

Background: Cerebral elevation and accumulation of amyloid β-peptide is an invariant feature of Alzheimer disease.
Results: Natural compound (20S)-Rg3, a PI4KIIα activator, modulates γ-secretase activity in lipid rafts by increasing levels of phosphoinositides.
Conclusion: Activation of a key phospholipid synthetic pathway by a natural product regulates γ-secretase activity.
Significance: We identify a novel molecular mechanism for the regulation of γ-secretase activity by (20S)-Rg3.
Keywords: Alzheimer Disease, Lipids, Lipid Raft, Natural Products, Phosphatidylinositol, Presenilin
Abstract
Amyloid β-peptide (Aβ) pathology is an invariant feature of Alzheimer disease, preceding any detectable clinical symptoms by more than a decade. To this end, we seek to identify agents that can reduce Aβ levels in the brain via novel mechanisms. We found that (20S)-Rg3, a triterpene natural compound known as ginsenoside, reduced Aβ levels in cultured primary neurons and in the brains of a mouse model of Alzheimer disease. The (20S)-Rg3 treatment induced a decrease in the association of presenilin 1 (PS1) fragments with lipid rafts where catalytic components of the γ-secretase complex are enriched. The Aβ-lowering activity of (20S)-Rg3 directly correlated with increased activity of phosphatidylinositol 4-kinase IIα (PI4KIIα), a lipid kinase that mediates the rate-limiting step in phosphatidylinositol 4,5-bisphosphate synthesis. PI4KIIα overexpression recapitulated the effects of (20S)-Rg3, whereas reduced expression of PI4KIIα abolished the Aβ-reducing activity of (20S)-Rg3 in neurons. Our results substantiate an important role for PI4KIIα and phosphoinositide modulation in γ-secretase activity and Aβ biogenesis.
Introduction
Elevation and accumulation of amyloid β-peptide (Aβ)3 in the brain are invariable events associated with Alzheimer disease (AD) (1, 2). Aβ is produced by sequential proteolytic cleavages of the β-amyloid precursor protein (APP) by a set of membrane-bound proteases termed β- and γ-secretases (3). Therapeutic strategies targeting AD currently under development include reducing Aβ generation, promoting Aβ clearance, and inhibiting Aβ aggregation (4). Despite initial promise, the majority of these approaches suffered major setbacks in late stage clinical trials due to toxicity issues. Recent imaging studies revealed that extensive Aβ pathology precedes any detectable clinical symptoms by at least a decade (5). Thus, early intervention is becoming increasingly important, and there is a significant unmet need for effective and safe prophylactic as well as therapeutic approaches in AD (6, 7).
Cholesterol and a growing number of other lipids, such as phosphoinositides, have been extensively implicated in AD pathogenesis (8). We have previously reported that phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2), a phospholipid that regulates key aspects of neuronal function (9), is involved with cellular mechanisms underlying AD, such as the biogenesis of Aβ production as well as the synaptic action of Aβ (10, 11). Cellular levels of PI(4,5)P2 are inversely correlated with levels of secreted Aβ in cultured cells (10). Conversely, overexpression of familial AD presenilin and APP mutants and exposure to Aβ oligomers caused a decrease in levels of PI(4,5)P2 (10–12).
Synthesis of PI(4,5)P2 in the brain is predominantly mediated by phosphatidylinositol-phosphate kinase type 1γ, which phosphorylates PI(4)P on the D-5 position of the inositol ring (9, 13, 14). In mammalian cells, phosphatidylinositol (PI) 4-kinase generates PI(4)P, the main precursor for PI(4,5)P2 (15). PI4KIIα is enriched in brain and has also been implicated in neurodegeneration (16, 17). Overexpression of this enzyme was shown to promote the synthesis of PI(4)P as well as PI(4,5)P2, suggesting that PI4KIIα may be the rate-limiting enzyme in PI(4,5)P2 synthesis (18, 19). Critically, PI4KIIα was shown to localize subcellularly to lipid rafts (20, 21), a major site of Aβ biogenesis.
The γ-secretase is a membrane-embedded, multicomponent proteolytic enzyme consisting of four essential components, including PS1 or -2, nicastrin, Aph-1, and PEN-2 (3, 22). Brain lipids have been shown to be essential for the reconstitution of γ-secretase activity (23, 24), and changes in lipid composition or membrane fluidity and thickness directly influence γ-secretase activity (8, 25–29), indicating that brain lipids are important regulatory factors for Aβ production. Several studies indicate that the presenilins, the catalytic components of the γ-secretase complex, may be enriched in detergent-resistant membrane domains (DRMs), which are also referred to as lipid rafts (25, 26, 30). Our previous study suggested that phosphoinositides may inhibit γ-secretase activity by regulating the interaction between the lipid substrate and enzyme complex (27, 29). Thus, it is conceivable that PI(4,5)P2 influences the properties of the γ-secretase by either modification of the lipid environment or sequestration of γ-secretase component(s) in DRMs.
Natural compounds have been the basis for traditional remedies but also have been a major source for diverse drug leads for modern medicine (31, 32). Natural compounds often produce cell or system level phenotypes via pleiotropic mechanisms, which may be ideal for the prevention and/or treatment of complex, chronic disorders, such as AD. However, elucidating the mechanisms of action for bioactive natural compounds has been one of the greatest challenges facing the field (33, 34). Ginseng (Panax ginseng) is one of the most common ingredients in herbal medicine throughout the world and has been used to treat multiple ailments by people in Eastern Asia for thousands of years (35, 36). Several studies indicate that ginseng improves memory and cognitive function in human (37, 38). Cognitive benefits in AD in several human clinical trials have also been reported (39–41). Ginsenosides, the active components of ginseng, are triterpene glycoside compounds that exhibit structural variation (42) allowing for diverse biological activities, including antitumor (43) and neuroprotective activities (44). More than 30 different ginsenosides have been identified to date, and a number of medical applications have been reported (35, 42). Some ginsenosides have been shown to reduce the levels of Aβ; however, the mechanism underlying Aβ-lowering activity is not known (45, 46).
In the present work, we identified a number of ginsenosides that have Aβ-reducing activity, including (20S)-Rg3, a component of heat-processed ginseng. We investigated the mechanism underlying the Aβ-reducing activity of (20S)-Rg3 and determined that it modulates the phosphoinositide synthetic pathway by increasing PI4KIIα activity. Our work provides evidence that the activity/levels of PI4KIIα and therefore modulation of PI(4)P and PI(4,5)P2 levels in the neuronal membrane reduce Aβ biogenesis. Thus, our studies reveal a novel association of Aβ-producing machinery and the phosphoinositide pathway.
EXPERIMENTAL PROCEDURES
Compounds
Individual ginsenosides were purified from white ginseng or heat-processed ginseng as described previously (47, 48). Sulindac sulfide and naproxen were purchased from Biomol, and Compound E (49) was kindly provided by Dr. T. Golde (Mayo Clinic, Jacksonville, FL). All other chemicals were from Sigma-Aldrich and Bio-Rad except where indicated.
Cell Lines and Primary Neuron Cultures
CHO cells stably expressing wild-type human APP751 (CHO-APP) were provided by Dr. D. J. Selkoe (50). These cells are the parental cell lines for all stably transfected CHO cell lines used in this study. 7WD4 cells were stably transfected with 10 μg of each plasmid of wild-type PS1 using the SuperFect (Qiagen) transfection reagent according to the manufacturer's protocol. Individual Zeocin-resistant colonies were isolated and screened for PS1 expression by Western blotting. Stable cell lines (CHO-APP/PS1WT) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and penicillin/streptomycin in the presence of 250 μg/ml Zeocin (Invitrogen) and 400 μg/ml G418 (Calbiochem). For transient transfection, cells were transfected in 100-mm dishes with 10 μg of plasmid, and protein expression was verified after 24 h by Western blotting. For drug treatments, serum-containing medium was changed to a reduced serum medium (Opti-MEM, Invitrogen) containing a 50 μm concentration of each compound. Stable PS1 Neuro2a APPsw transfectants were kindly provided by S. H. Kim and S. Sisodia (University of Chicago). Primary hippocampal cultures were prepared from 1-day-old B6SJLF1/J mouse pups as described previously (11). Cells were grown in Eagle's minimum essential medium supplemented with 10% heat-inactivated fetal calf serum, 45 mm glucose, 1% Eagle's minimum essential medium vitamin solution, and 2 mm glutamine. After 24 h, this medium was replaced by Neurobasal A medium supplemented with 2% B27 nutrient mixture, 1% heat-inactivated fetal calf serum, 0.4 mm glutamine, and 6.6 ng/ml 5-flurodioxyuridine and 16.4 ng/ml uridine to suppress cell division. To prevent the induction of plasticity by spontaneous synaptic activity, kynurenic acid (0.5 mm) was also included in the culture medium.
