contributes to cell proliferation (25, 27a). With TPN, no colocalization was observed between β-catenin and 4EBP1 in immunofluorescent images. This suggested that both Wnt/β-catenin and mTOR pathways are adversely affected in the TPN mice model, and that inhibition of both pathways contributes to poor EC proliferation in our TPN model.
Although the mTOR pathway has not been previously explored in TPN model, because of the known association of mTOR with the modulation of metabolism, such changes are intriguing. The rictor-mTOR complex phosphorylates Akt/PKB on Ser473 in the hydrophobic motif, which may facilitate the phosphorylation by PDK1 of the activation loop of Akt/PKB on Thr308. How the rictor-mTOR complex is regulated is unknown. In its nonphosphorylated state 4EBP1 represses cap-dependent mRNA translation by binding to the eIF-4E cap binding protein and preventing it from interacting with the eIF-4G protein. Phosphorylation of 4EBP1 by raptor-mTOR releases eIF-4E to restore cap-dependent translation, which is particularly important for the translation of mRNA (10). It has been reported that inhibition of mTOR by rapamycin markedly impairs 4EBP1 phosphorylation and blocks its release from eIF4E(2). Furthermore, phosphorylation of 4EBP1 is strongly modulated by the PI3K/Akt pathway as well as by nutrients/amino acids, both upstream regulators of mTOR signaling (11, 24, 32). In our study, both of mTOR downstream factors were inhibited with TPN administration, and stimulation of p-AKT activity prevented these changes.
It has been reported that PI3K elicits its antiapoptotic activity through the activation of the serine/threonine kinase Akt. Presently, the precise downstream mechanisms by which the activation of p-Akt leads to a decline in epithelial cell apoptosis is unknown. Activated Akt can phosphorylate the proapoptotic protein Bad, allowing Bad to associate with 14–3-3 and dissociation from Bcl-XL, thereby freeing it to exert its antiapoptotic function (35). PI3K/Akt signals can also exert an antiapoptotic action on gastric epithelial cells in vitro and in vivo through Bax, Bcl-2, and inhibition of caspase-3 activity(36). In our present study, the TAT-TCL1 peptide had a significant antiapoptotic action presumably by promoting p-Akt activity. Use of this peptide led to a decrease in Bad and Bax abundance and prevented a rise in active caspase-3. A rebalance of cell proliferation and apoptosis through p-Akt is critical for tissue homeostasis in our TPN mice model.
In conclusion, TPN led to a marked decline in p-Akt abundance. By sustaining p-Akt with TAT-TCL1, a partial prevention in the loss of downstream signaling, including preservation of EC proliferation, reduction in apoptosis, and preserved mTOR signaling was observed in a TPN model. These results suggest that changes in the p-Akt signaling pathway may well be a major mechanism that contributes to many of the intestinal EC derangements associated with TPN administration. New strategies that can sustain phosphorylation of Akt may have clinical relevance to patients receiving parenteral nutrition.
GRANTS
This work was supported by NIH grant 5R01 AI044076–11 (to D. H. Teitelbaum). The work was also supported with the help of the National Cancer Institute through the University of Michigan's Cancer Center Support Grant (5 P03 CA46592).
DISCLOSURES
No conflicts of interest are declared by the authors.
ACKNOWLEDGMENTS
Jonathan E. McDunn is presently at Metabolon, Durham, NC.