Analyses of Aβ and APP
Quantitation and detection of all Aβ species were performed using either Western blot analysis or sandwich ELISA (Invitrogen). Media were collected after 6–8 h of drug treatment, and cell debris was removed by centrifugation. Conditioned media (1.0 ml) were incubated at 4 °C overnight with 7N22 (1:500; Invitrogen) in a rotator. Protein G Plus/A-agarose beads (Calbiochem) were added, and rotational incubation was continued for an additional 4 h. The beads were then washed twice with ice-cold immunoprecipitation buffer (0.1% Triton X-100, 140 mm NaCl, 0.025% sodium azide, 10 mm Tris-HCl, pH 8.0; Ref. 84) and once with 10 mm Tris-HCl, pH 8.0 containing 0.025% sodium azide. Samples were collected by centrifugation at 8,000 × g for 2 min using Spin-X (Costar). Samples were resolved by Tricine/urea gels (85) and subjected to Western blot analyses using 6E10. Aβ sandwich ELISA was performed using commercial Aβ40 and Aβ42 ELISA kits (Invitrogen) according to the manufacturer's protocol. Full-length APP was detected by Western blot analysis using the APP-CTmax antibody (10). Media were collected after 8 h of treatment, and cell debris was removed by centrifugation. A volume of 400 μl of conditioned medium was immunoprecipitated with 7N22 (for α-secretase-derived soluble APP (sAPPα); Invitrogen) and 800 μl of medium with sβWT (for β-secretase-derived soluble APP (sAPPβ)). sβWT antibody was described previously (51). Briefly, the antibody was generated by immunizing rabbits with keyhole limpet hemocyanin-conjugated peptides corresponding to the C-terminal region of secreted APPβ, (C)GGGISEVKM. An additional cysteine residue, indicated as (C), was inserted as a spacer. Samples were separated on Tris/glycine gels (4–20%). Either sAPPα or sAPPβ was detected by Western blot analysis using LN27 (which recognizes the first 200 amino acids in APP N terminus; Invitrogen).
Lipid Raft Fractionation
CHO-APP/PS1 and brain tissue from APP/PS1 mice 65 were used for isolation of lipid rafts as described previously (26) with minor modifications: Lubrol WX was replaced with Brij98, the homogenization step was skipped, and different discontinuous sucrose gradient steps were performed (5–40%). Cells grown to confluence in two 150-mm dishes were washed twice with ice-cold phosphate buffer saline and scraped into 1 ml of lysis buffer (1% Brij98 in 25 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5 mm EDTA) supplemented with a protease inhibitor tablet (Roche Applied Science), and lysates were incubated at 37 °C for 10 min. Solubilized cell lysate was then adjusted to contain 40% sucrose by the addition of an equal volume of 80% sucrose in lysis buffer and transferred to a 12-ml ultracentrifuge tube. A discontinuous sucrose gradient was then formed by the addition of 35% sucrose (6 ml) and 5% sucrose (4 ml) and centrifuged at 39,000 rpm for 18 h in an SW41 rotor (Beckman) at 4 °C (fraction 1 (top) to fraction 12 (bottom)). Twelve 1-ml fractions were collected starting with the top of the gradient, and equal volumes of each fraction were analyzed by Western blotting.
Immunocytochemistry
CHO-APP-PS1WT cells were transiently transfected with GFP-PI4KIIα. After an overnight incubation in the presence or absence of (20S)-Rg3, cells were fixed in 4% paraformaldehyde, permeabilized, and blocked for 1 h in 10% normal goat serum. Primary antibodies (PS1-NT and flotillin-1) were diluted in 10% normal goat serum containing 0.2% Triton X-100 and incubated overnight at 4 °C. Organelle localization was detected using anti-flotillin-1 (BD Biosciences). Following incubation of primary antibody, cells were washed and incubated for 1 h at room temperature with Alexa Fluor-conjugated secondary antibody (Invitrogen). Immunostained cells were mounted using Vectashield mounting medium (Vector Laboratories) and imaged using a Nikon C1 digital confocal system. Quantification analysis using ImageJ image analysis software was performed to measure the co-localization of PS1 or PI4KIIα with flotillin-1. The percent co-localization was calculated as the area of the merged signal divided by the total area of the flotillin-1 signal in the image. This analysis was performed on 10 cells. The co-localization data sets of (20S)-Rg3-treated and non-treated cells were compared using Student's t test to measure statistical significance.
Lipid Analysis
The analysis of anionic phospholipid content of cells was carried out as described (11, 52). Briefly, cells grown in 100-mm dishes were scraped into 0.75 ml of ice-cold MeOH, 1 m HCl (1:1) supplemented with 2 mm AlCl3. Lipids were then extracted and deacylated by incubation with 0.5 ml of methylamine reagent (MeOH, 40% methylamine in water, n-butyl alcohol, water (47:36:9:8)) at 50 °C for 45 min. The aqueous phase was dried under N2, resuspended in 0.5 ml of n-butyl alcohol:petroleum ether:ethyl formate (20:40:1), and extracted twice with an equal volume of water. Aqueous extracts were dried, resuspended in water, and subjected to anion-exchange HPLC on an Ionpac AS11-HC column (Dionex). Negatively charged glycerol headgroups were eluted with a 1.5–86 mm KOH gradient and detected on line by suppressed conductivity in a Dionex ion chromatography system equipped with an ASRS ULTRA II self-regenerating suppressor. Individual peaks were identified, and peak areas were calculated using Chromeleon software (Dionex). Using deacylated anionic phospholipids as standards, lipid masses were calculated and expressed as molar fractions of total anionic phospholipids present in the sample.
Preparation of Lipid Kinases, Lipid Kinase Activity Assay, and Thin Layer Chromatography (TLC)
Stable PS1 Neuro2a APPsw cells cultured on 15-cm culture dishes were transfected with PI4KIIα or the kinase-inactive mutant (D308A) of PI4KIIα containing a C-terminal HA epitope tag (kindly provided by Dr. Tamas Balla, National Institute of Child Health and Human Development, National Institutes of Health; Ref. 18) for 48 h. Cells were lysed in 1 ml of lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1 mm dithiothreitol, 1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride, protease inhibitor tablet), and the lysates were cleared by centrifugation (14,000 × g, 15 min). After preclearing with 20 μl (1:1 slurry) of protein G Plus/A-agarose resin (Calbiochem) for 30 min, 10 μg of monoclonal anti-HA antibody (MMS 101P, Covance) was added to the lysates, and the samples were incubated on a rotating platform at 4 °C overnight. The antibody was then collected on 50 μl of Protein G Plus/A-agarose resin, and the complex was washed three times with 1 ml of lysis buffer before a final wash in the PI kinase buffer. The enzyme was then analyzed by Western blotting, or its activity was assayed on the resin as described below. PIPK1γ90 was purified using glutathione-Sepharose resin (GE Healthcare) as described previously (53). For the lipid kinase assay, stable PS1 Neuro2a APPsw cells were washed twice in ice-cold PBS, gently scraped, and centrifuged at 3,000 rpm (4 °C, 5 min). Cell pellets were resuspended in 0.5 ml of HB buffer (250 mm sucrose, 5 mm Hepes, protease inhibitors) and centrifuged again at 3,000 rpm (4 °C, 5 min). Cell pellets were resuspended in 0.125 ml of HB and homogenized by passing through a syringe/needle followed by centrifugation at 3,000 rpm (4 °C, 5 min). 100 μg of protein from the supernatant were used for TLC as described below. Either membrane preparations or partially purified kinase was mixed with 2.5 μl of ATP (final concentration, 50 μm), 2.5 μl of CaCl2 (final concentration, 50 μm), 27.0 μl of EGTA-free kinase buffer (25 mm Hepes, 100 mm KCl, 2.5 mm MgCl2), and 0.5 μl of [γ-32P]ATP (final concentration, 100 nCi/μl), and samples were incubated at 37 °C for 15 min. Reactions were stopped with 700 μl of CHCl3:MeOH (2:1) supplemented with 10 μg/ml brain-derived phosphoinositides. 400 μl of 1 n HCl were added, and samples were vortexed 2 × 10 s and centrifuged for 1 min at 10,000 rpm in a microcentrifuge. The organic phase was washed with 500 μl of MeOH:HCl:H2O (20:20:1). The solvent phase was dried under nitrogen and resuspended in 40 μl of CHCl3:MeOH (2:1). 8 μl of sample were spotted onto a TLC silica plate primed with oxalate solution. TLC was run in chloroform:acetone:methanol:acetic acid:water (64:30:24:32:14, v/v) and developed on film or a phosphorimaging screen.
Neuronal Differentiation of Mouse Embryonic Stem (ES) Cells
Mouse ES cells that harbor the heterozygote knock-out of the PI4KIIα allele (PI4KIIα+/−, AD0414, and AC0537) and two wild-type (E14 p20 and nn13 p19) cell lines were purchased from Sanger Institute Gene Trap Resource. Direct differentiation of pyramidal neurons from ES cells was performed as described previously (54) with some modifications. ES cells were thawed and subsequently feeder cell-deprived for at least two passages in ES medium (DMEM, 15% ES grade FBS, non-essential amino acids, nucleosides, β-mercaptoethanol) supplemented with leukemia inhibitory factor (1,000 units/ml). Prior to plating, cell culture dishes were coated with 0.1% gelatin solution for at least 30 min. ES medium was changed every day. For embryoid body formation, 3 × 106 ES cells were plated onto non-adherent bacterial dishes in EB medium (ES medium with only 10% FBS and without leukemia inhibitory factor) and cultured for 8 days. Medium was changed every 2 days, and 5 mm retinoic acid was added at day 4 for 4 days. Embryoid bodies were dissociated and plated on poly-d-lysine/laminin-coated dishes in N2 medium. Medium was changed after 2 h and again after 24 h. B27-containing differentiation medium was added after 48 h. Subsequently, B27 medium was changed every 2 days. At 8 days postplating, B27 medium was supplemented with β-mercaptoethanol (25 μm). To measure changes in Aβ level, we used a lentiviral system for expression of APP. The Lenti-APPsw construct was made by PCR amplification followed by ligation into pLenti6/V5-D-TOPO (Invitrogen) using the manufacturer's protocol. The following primers were used for the amplification: APPLenti-F, 5′-CACCATGCTGCCCGGTTTGGCACTGCTCCTGC-3′; APPLenti-R, 5′-GATATCTCTAGTTCTGCATCTGCTCAAAGAAC-3′. This vector was co-transfected into HEK293 T cells with ViraPower packaging mixture (Invitrogen) to generate the lentivirus following the manufacturer's protocols. Differentiated ES cell-derived neurons on glass coverslips were fixed with 4% paraformaldehyde for 15 min. After removal of paraformaldehyde, three washes in PBS, and permeabilization with 0.2% (v/v) Triton X-100 for 3 min, cells were blocked in 10% normal goat serum for 3 h at room temperature. Cells were incubated with the primary antibody against neuronal β-tubulin (TUJ1, Covance) in 10% normal goat serum and PBS with 0.2% Triton X-100 overnight at 4 °C. Detection was achieved using fluorescent Alexa Fluor (Invitrogen/Molecular Probes) secondary antibodies (1:500). Cell nuclei were stained with DAPI.