REFERENCES
- 1. Auguin D, Barthe P, Royer C, Stern M, Noguchi M, Arold S, Roumestand C. Structural basis for the co-activation of protein kinase B by T-cell Leukemia-1 (TCL1) family proto-oncoproteins. J Cell Biol 27: 35890–35902, 2004 [DOI] [PubMed] [Google Scholar]
- 2. Beretta L, Gingras A, Svitkin Y, Hall M, Sonenberg N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J 15: 658–664, 1996 [PMC free article] [PubMed] [Google Scholar]
- 3. Blachier F, Boutry C, Bos C, Tomé D. Metabolism and functions of L-glutamate in the epithelial cells of the small and large intestines. Am J Clin Nutr 90: 814S–821S, 2009 [DOI] [PubMed] [Google Scholar]
- 4. Chiu T, Santiskulvong C, Rozengurt E. EGF receptor transactivation mediates ANG II-stimulated mitogenesis in intestinal epithelial cells through the PI3-kinase/Akt/mTOR/p70S6K1 signaling pathway. Am J Physiol Gastrointest Liver Physiol 288: G182–G194, 2005 [DOI] [PubMed] [Google Scholar]
- 5. Clark J, Gan H, Samocha A, Fox A, Buchman T, Coopersmith C. Enterocyte-specific epidermal growth factor prevents barrier dysfunction and improves mortality in murine peritonitis. Am J Physiol Gastrointest Liver Physiol 297: G471–G479, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. El-Kadi S, Baldwin RT, McLeod K, Sunny N, Bequette B. Glutamate is the major anaplerotic substrate in the tricarboxylic acid cycle of isolated rumen epithelial and duodenal mucosal cells from beef cattle. J Nutr 139: 869–875, 2009 [DOI] [PubMed] [Google Scholar]
- 7. Evans M, Jones D, Ziegler T. Glutamine inhibits cytokine-induced apoptosis in human colonic epithelial cells via the pyrimidine pathway. Am J Physiol Gastrointest Liver Physiol 289: G388–G396, 2005 [DOI] [PubMed] [Google Scholar]
- 8. Feng Y, Sun X, Yang H, Teitelbaum D. Dissociation of E-Cadherin and beta-catenin in a mouse model of total parenteral nutrition: a mechanism for the loss of epithelial cell proliferation and villus atrophy. J Physiol 587: 641–654, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Franke T. PI3K/Akt: getting it right matters. Oncogene 27: 6473–6488, 2008 [DOI] [PubMed] [Google Scholar]
- 10. Gingras A, Raught B, Gygi S, Niedzwiecka A, Miron M, Burley S, Polakiewicz R, Wyslouch-Cieszynska A, Aebersold R, Sonenberg N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev 15: 2852–2864, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gingras A, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev 15: 807–826, 2001 [DOI] [PubMed] [Google Scholar]
- 13. Grossmann J, Maxson J, Whitacre C, Orosz D, Berger N, Fiocchi C, Levine A. New isolation technique to study apoptosis in human intestinal epithelial cells. Am J Pathol 153: 53–62, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. King B, Kudsk K, Li J, Wu Y, Renegar K. Route and type of nutrition influence mucosal immunity to bacterial pneumonia. Ann Surg 229: 272–278, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kiristioglu I, Teitelbaum DH. Alteration of the intestinal intraepithelial lymphocytes during total parenteral nutrition. J Surg Res 79: 91–96, 1998 [DOI] [PubMed] [Google Scholar]
- 16. Kudsk K, Croce M, Fabian T. Enteral versus parenteral feeding: effects on septic morbidity after blunt and penetrating abdominal trauma. Ann Surg 215: 503–513, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kudsk KA, Li J, Renegar KB. Loss of upper respiratory tract immunity with parenteral feeding. Ann Surg 223: 629–635; discussion 635–628, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Larson SD, Li J, Chung DH, Evers BM. Molecular mechanisms contributing to glutamine-mediated intestinal cell survival. Am J Physiol Gastrointest Liver Physiol 293: G1262–G1271, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin J, Li P, Snook A, Schulz S, Dasgupta A, Hyslop T, Gibbons A, Marszlowicz G, Pitari G, Waldman S. The hormone receptor GUCY2C suppresses intestinal tumor formation by