Compound Treatment for AD Mouse Models and Aβ Analysis of the Brain Tissue
The mice used were heterozygous, double transgenic animals expressing both human APP(K670N/M671L) and PS1(M146L) proteins as described previously (55). These Alzheimer disease model mice were age-matched (3 months old) in all experiments with wild-type littermates. Both sets of mice were produced by crossing heterozygous APP mice with heterozygous PS1 mice and were weaned at 3 weeks and genotyped by PCR of digested tail samples. (20S)-Rg3 was prepared in a saline solution containing 0.01% DMSO at a concentration of 10 mg/kg of body weight. This compound (or saline with 0.01% DMSO for controls) was administered daily via intraperitoneal injection. After sacrifice, one hemibrain from each mouse was frozen on dry ice, homogenized in sucrose buffer, and extracted via formic acid for Aβ quantification (86) using a commercial sandwich ELISA kit (Invitrogen). ELISA results are reported as the mean ± S.D. in pmol of Aβ/g of wet brain.
Expression and Purification of Recombinant PI4KIIα
Full-length PI4KIIα was expressed in Escherichia coli as a fusion product with glutathione S-transferase (GST) using pGEX-KG vector. The GST-PI4KIIα fusion protein was generated using the EcoRI-StyI sites of the pGEX-KG vector, which contains human PI4KIIα from pET-GST-PI4KIIα (kindly provided by Dr. T. Balla). pGEX-KG vector was used as a GST control. GST fusion proteins were expressed in E. coli BL21(DE3). Cultures were grown in YTA medium (16 g/liter Tryptone, 10 g/liter yeast extract, 5 g/liter NaCl, pH 7.0) supplemented with ampicillin to an OD of 0.8 and induced overnight at 28 °C by adding isopropyl 1-thio-β-d-galactopyranoside to 1 mm. Cells were harvested and lysed in 25 ml of lysis buffer (50 mm Hepes, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 10% glycerol, 5 mm DTT, 1 mm ATP, EDTA-free protease inhibitor tablet (Roche Applied Science))/1 liter of culture using a high pressure homogenizer (Emulsiflex C-5, Avestin). Triton X-100 was added to 1% to the lysate at this stage. Lysates were cleared by centrifugation (MLA-80, Beckman) at 50,000 rpm (20 min, 4 °C) and applied to a 5-ml glutathione-Sepharose column (GSTrap HP, GE Healthcare). The column was washed with 10 column volumes of lysis buffer with 500 mm NaCl and 1 mm DTT (no ATP) followed by 10 column volumes of lysis buffer with 150 mm NaCl and 1 mm DTT (no ATP). Protein was eluted using a gradient of 1–50 mm glutathione in lysis buffer with 1 mm DTT and no ATP. Aliquots of the resulting fractions were subjected to SDS-PAGE under denaturing conditions. The fractions containing GST-PI4KIIα were pooled and kept frozen at −80 °C.
Reconstituted γ-Secretase Assay
The γ-secretase assay was performed as described previously (27, 29). Membrane fractions isolated from CHO cells were solubilized with 1% CHAPSO and cleared by centrifugation at 100,000 × g for 1 h. The resulting supernatant was incubated with 100 nm C99-FLAG substrate for 4 h in the presence of the indicated phospholipids. Aβ40 and Aβ42 were visualized by Western blot analyses using BA27 and BC05 antibodies, respectively.
Statistical Analysis
Statistical analyses were performed using Student's t test by comparison with control (DMSO) data. Data are presented as means ± S.D. Data for each condition were acquired from at least three independent experiments.
RESULTS
Identification of (20S)-Rg3 as an Aβ-lowering Natural Compound
We initially tested a number of natural products for their ability to reduce Aβ42 production. We treated CHO cells stably expressing human wild-type APP (CHO-APP) (10) with the extracts prepared from either unprocessed ginseng (“ginseng extracts”), heat-processed ginseng extracts, or chromatographic fractions (fractions 1 and 2) enriched with ginsenosides (bioactive components of ginseng extracts) prepared from heat-processed ginseng (Fig. 1A). Historically, prior to ingestion, ginseng roots are often subjected to heat processing, which leads to changes in the chemical structure of active components, resulting in enhanced pharmacological activity and reduced toxicity (56). We found that extracts and one of the fractions prepared from heat-processed ginseng, but not the extracts from unprocessed ginseng, reduced the secretion of Aβ42 (Fig. 1A). To further identify the active compound(s) responsible for the Aβ42-lowering activity, we next tested the effects of individual ginsenosides on Aβ production. The ginsenosides are a family of active components of ginseng that differ by the side chain and sugar moiety attached to a common structural backbone (see Table 1 for chemical structures). We found that several ginsenosides, including 20R and 20S epimers of Rg3, Rk1, and Rg5, inhibited the secretion of Aβ42 (Fig. 1B). Other ginsenosides, including Rb1, Rb2, Rc, Rd, Re, Re, Rg1, and Rg2, had no significant effect on Aβ secretion in the CHO cells (Fig. 1C).
FIGURE 1.

Identification of active compounds that contribute to Aβ42-lowering activity of heat-processed ginseng. A, effects of ginseng extracts on Aβ42 production. CHO cells stably transfected with human APP (CHO-APP cells) were treated with a ginsenoside (saponin)-enriched fraction derived from either unprocessed ginseng (lane 2) or heat-processed ginseng (lane 5) along with fractions 1 and 2, which resulted from further chromatographic separation of heat-processed ginseng extracts. Fraction 2 is enriched for ginsenosides that are uniquely present in heat-processed ginseng but not in unprocessed ginseng (lane 4). Secreted Aβ42 was immunoprecipitated using Aβ42-specific antibody (FCA42) and probed with anti-Aβ antibody 6E10 (top panel, Media). Cell-associated holoAPP (APP-FL) and C-terminal fragments (APP-CTF) were analyzed by Western blot analysis using antibody specific to the APP C-terminal end (R1). B and C, effects of individual ginsenosides from white (B) or heat-processed (C) ginseng on the production of Aβ40 and Aβ42. CHO-APP cells were treated with the indicated compounds at 50 μg/ml for 8 h. Levels of secreted Aβ40 and Aβ42 were determined by ELISA and normalized to cell-associated full-length APP. In CHO-APP cells, average Aβ amounts in control samples were 320 pm for Aβ40 and 79 pm for Aβ42. The relative levels of Aβ40 and Aβ42 were normalized to values obtained from non-treated and vehicle-treated cells and are shown as percent of control. One of three representative experiments is shown. D, chemical structure of (20S)-Rg3. E, dose-dependent inhibition of Aβ secretion by (20S)-Rg3. CHO cells stably expressing human APP and PS1WT (CHO-APP/PS1WT cells) were treated with the indicated concentrations of (20S)-Rg3 for 8 h. Levels of secreted Aβ40 and Aβ42 were determined by ELISA and normalized to cell-associated full-length APP. The relative levels of Aβ40 and Aβ42 were normalized to values from vehicle-treated cells and are shown as percentage of control (data are expressed as mean ± S.D. (error bars); n = 4; *, p < 0.005). F, steady-state levels of full-length APP and APP C-terminal fragments (two upper panels) were examined by Western blot analysis using APP-CTmax antibody. Note that (20S)-Rg3 treatment resulted in a dose-dependent increase in APP C-terminal fragments. F, the effects of (20S)-Rg3 treatment on levels of secreted APP ectodomain. Soluble APP fragments that resulted from α-secretase cleavage of APP (sAPPα) were detected by immunoprecipitation (6E10) and Western blot analyses (LN27) in CHO-APP/PS1WT cells. sAPPβ was detected by immunoprecipitation (sAPPβ antibody) and Western blot analyses (LN27) (two lower panels). G, quantification of Western blot data in F. Note that (20S)-Rg3 treatment did not change the levels of either full-length APP (APP-FL) or secreted APP ectodomains (sAPPα and sAPPβ).
TABLE 1.
Structure-activity relationship between the chemical structure of ginsenosides and their effects on Aβ42 generation
Ginsenosides differ at their three (or two) side chains attached to the common triterpene backbone known as dammarane. The common structure skeleton for each group of ginsenosides is shown at left. CHO-APP cells were treated with the indicated ginsenosides and analyzed for Aβ42 as described in Fig. 1 under the condition of 50 μm, 8-h treatment. Based on the results of three independent experiments, ginsenosides that harbor Aβ42-lowering activity are indicated as either Yes or No. Ginsenosides that affected cell viability at 50 μm are indicated by (a). Glc, d-glucopyranosyl; Ara (pyr), l-arabinopyranosyl; Ara (fur), l-arabinofuranosyl; Rha, l-rhamnopyranosyl; Xyl, d-xylopyranosyl.
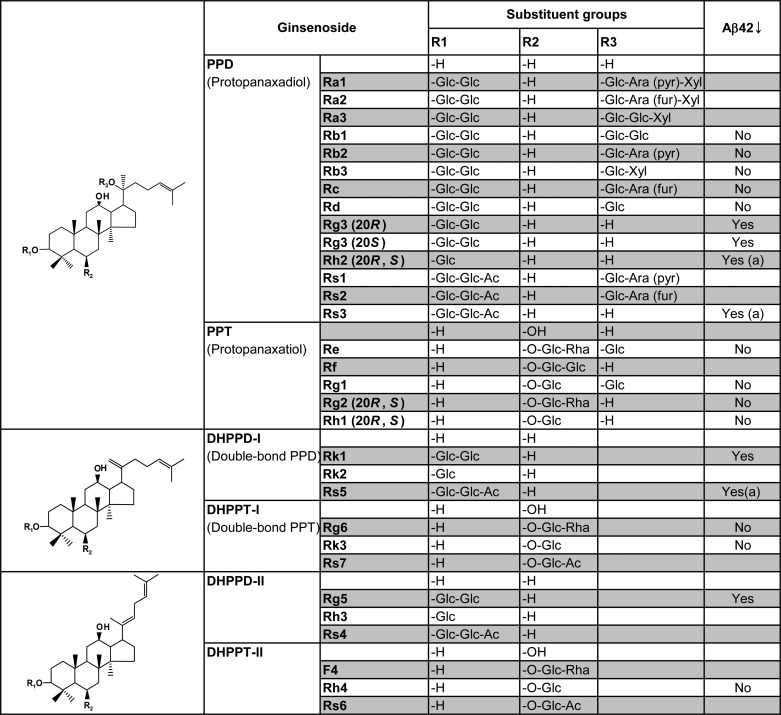
We selected (20S)-Rg3 for further mechanistic studies (Fig. 1D) because this compound was one of the most potent ginsenosides among the tested compounds (Fig. 1B) and is also relatively abundant compared with other trace ginsenosides present in heat-processed ginseng (∼3.6%, w/w; Ref. 57). In these cells, (20S)-Rg3 lowered Aβ42 more potently than Aβ40 in a dose-dependent manner (Fig. 1E). Aβ40-reducing activity was observed only at higher concentrations. (20S)-Rg3 treatment did not affect the steady-state levels of full-length APP (Fig. 1, F and G). In contrast, levels of the C-terminal APP fragments were up-regulated by the treatment. The levels of secreted soluble APP ectodomains resulting from either α- or β-secretase-mediated APP cleavages (sAPPα or sAPPβ) were not affected by the (20S)-Rg3 treatment (Fig. 1, F and G).
Effects of (20S)-Rg3 on Aβ Generation in Cultured Primary Neurons
To validate the Aβ-reducing activity of (20S)-Rg3 in neurons, we next tested its effectiveness in cultured hippocampal neurons derived from Tg2576 mice (58) expressing the Swedish familial AD mutant form of human APP. Treatment of these neurons with (20S)-Rg3 led to the inhibition of both Aβ40 and Aβ42 secretion in a dose-dependent manner (Fig. 2A). In contrast to the results obtained from cell lines (Fig. 1E), the Aβ-lowering potency was similar for both the Aβ40 and Aβ42 species. (20S)-Rg3 treatment did not lead to changes in the steady-state levels of full-length APP, sAPPα, or sAPPβ (Fig. 2B). The C-terminal fragments were elevated in the samples from neurons treated with (20S)-Rg3, suggesting a potential involvement of γ-secretase inhibition in the Aβ-lowering effects of (20S)-Rg3.
FIGURE 2.

(20S)-Rg3 reduces Aβ42 secretion in cultured primary neurons. A, primary hippocampal neurons derived from Tg2575 APP transgenic mice were treated with (20S)-Rg3 for 6 h (at the indicated concentrations). The levels of secreted Aβ42 were measured by ELISA, and the values were normalized to the levels of full-length APP (APP-FL) (data are expressed as mean ± S.D. (error bars); n = 3; *, p < 0.05; **, p < 0.005). B, analysis of APP processing after (20S)-Rg3 treatment. Steady-state levels of full-length APP and APP C-terminal fragments were examined by Western blot analysis using APP-CTmax antibody. Soluble APP fragments that resulted from α-secretase cleavage of APP (sAPPα) were detected by immunoprecipitation (6E10) and Western blot analyses (LN27) in CHO-APP/PS1WT cells. sAPPβ was detected by immunoprecipitation (sAPPβ antibody) and Western blot analyses (LN27).
Effects of (20S)-Rg3 on the Generation of Intracellular Domains (ICDs) from Notch and p75NTR
In addition to its role in Aβ biogenesis, γ-secretase mediates the intramembrane cleavage of other cellular substrates (e.g. Notch, ErbB4, and p75 neurotrophin receptor; for a review, see Ref. 59).
Using a cell-free γ-secretase assay, we tested whether (20S)-Rg3 affected other γ-secretase substrates. γ-Secretase-mediated intramembrane cleavage of Notch1 or the p75 neurotrophin receptor to yield Notch1 or p75 neurotrophin receptor intracellular domains (NICD or p75-ICD, respectively; Refs. 60 and 61) was also unaffected by treatment with (20S)-Rg3 (Fig. 3). Under these conditions, a γ-secretase inhibitor, Compound E (49), effectively inhibited cell-free generation of NICD or p75-ICD, whereas sulindac sulfide, an Aβ42-lowering nonsteroidal anti-inflammatory drug (63), did not affect ICD generation (Fig. 3). Accordingly, it is plausible that normal physiological pathways that require γ-secretase processing of these substrates are not likely to be adversely affected by (20S)-Rg3 treatment.
FIGURE 3.
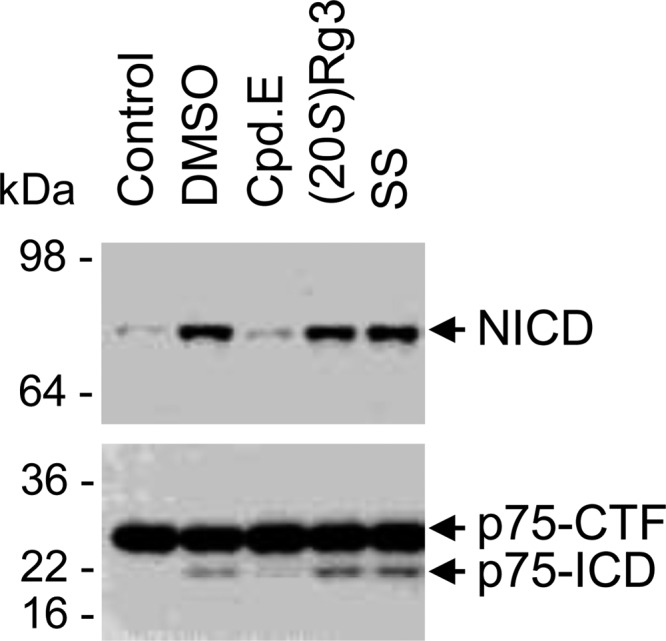
(20S)-Rg3 does not significantly affect the production of the ICDs of Notch1 (NICD) or p75NTR (p75-ICD). Membrane fractions isolated from 293 cells expressing either Notch-ΔE (upper) or p75-ΔE (lower) were incubated in the presence of the following compounds: Compound E (Cpd.E; general γ-secretase inhibitor), sulindac sulfide (SS; an Aβ42-lowering nonsteroidal anti-inflammatory drug), and (20S)-Rg3. p75-ICD was detected using antibodies raised against its cytoplasmic domains, and NICD was detected using a cleavage site-specific NICD antibody. Very low amounts of NICD and p75-ICD were detected in starting material (Control) or in samples treated with Compound E. Generation of NICD and p75-ICD was preserved in samples incubated with DMSO, (20S)-Rg3, and sulindac sulfide.
(20S)-Rg3 Reduces the Association of PS1 with Lipid Rafts
Lipid rafts are DRMs that consist of a characteristic subset of lipids and proteins (64). Abundant evidence indicates that the majority of components in amyloidogenic APP processing machinery, including BACE1 and PS1, are enriched in biochemically isolated DRMs (25, 26, 30, 65–67). Given the predicted lipophilic nature of the steroidal triterpene structure of (20S)-Rg3 (Fig. 1D), we suspected that (20S)-Rg3 may modulate γ-secretase activity by influencing the membrane environment (e.g. lipid rafts). To detect the raft-associated pool of PS1 and other γ-secretase components, we first performed biochemical analyses of PS1-harboring DRMs (25, 26, 30). It has been shown that the association of PS1 with lipid rafts is differentially sensitive to detergents, such as Triton X-100 and Lubrol WX (26). Although Lubrol WX was found to be an optimal detergent for the isolation of PS1-bearing DRMs, it is no longer commercially available. We therefore tested a number of alternate detergents for their ability to separate PS1-enriched DRMs by density gradient centrifugation (data now shown) and identified Brij98 as an optimal detergent that could efficiently separate PS1-harboring lipid rafts (Fig. 4A). Both PS1 C- and N-terminal fragments, which are active forms of the γ-secretase complex (68), were detected in fractions positive for the raft maker flotillin-1 (Fig. 4A). Full-length and C-terminal fragments of APP as well as mature forms of nicastrin and PEN-2 were also detected in the lipid raft fractions (Fig. 4C). In cells treated with (20S)-Rg3, we found that the levels of lipid raft-associated PS1 C-terminal fragments (CTF) and -NTF were significantly reduced (Fig. 4, B–E), whereas the levels of the corresponding fragments in total cell extracts remained unchanged (Fig. 4C). We next performed confocal microscopy to assess the subcellular distribution of PS1 and the lipid raft marker flotillin-1 in intact cells. PS1 and flotillin-1 substantially co-localized in puncta throughout the cytoplasm (Fig. 4F). Treatment of cells with (20S)-Rg3 decreased the co-localization of PS1 with flotillin-1 (Fig. 4G and Ref. 69). In contrast, co-localization of PI4KIIα, another raft-associated protein, with flotillin-1 was not affected by (20S)-Rg3 treatment (data not shown). Collectively, these data suggest that (20S)-Rg3 causes the redistribution of PS1 from lipid rafts to a non-lipid raft membrane compartment.