inhibiting AKT signaling. Gastroenterology 138: 241–254, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mandal C, Ghosh-Choudhury G, Ghosh-Choudhury N. Phosphatidylinositol 3 kinase/Akt signal relay cooperates with smad in bone morphogenetic protein-2-induced colony stimulating factor-1 expression and osteoclast differentiation. Endocrinology 150: 4989–4998, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matheny RJ, Adamo M. Effects of PI3K catalytic subunit and Akt isoform deficiency on mTOR and p70S6K activation in myoblasts. Biochem Biophys Res Commun 390: 252–257, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McDunn J, Muenzer J, Dunne B, Zhou A, Yuan K, Hoekzema A, Hilliard C, Chang K, Davis C, McDonough J, Hunt C, Grigsby P, Piwnica-Worms D, Hotchkiss R. An anti-apoptotic peptide improves survival in lethal total body irradiation. Biochem Biophys Res Commun 382: 657–662, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McDunn J, Muenzer J, Rachdi L, Chang K, Davis C, Dunne W, Piwnica-Worms D, Bernal-Mizrachi E, Hotchkiss R. Peptide-mediated activation of Akt and extracellular regulated kinase signaling prevents lymphocyte apoptosis. FASEB J 22: 561–568, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Proud C. Regulation of protein synthesis by insulin. Biochem Soc Trans 34: 213–216, 2006 [DOI] [PubMed] [Google Scholar]
- 25. Schmelzle T, Hall M. mTOR, a central controller of cell growth. Cell 103: 253–262, 2000 [DOI] [PubMed] [Google Scholar]
- 26. Schwarz B, Wang F, Shen L, Clayburgh D, Su L, Wang Y, Fu Y, Turner J. LIGHT signals directly to intestinal epithelia to cause barrier dysfunction via cytoskeletal and endocytic mechanisms. Gastroenterology 132: 2383–2394, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schwarze S, Ho A, Vocero-Akbani A, Dowdy S. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285: 1569–1572, 1999 [DOI] [PubMed] [Google Scholar]
- 27a. Shao J, Evers BM, Sheng H. Roles of phosphatidylinositol 3′-kinase and mammalian target of rapamycin/p70 ribosomal protein S6 kinase in K-Ras-mediated transformation of intestinal epithelial cells. Cancer Res 64: 229–235, 2004 [DOI] [PubMed] [Google Scholar]
- 28. Sheng H, Shao J, Townsend CM, Jr, Evers BM. Phosphatidylinositol 3-kinase mediates proliferative signals in intestinal epithelial cells. Gut 52: 1472–1478, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stern LE, Erwin CR, O'Brien DP, Huang F, Warner BW. Epidermal growth factor is critical for intestinal adaptation following small bowel resection. Microsc Res Tech 51: 138–148, 2000 [DOI] [PubMed] [Google Scholar]
- 30. Sukhotnik I, Mogilner J, Karry R, Shamian B, Lurie M, Kokhanovsky N, Ure B, Coran A. Effect of oral glutamine on enterocyte turnover during methotrexate-induced mucositis in rats. Digestion 79: 5–13, 2009 [DOI] [PubMed] [Google Scholar]
- 31. Vaslin A, Puyal J, Clarke P. Excitotoxicity-induced endocytosis confers drug targeting in cerebral ischemia. Ann Neurol 65: 337–347, 2009 [DOI] [PubMed] [Google Scholar]
- 32. Wang X, Beugnet A, Murakami M, Yamanaka S, Proud C. Distinct signaling events downstream of mTOR cooperate to mediate the effects of amino acids and insulin on initiation factor 4E-binding proteins. Mol Cell Biol 25: 2558–2572, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang H, Fan Y, Teitelbaum DH. Intraepithelial lymphocyte-derived interferon-gamma evokes enterocyte apoptosis with parenteral nutrition in mice. Am J Physiol Gastrointest Liver Physiol 284: G629–G637, 2003 [DOI] [PubMed] [Google Scholar]
- 34. Zhang M, Li Z, Fang Y, Zhu H, Xue J, Chen J, Jia W. TAT-phiC31 integrase mediates DNA recombination in mammalian cells. J Biotechol 142: 107–113, 2009 [DOI] [PubMed] [Google Scholar]
- 35. Ziegler TR, Bazargan N, Leader LM, Martindale RG. Glutamine and the gastrointestinal tract. Curr Opin Clin Nutr Metab Care 3: 355–362, 2000 [DOI] [PubMed] [Google Scholar]