FIGURE 4.
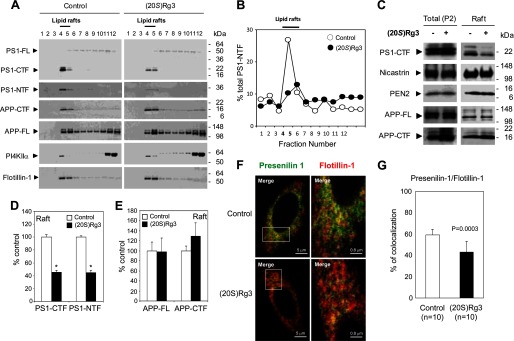
(20S)-Rg3 modulates the association of PS1 with lipid rafts. A, CHO-APP/PS1WT cells were treated with 50 μm (20S)-Rg3 at 37 °C for 6 h and compared with untreated cells. CHO-APP/PS1WT cells were solubilized with 1% Brij98-containing buffer at 37 °C for 10 min and subjected to discontinuous sucrose gradients. The distributions of PS1, APP, PI4KIIα, and flotillin-1 were determined by subjecting equal volumes of each fraction to SDS-PAGE followed by Western blot analysis using specific antibodies. Fractions 4 and 5 contain the detergent-insoluble proteins with light buoyant density and are enriched with a lipid raft marker, flotillin-1 (“Lipid rafts”). B, relative distribution of PS1-NTF in each sucrose density fraction is quantified and plotted as percent of total intensity. Note that the peak of PS1-NTF association in lipid raft is reduced by treatment with (20S)-Rg3 (50 μm) (closed symbol) as compared with control (open symbol). C, Western blot analysis of γ-secretase components and APP in total membrane preparation (P2) compared with pooled lipid raft fractions 4 and 5 from B in the presence or absence of (20S)-Rg3. D and E, quantification of PS1-CTF (D) and PS1-NTF (E) levels from Western blot is shown as percent of control (data are expressed as mean ± S.D. (error bars); n = 3; *, p < 0.005). F, confocal microscopic analysis of the subcellular distribution of PS1 and flotillin-1. CHO-APP/PS1WT cells were incubated in the presence or absence of (20S)-Rg3 and subjected to immunocytochemical analysis. Cells were double labeled with α-PS1-NT (green) and α-flotillin-1 (red) antibodies followed by Alexa Fluor 488-conjugated and Alexa Fluor 568-conjugated secondary antibodies, respectively. Magnification, 100×. Far right panels are 6× magnification of squares shown in the left panels. G, quantification of percent co-localization between PS1 and flotillin-1 using ImageJ. FL, full length.
(20S)-Rg3 Reduces Aβ Levels and the Lipid Raft Association of PS1 in Vivo
To test the effects of (20S)-Rg3 in vivo, we administered (20S)-Rg3 to a mouse model for Aβ pathology (APP/PS1 mouse) at 3 months of age, a stage at which the Aβ burden is significant (55, 70). APP/PS1 mice were treated with (20S)-Rg3 once a day for 4 weeks by intraperitoneal injection (10 mg/kg/day). Aβ ELISA analysis of brain tissues revealed that (20S)-Rg3 treatment resulted in a significant reduction of Aβ40 and Aβ42 in the brain (Fig. 5, A and B), indicating that (20S)-Rg3 harbors Aβ-lowering activity in vivo. To evaluate the effects of (20S)-Rg3 on the association of PS1 with lipid rafts in vivo, forebrain samples from (20S)-Rg3-treated or control APP/PS1 mice were subjected to lipid raft fractionation. Both PS1 C- and N-terminal fragments were detected in the lipid raft fractions (Fig. 5C). The levels of lipid raft-associated C- and N-terminal PS1 fragments were substantially reduced in the samples from the (20S)-Rg3-treated groups compared with controls (Fig. 5D).
FIGURE 5.

Effect of (20S)-Rg3 on Aβ levels and the levels of PS1 fragments associated with lipid rafts in APP/PS1 mice. A and B, (20S)-Rg3 (n = 5) or control (n = 5) was administered daily via intraperitoneal injection into APP/PS1 mice (3 months old) for 3 weeks (10 mg/kg/day). Following treatment, the half-hemisphere of brain tissues from (20S)-Rg3- or vehicle-treated mice was extracted using formic acid, and the relative levels of Aβ40 and Aβ42 were determined by sandwich ELISA. Horizontal bars represent the average. C, the brain tissues were homogenized, solubilized by 1% Brij98 at 37 °C, and subjected to sucrose density gradient centrifugation. Lipid raft association of PS1-CTF, PS1-NTF, and flotillin-1 was analyzed by Western blotting. D, quantification of the Western blot data in C is shown as percent of control (data are expressed as mean ± S.D. (error bars); n = 3; *, p < 0.005).
Modulation of Phosphoinositide Metabolism by (20S)-Rg3
It has been reported previously in the literature that ginsenosides regulate the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB; also called AKT) pathway (71–74), suggesting a possible link between (20S)-Rg3 and phosphoinositide regulation. To explore the possible lipid-modulatory role of (20S)-Rg3, we examined the steady-state anionic phospholipid profile using conductance-based HPLC analysis (11, 12, 52). We found that treatment of mouse cortical neurons with (20S)-Rg3 led to increased levels of both PI(4)P and PI(4,5)P2 (Fig. 6A). In contrast, the levels of other phospholipids, such as phosphoinositol, diphosphatidylglycerol, and phosphatidylserine, were not affected. We next performed a lipid kinase activity assay using a detergent extract and [γ-32P]ATP followed by TLC analysis to determine whether (20S)-Rg3 influences the formation of PI(4)P and/or PI(4,5)P2 (Fig. 6, B and C) (10). Treatment of the cells with (20S)-Rg3 significantly enhanced the incorporation of radiolabeled phosphate into both PI(4)P and PI(4,5)P2, indicating that (20S)-Rg3 increases the formation of both PI(4)P and PI(4,5)P2.
FIGURE 6.

(20S)-Rg3 increases PI(4,5)P2 by activating PI4KIIα. A, treatment of cortical neurons with (20S)-Rg3 (50 μm) leads to increased steady-state levels of PI(4)P and PI(4,5)P2 (n = 6 per treatment; *, p < 0.05). B, lipid kinase and TLC analysis of membranes prepared from stable PS1 Neuro2a APPsw cells treated with vehicle (Control) or 50 μm (20S)-Rg3. C, quantification of the TLC data on PI(4)P and PI(4,5)P2 in B. Treatment with (20S)-Rg3 promotes both PI(4)P and PI(4,5)P2 synthesis (data are expressed as mean ± S.D. (error bars); n = 3; *, p < 0.005; **, p < 0.01). D, (20S)-Rg3 enhances the PI4KIIα activity. A lipid kinase activity assay and TLC analysis were performed using either purified wild-type (IIα) or kinase-dead mutant (IIα-KD) forms of PI4KIIα. The reaction was performed in the absence or presence of 50 μm (20S)-Rg3. E, quantification of the TLC data in D (data are expressed as mean ± S.D. (error bars); n = 3; **, p < 0.01). F, dose-response curve of PI4KIIα activation by (20S)-Rg3. Increasing amounts of (20S)-Rg3 were included in the PI4KIIα lipid kinase assay to monitor the PI(4)P formation. G and H, (20S)-Rg3 has no effect on PIPK1γ. A lipid kinase assay and TLC analysis were performed using 2 μg of recombinant PIPK1γ (data are expressed as mean ± S.D. (error bars); n = 3). I, TLC analysis using either purified wild-type PI4KIIα or PIPk1γ. The reaction was performed in the absence or presence of 50 μm (20S)-Rg3. M, lipid markers run in parallel. J, quantification of the TLC data on PI(4)P and PI(4,5)P2 in I (data are expressed as mean ± S.D. (error bars); n = 3). PS, phosphatidylserine; DPG, diphosphatidylglycerol.
(20S)-Rg3 Potentiates the Activity of PI4KIIα
Because (20S)-Rg3 led to increased steady-state levels and synthesis of both PI(4)P and PI(4,5)P2, we hypothesized that the compound may modulate the activity of a PI lipid kinase (e.g. PI 4-kinase and/or PI 5-kinase) that mediates the formation of PI(4)P, the rate-limiting precursor of PI(4,5)P2 synthesis (13, 15). Among three known isoforms of PI 4-kinases, we particularly focused on the type IIα PI 4-kinase (PI4KIIα) because PI4KIIα is enriched in the brain and has been shown to undergo palmitoylation and localize to lipid rafts where a significant pool of PS1 is concentrated (20, 21, 75). PI4KIIα and PS1 fragments co-localized in the DRM fractions (Fig. 4A). Because overexpression of PI4KIIα was shown to promote the synthesis of PI(4)P as well as PI(4,5)P2 (18), we attempted to determine whether overexpression of PI 4-kinase and subsequent increases in PI(4)P and PI(4,5)P2 could recapitulate the effects elicited by (20S)-Rg3 treatment. Constructs encoding either mammalian wild-type (IIα) or the kinase-dead mutant (IIα-KD) forms of PI4KIIα (HA-tagged) were transfected into stable PS1 Neuro2a APPsw cells. The expressed enzyme was immunoprecipitated using anti-HA affinity beads from cell extracts. Equal amounts of purified PI4KIIα and PI4IIα-KD were subjected to the lipid kinase activity assay using PI as a substrate followed by TLC analysis (Fig. 6D). PI4KIIα, but not the kinase-dead mutant version of this enzyme, induced the incorporation of [γ-32P]ATP into PI(4)P as expected. Incubation of (20S)-Rg3 with the immunoprecipitated enzyme enhanced PI4KIIα activity (Fig. 6, D and E) in a dose-dependent manner (Fig. 6F). In contrast, (20S)-Rg3 up to 50 μm did not confer any detectable effects on the activity of PIPK1γ, a major brain PI(4)P 5-kinase (Fig. 6, G and H) (75–77). The (20S)-Rg3-induced increase in PI4KIIα activity was also observed with recombinant PI4KIIα (data not shown). Co-incubation of (20S)-Rg3 with PI4KIIα and PIPK1γ led to both PI(4)P and PI(4,5)P2 formation, recapitulating the observed PI(4)P and PI(4,5)P2 elevation in (20S)-Rg3-treated cells (Fig. 6, I and J).
PI4KIIα Overexpression Recapitulates the Effects of (20S)-Rg3
Because (20S)-Rg3 potentiates PI4KIIα activity, we tested whether increasing PI4KIIα activity through overexpression exerted similar effects on Aβ42 generation as did (20S)-Rg3 treatment. Aβ42 generation was significantly down-regulated in stable PS1 Neuro2a cells transiently transfected with constructs encoding HA-tagged PI4KIIα (Fig. 7A). The secreted Aβ42 levels were also reduced in CHO cells stably expressing APP and transiently transfected with wild-type PI4KIIα but not in cells transfected with the KD mutant form of PI4KIIα. This indicates that the catalytic activity of PI4KIIα is required for the observed reduction in Aβ42 generation (Fig. 7A). In the PI4KIIα-overexpressing cells, the steady-state level of full-length APP was not affected; however, we observed an increased accumulation of APP C-terminal fragments (Fig. 7B) congruent with the effects of (20S)-Rg3 (Figs. 1F and 2B).
FIGURE 7.

PI4KIIα modulates Aβ production and the association of PS1 with lipid rafts. A, overexpression of active PI4KIIα but not the kinase-dead mutant form of PI4KIIα (PI4KIIα-KD), leads to a reduction in Aβ42 secretion in stable PS1 Neuro2a APPWT cells. Aβ42 levels were normalized to APP full length (data are expressed as mean ± S.D. (error bars); n = 3; **, p < 0.01). B, effect of overexpression of PI4KIIα on the steady-state levels of APP full length (APP-FL) and C-terminal fragments (APP-CTF). C, the effects observed on the association of PS1-CTF with lipid rafts after the overexpression of PI4KIIα resemble the effects of (20S)-Rg3 treatment. CHO-APP/PS1WT cells were transiently transfected with HA-tagged PI4KIIα or PI4KIIα-KD. After a 24-h incubation, cells were solubilized with 1% Brij98 at 37 °C and subjected to sucrose density gradient centrifugation. Lipid raft association of PS1-CTF, PI4KIIα, and flotillin-1 was analyzed by Western blotting using anti-PS1-loop, anti-HA, and anti-flotillin antibodies. PI4KIIα and PI4KIIα-KD co-distribute with PS1-CTF and flotillin-1. D, Western analysis of pooled fractions 4 and 5 from C. Equal amounts of protein were analyzed per lane (20 μg). E, quantification of PS1-CTF levels from Western blot is shown as percent of control (data are expressed as mean ± S.D. (error bars); n = 3; **, p < 0.01). N/S, not significant.
We next determined whether increasing PI4KIIα levels could influence the association of PS1 with lipid rafts. We found that overexpression of PI4KIIα, but not the KD mutant, reduced the association of PS1-CTF with the lipid raft fraction (Fig. 7, C, D, and E), indicating that PI 4-kinase activity is likely to be responsible for the changes in lipid raft localization of PS1. Collectively, our results suggest that both (20S)-Rg3 treatment and PI4KIIα overexpression may modulate both Aβ42 secretion and the association of PS1-CTF with lipid rafts through a mechanism involving PI4KIIα-mediated phosphoinositide modulation.
Phosphoinositide Modulation of γ-Secretase Activity
Our data support the model that (20S)-Rg3 mediates an increase in PI4KIIα activity and a subsequent increase in PI(4)P and PI(4,5)P2 levels. To examine the consequences of elevated PI(4)P and PI(4,5)P2 on γ-secretase activity, we next performed a γ-secretase assay using CHO cell extracts in the presence of four different phosphoinositides (Fig. 8, A and B) (27). The incubation of PI or phosphatidylcholine did not yield any detectable effects on the γ-secretase-mediated generation of both Aβ40 and Aβ42 from the exogenous γ-secretase substrate, C-terminal APP fragments. In contrast, Aβ generation was substantially reduced in the presence of PI(4)P and PI(4,5)P2.
FIGURE 8.

Effects of PI(4)P and PI(4,5)P2 on γ-secretase activity. A, CHAPSO extracts of CHO cell membrane fractions were subjected to incubation with C99-FLAG for 4 h in the presence of the indicated phospholipids at the concentrations shown. Aβ40 and Aβ42 production was detected by Western blot analysis using BA27 and BC05, respectively. B, quantification of the data shown in A as percent of control. PC, phosphatidylcholine.
PI4KIIα Is Necessary for the Aβ-lowering Activity of (20S)-Rg3
To further substantiate the role of PI4KIIα in Aβ generation in neurons, we next used mouse ES cell-derived primary neurons harboring a heterozygous knock-out of the PI4KIIα allele (PI4KIIα+/−; obtained from Sanger Institute Gene Trap Resource). Pyramidal neurons were generated by directed differentiation of ES cells using a modified version of a published protocol (54). The differentiation and morphology of pyramidal neurons from PI4KIIα heterozygous (+/−) ES cells were indistinguishable from neurons derived from wild-type ES cells (Fig. 9A). Among several putative ES cell clones, we identified two clonal lines (AC0537 and AD0414) that expressed significantly reduced amounts of PI4KIIα protein as detected by Western blot analysis (Fig. 9B). The lipid kinase activity in homogenates from the differentiated pyramidal neurons revealed that both basal and (20S)-Rg3-augmented PI 4-kinase activity was significantly reduced in the heterozygous deleted lines (Fig. 9, C and D). Subsequently, the PI4KIIα+/− neurons were infected with lentivirus carrying human APP harboring the Swedish mutation (Fig. 9B) to examine Aβ42 generation. Secreted Aβ42 levels were substantially elevated in neurons derived from PI4KIIα+/− ES cells (Fig. 9E) in contrast to the reduced Aβ42 levels in PI4KIIα-overexpressing cells (Fig. 7A). Thus, our data suggest that PI4KIIα activity may be inversely correlated with Aβ42 production in neurons. Importantly, the PI4KIIα+/− ES cell-derived neurons did not exhibit the pharmacological response to (20S)-Rg3 and were no longer able to reduce Aβ42 (Fig. 9E), indicating that the Aβ42-lowering activity of (20S)-Rg3 requires normal expression of PI4KIIα. These data further support our model that (20S)-Rg3 lowers Aβ42 generation by enhancing the activity of PI4KIIα.
FIGURE 9.
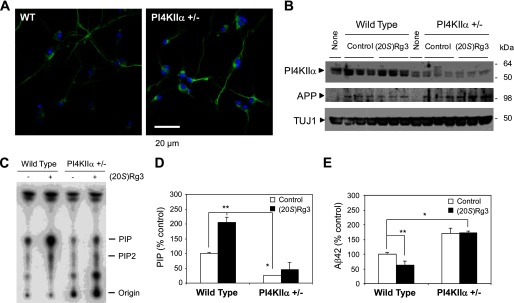
Reduced PI4KIIα leads to elevated Aβ42 production and abolishes the Aβ42-lowering activity of (20S)-Rg3 in neurons. A, directed differentiation of mouse ES cells into pyramidal neurons. Neuronal marker (TUJ1) reactivity and DAPI nuclear stain are shown in representative neurons derived from either wild-type (WT) or PI4KIIα heterozygous knock-out ES cells (+/−). B, Western blot analysis of lysates prepared from pyramidal neurons derived from WT or PI4KIIα+/− ES cells. Human APP was introduced by lentiviral infection of human APP (“None ” denotes no infection of APP virus). C and D, reduced PI 4-kinase activity in ES cell-derived neurons derived from WT or PI4KIIα+/− ES cells. A lipid kinase activity assay and TLC analysis were performed in neurons that were incubated with or without (20S)-Rg3. The quantification of [γ-32P]ATP incorporation into PI(4)P (representing PI4K activity) is shown in D (data are expressed as mean ± S.D. (error bars); n = 3; *, p < 0.005; **, p < 0.01). E, reduction of PI4KIIα results in elevated Aβ42 generation in neurons (white bars). (20S)-Rg3 reduces Aβ42 secretion in wild-type neurons but fails to reduce Aβ42 secretion in the neurons with reduced PI4KIIα (black bars) (data are expressed as mean ± S.D. (error bars); n = 3; *, p < 0.005; **, p < 0.01).
DISCUSSION
In the present study, we identified several triterpenoid natural compounds derived from heat-processed ginseng, including ginsenoside (20S)-Rg3, that reduced Aβ levels in cultured cell lines, neurons, and the brains of an AD mouse model. Our studies also provide evidence that activation of a lipid kinase, PI4KIIα, underlies the mechanism for the Aβ-lowering activity of (20S)-Rg3. Thus, our study reveals a novel mechanism for γ-secretase modulation by a small molecule activator of a key phospholipid synthetic pathway.
We showed that (20S)-Rg3 treatment can increase the levels of PI(4)P and PI(4,5)P2 in cultured cells and enhance the activity of PI4KIIα directly in a cell-free system (Fig. 6). Because PI(4)P is the rate-limiting substrate in the PI(4)P 5-kinase-mediated synthesis of PI(4,5)P2 (18, 19), (20S)-Rg3-evoked activation of PI4KIIα in the presence of PIPK1γ led to increased formation of both PI(4)P and PI(4,5)P2 (Fig. 6, I and J).
The changes in levels of PI(4,5)P2 or PI(4)P can affect γ-secretase activity through several different mechanisms. PI(4,5)P2 has been shown to contribute to regulation of the lateral mobility of membrane proteins potentially in lipid rafts (78–80). Accordingly, PI(4,5)P2-evoked changes in the membrane lipid microenvironment may result in alterations in fluidity and thickness of the membrane, which are the parameters that influence properties of the γ-secretase enzyme, including potency and substrate cleavage patterns (81, 82). We showed that PI(4)P and PI(4,5)P2 confer inhibitory effects on γ-secretase-mediated generation of both Aβ40 and Aβ42 (Fig. 8). Thus, changes in membrane composition by augmentation of PI(4)P and PI(4,5)P2 decreased the γ-secretase-dependent production of Aβ, which is consistent with recent studies on the γ-secretase activity dependence on the lipid environment (27–29). Additionally, increases in PI(4,5)P2 levels can recruit specific PI(4,5)P2-binding molecules to lipid rafts (78–80), which could subsequently influence the γ-secretase activity. Because ginsenosides, including (20S)-Rg3, have steroid-like structures (Table 1), these Aβ-lowering ginsenoside compounds may promote the activity of PI4KIIα by serving as membrane lipid mimics, thereby interacting with PI4KIIα and stabilizing its structure. It is interesting to note that membrane cholesterol levels correlate with PI4KIIα activity (20, 83), and 25-hydroxycholesterol promotes PI4KIIα activity (62). Thus, it is conceivable that (20S)-Rg3 modulates PI4KIIα in a manner similar to cholesterol-dependent regulation of PI4KIIα activity and PI(4)P production.
In addition to modulating phosphoinositide metabolism and Aβ42 secretion, both (20S)-Rg3 treatment and PI4KIIα expression reduced association of PS1 fragments, the catalytic components of γ-secretase complex, with lipid raft fractions (Figs. 4 and 7). (20S)-Rg3-induced reduction of PS1 fragments in lipid rafts is unlikely due to disruption of the integrity of lipid rafts because flotillin levels and PI4KIIα association with rafts were not affected by the treatment. Because overall levels of PS1 fragments were not affected by (20S)-Rg3, the reduced association of PS1 fragments with lipid rafts and subsequent change in γ-secretase activity in (20S)-Rg3-treated cells and brain tissue is likely due to redistribution of PS1 fragments from lipid rafts to a non-lipid raft membrane compartment.
Acknowledgments
We thank P. De Camilli for the anti-PI4KIIα antibody and GST-PIPK1γ, G. Thinakaran for PS1 antibodies, T. Balla for PI4KIIα cDNAs, B. Rosenthal and J. Trufant for experimental assistance, S. Voronov for help with lipid analyses, and L. B. J. McIntire for critical comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 NS074536 (to T.-W. K.) and R01 NS056049 (to G. D. P.). This work was also supported by a grant from the Korea Society of Ginseng (to T.-W. K.).
- Aβ
- amyloid β-peptide
- PS
- presenilin
- AD
- Alzheimer disease
- PI4K
- phosphatidylinositol 4-kinase
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- APP
- β-amyloid precursor protein
- PI(4)P
- phosphatidylinositol 4-phosphate
- PI
- phosphatidylinositol
- DRM
- detergent-resistant membrane domain
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- PIPK1γ
- phosphatidylinositol-phosphate kinase type 1γ
- CHAPSO
- 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonic acid
- ICD
- intracellular domain
- NICD
- Notch1 intracellular domain
- CTF
- C-terminal fragments
- NTF
- N-terminal fragments
- KD
- kinase-dead
- sAPPα
- α-secretase-derived soluble APP
- sAPPβ
- β-secretase-derived soluble APP.
REFERENCES
- 1. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 2. Tanzi R. E., Bertram L. (2005) Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555 [DOI] [PubMed] [Google Scholar]
- 3. O'Brien R. J., Wong P. C. (2011) Amyloid precursor protein processing and Alzheimer's disease. Annu. Rev. Neurosci. 34, 185–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mangialasche F., Solomon A., Winblad B., Mecocci P., Kivipelto M. (2010) Alzheimer's disease, clinical trials and drug development. Lancet Neurol. 9, 702–716 [DOI] [PubMed] [Google Scholar]
- 5. Bateman R. J., Xiong C., Benzinger T. L., Fagan A. M., Goate A., Fox N. C., Marcus D. S., Cairns N. J., Xie X., Blazey T. M., Holtzman D. M., Santacruz A., Buckles V., Oliver A., Moulder K., Aisen P. S., Ghetti B., Klunk W. E., McDade E., Martins R. N., Masters C. L., Mayeux R., Ringman J. M., Rossor M. N., Schofield P. R., Sperling R. A., Salloway S., Morris J. C.; Dominantly Inherited Alzheimer Network (2012) Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N. Engl. J. Med. 367, 795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Golde T. E., Schneider L. S., Koo E. H. (2011) Anti-Aβ therapeutics in Alzheimer's disease, the need for a paradigm shift. Neuron 69, 203–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karran E., Mercken M., De Strooper B. (2011) The amyloid cascade hypothesis for Alzheimer's disease, an appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698–712 [DOI] [PubMed] [Google Scholar]
- 8. Di Paolo G., Kim T. W. (2011) Linking lipids to Alzheimer's disease, cholesterol and beyond. Nat. Rev. Neurosci. 12, 284–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Di Paolo G., De Camilli P. (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657 [DOI] [PubMed] [Google Scholar]
- 10. Landman N., Jeong S. Y., Shin S. Y., Voronov S. V., Serban G., Kang M. S., Park M. K., Di Paolo G., Chung S., Kim T. W. (2006) Presenilin mutations linked to familial Alzheimer's disease cause an imbalance in phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. U.S.A. 103, 19524–19529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berman D. E., Dall'Armi C., Voronov S. V., McIntire L. B., Zhang H., Moore A. Z., Staniszewski A., Arancio O., Kim T. W., Di Paolo G. (2008) Oligomeric amyloid-β peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat. Neurosci. 11, 547–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McIntire L. B., Berman D. E., Myaeng J., Staniszewski A., Arancio O., Di Paolo G., Kim T.-W. (2012) Reduction of synaptojanin 1 ameliorates synaptic and behavioral impairments in a mouse model of Alzheimer's disease. J. Neurosci. 32, 15271–15276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Loijens J. C., Anderson R. A. (1996) Type I phosphatidylinositol-4-phosphate 5-kinases are distinct members of this novel lipid kinase family. J. Biol. Chem. 271, 32937–32943 [DOI] [PubMed] [Google Scholar]
- 14. Frere S. G., Chang-Ileto B., Di Paolo G. (2012) Role of phosphoinositides at the neuronal synapse. Subcell. Biochem. 59, 131–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Balla A., Balla T. (2006) Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol. 16, 351–361 [DOI] [PubMed] [Google Scholar]
- 16. Guo J., Wenk M. R., Pellegrini L., Onofri F., Benfenati F., De Camilli P. (2003) Phosphatidylinositol 4-kinase type IIα is responsible for the phosphatidylinositol 4-kinase activity associated with synaptic vesicles. Proc. Natl. Acad. Sci. U.S.A. 100, 3995–4000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Simons J. P., Al-Shawi R., Minogue S., Waugh M. G., Wiedemann C., Evangelou S., Loesch A., Sihra T. S., King R., Warner T. T., Hsuan J. J. (2009) Loss of phosphatidylinositol 4-kinase 2α activity causes late onset degeneration of spinal cord axons. Proc. Natl. Acad. Sci. U.S.A. 106, 11535–11539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Balla A., Tuymetova G., Barshishat M., Geiszt M., Balla T. (2002) Characterization of type II phosphatidylinositol 4-kinase isoforms reveals association of the enzymes with endosomal vesicular compartments. J. Biol. Chem. 277, 20041–20050 [DOI] [PubMed] [Google Scholar]
- 19. Pan W., Choi S. C., Wang H., Qin Y., Volpicelli-Daley L., Swan L., Lucast L., Khoo C., Zhang X., Li L., Abrams C. S., Sokol S. Y., Wu D. (2008) Wnt3a-mediated formation of phosphatidylinositol 4,5-bisphosphate regulates LRP6 phosphorylation. Science 321, 1350–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waugh M. G., Minogue S., Chotai D., Berditchevski F., Hsuan J. J. (2006) Lipid and peptide control of phosphatidylinositol 4-kinase IIα activity on Golgi-endosomal Rafts. J. Biol. Chem. 281, 3757–3763 [DOI] [PubMed] [Google Scholar]
- 21. Barylko B., Mao Y. S., Wlodarski P., Jung G., Binns D. D., Sun H. Q., Yin H. L., Albanesi J. P. (2009) Palmitoylation controls the catalytic activity and subcellular distribution of phosphatidylinositol 4-kinase IIα. J. Biol. Chem. 284, 9994–10003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haass C. (2004) Take five-BACE and the γ-secretase quartet conduct Alzheimer's amyloid β-peptide generation. EMBO J. 23, 483–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Urban S., Wolfe M. S. (2005) Reconstitution of intramembrane proteolysis in vitro reveals that pure rhomboid is sufficient for catalysis and specificity. Proc. Natl. Acad. Sci. U.S.A. 102, 1883–1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wrigley J. D., Schurov I., Nunn E. J., Martin A. C., Clarke E. E., Ellis S., Bonnert T. P., Shearman M. S., Beher D. (2005) Functional overexpression of γ-secretase reveals protease-independent trafficking functions and a critical role of lipids for protease activity. J. Biol. Chem. 280, 12523–12535 [DOI] [PubMed] [Google Scholar]
- 25. Wada S., Morishima-Kawashima M., Qi Y., Misono H., Shimada Y., Ohno-Iwashita Y., Ihara Y. (2003) γ-Secretase activity is present in rafts but is not cholesterol-dependent. Biochemistry 42, 13977–13986 [DOI] [PubMed] [Google Scholar]
- 26. Vetrivel K. S., Cheng H., Lin W., Sakurai T., Li T., Nukina N., Wong P. C., Xu H., Thinakaran G. (2004) Association of γ-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 279, 44945–44954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Osawa S., Funamoto S., Nobuhara M., Wada-Kakuda S., Shimojo M., Yagishita S., Ihara Y. (2008) Phosphoinositides suppress γ-secretase in both the detergent-soluble and -insoluble states. J. Biol. Chem. 283, 19283–19292 [DOI] [PubMed] [Google Scholar]
- 28. Osenkowski P., Ye W., Wang R., Wolfe M. S., Selkoe D. J. (2008) Direct and potent regulation of γ-secretase by its lipid microenvironment. J. Biol. Chem. 283, 22529–22540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Holmes O., Paturi S., Ye W., Wolfe M. S., Selkoe D. J. (2012) Effects of membrane lipids on the activity and processivity of purified γ-secretase. Biochemistry 51, 3565–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wahrle S., Das P., Nyborg A. C., McLendon C., Shoji M., Kawarabayashi T., Younkin L. H., Younkin S. G., Golde T. E. (2002) Cholesterol-dependent γ-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 9, 11–23 [DOI] [PubMed] [Google Scholar]
- 31. Koehn F. E., Carter G. T. (2005) The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 4, 206–220 [DOI] [PubMed] [Google Scholar]
- 32. Newman D. J., Cragg G. M. (2012) Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 75, 311–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim J., Lee H. J., Lee K. W. (2010) Naturally occurring phytochemicals for the prevention of Alzheimer's disease. J. Neurochem. 112, 1415–1430 [DOI] [PubMed] [Google Scholar]
- 34. Williams P., Sorribas A., Howes M. J. (2011) Natural products as a source of Alzheimer's drug leads. Nat. Prod. Rep. 28, 48–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lü J. M., Yao Q., Chen C. (2009) Ginseng compounds, an update on their molecular mechanisms and medical applications. Curr. Vasc. Pharmacol. 7, 293–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim S. K., Park J. H. (2011) Trends in ginseng research in 2010. J. Ginseng Res. 35, 389–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wesnes K. A., Ward T., McGinty A., Petrini O. (2000) The memory enhancing effects of a Ginkgo biloba/Panax ginseng combination in healthy middle-aged volunteers. Psychopharmacology 152, 353–361 [DOI] [PubMed] [Google Scholar]
- 38. Reay J. L., Kennedy D. O., Scholey A. B. (2005) Single doses of Panax ginseng (G115) reduce blood glucose levels and improve cognitive performance during sustained mental activity. J. Psychopharmacol. 19, 357–365 [DOI] [PubMed] [Google Scholar]
- 39. Lee S. T., Chu K., Sim J. Y., Heo J. H., Kim M. (2008) Panax ginseng enhances cognitive performance in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 22, 222–226 [DOI] [PubMed] [Google Scholar]
- 40. Heo J. H., Lee S. T., Chu K., Oh M. J., Park H. J., Shim J. Y., Kim M. (2008) An open-label trial of Korean red ginseng as an adjuvant treatment for cognitive impairment in patients with Alzheimer's disease. Eur. J. Neurol. 15, 865–868 [DOI] [PubMed] [Google Scholar]
- 41. Heo J. H., Lee S. T., Oh M. J., Park H. J., Shim J. Y., Chu K., Kim M. (2011) Improvement of cognitive deficit in Alzheimer's disease patients by long term treatment with Korean red ginseng. J. Ginseng Res. 35, 457–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baek S. H., Bae O. N., Park J. H. (2012) Recent methodology in ginseng analysis. J. Ginseng Res. 36, 119–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shibata S. (2001) Chemistry and cancer preventing activities of ginseng saponins and some related triterpenoid compounds. J. Korean Med. Sci. 16, S28-S37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim Y. C., Kim S. R., Markelonis G. J., Oh T. H. (1998) Ginsenosides Rb1 and Rg3 protect cultured rat cortical cells from glutamate-induced neurodegeneration. J. Neurosci. Res. 53, 426–432 [DOI] [PubMed] [Google Scholar]
- 45. Chen F., Eckman E. A., Eckman C. B. (2006) Reductions in levels of the Alzheimer's amyloid β peptide after oral administration of ginsenosides. FASEB J. 20, 1269–1271 [DOI] [PubMed] [Google Scholar]
- 46. Kang M. S., Chung S., Kim T. W. (2006) Ginseng-derived compounds ameliorate familial Alzheimer's disease pathological phenotypes via a phospholipase C-dependent mechanism. Alzheimers Dement. 2, S490 [Google Scholar]
- 47. Park I. H., Kim N. Y., Han S. B., Kim J. M., Kwon S. W., Kim H. J., Park M. K., Park J. H. (2002) Three new dammarane glycosides from heat processed ginseng. Arch. Pharm. Res. 25, 428–432 [DOI] [PubMed] [Google Scholar]
- 48. Park I. H., Han S. B., Kim J. M., Piao L., Kwon S. W., Kim N. Y., Kang T. L., Park M. K., Park J. H. (2002) Four new acetylated ginsenosides from processed ginseng (sun ginseng). Arch. Pharm. Res. 25, 837–841 [DOI] [PubMed] [Google Scholar]
- 49. Seiffert D., Bradley J. D., Rominger C. M., Rominger D. H., Yang F., Meredith J. E., Jr., Wang Q., Roach A. H., Thompson L. A., Spitz S. M., Higaki J. N., Prakash S. R., Combs A. P., Copeland R. A., Arneric S. P., Hartig P. R., Robertson D. W., Cordell B., Stern A. M., Olson R. E., Zaczek R. (2000) Presenilin-1 and -2 are molecular targets for γ-secretase inhibitors. J. Biol. Chem. 275, 34086–34091 [DOI] [PubMed] [Google Scholar]
- 50. Koo E. H., Squazzo S. L. (1994) Evidence that production and release of amyloid β-protein involves the endocytic pathway. J. Biol. Chem. 269, 17386–17389 [PubMed] [Google Scholar]
- 51. Okada H., Zhang W., Peterhoff C., Hwang J. C., Nixon R. A., Ryu S. H., Kim T. W. (2010) Proteomic identification of sorting nexin 6 as a negative regulator of BACE1-mediated APP processing. FASEB J. 24, 2783–2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nasuhoglu C., Feng S., Mao J., Yamamoto M., Yin H. L., Earnest S., Barylko B., Albanesi J. P., Hilgemann D. W. (2002) Nonradioactive analysis of phosphatidylinositides and other anionic phospholipids by anion-exchange high-performance liquid chromatography with suppressed conductivity detection. Anal. Biochem. 301, 243–254 [DOI] [PubMed] [Google Scholar]
- 53. Lee S. Y., Voronov S., Letinic K., Nairn A. C., Di Paolo G., De Camilli P. (2005) Regulation of the interaction between PIPKIγ and talin by proline-directed protein kinases. J. Cell Biol. 168, 789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bibel M., Richter J., Schrenk K., Tucker K.L., Staiger V., Korte M., Goetz M., Barde Y. A. (2004) Differentiation of mouse embryonic stem cells into a defined neuronal lineage. Nat. Neurosci. 7, 1003–1009 [DOI] [PubMed] [Google Scholar]
- 55. Trinchese F., Liu S., Battaglia F., Walter S., Mathews P. M., Arancio O. (2004) Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann. Neurol. 55, 801–814 [DOI] [PubMed] [Google Scholar]
- 56. Kim W. Y., Kim J. M., Han S. B., Lee S. K., Kim N. D., Park M. K., Kim C. K., Park J. H. (2000) Steaming of ginseng at high temperature enhances biological activity. J. Nat. Prod. 63, 1702–1704 [DOI] [PubMed] [Google Scholar]
- 57. Kwon S. W., Han S. B., Park I. H., Kim J. M., Park M. K., Park J. H. (2001) Liquid chromatographic determination of less polar ginsenosides in processed ginseng. J. Chromatogr. A 921, 335–339 [DOI] [PubMed] [Google Scholar]
- 58. Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S., Yang F., Cole G. (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274, 99–102 [DOI] [PubMed] [Google Scholar]
- 59. Landman N., Kim T. W. (2004) Got RIP? Presenilin-dependent intramembrane proteolysis in growth factor receptor signaling. Cytokine Growth Factor Rev. 15, 337–351 [DOI] [PubMed] [Google Scholar]
- 60. Schroeter E. H., Kisslinger J. A., Kopan R. (1998) Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393, 382–386 [DOI] [PubMed] [Google Scholar]
- 61. Jung K. M., Tan S., Landman N., Petrova K., Murray S., Lewis R., Kim P. K., Kim D. S., Ryu S. H., Chao M. V., Kim T. W. (2003) Regulated intramembrane proteolysis of the p75 neurotrophin receptor modulates its association with the TrkA receptor. J. Biol. Chem. 278, 42161–42169 [DOI] [PubMed] [Google Scholar]
- 62. Banerji S., Ngo M., Lane C. F., Robinson C. A., Minogue S., Ridgway N. D. (2010) Oxysterol binding protein-dependent activation of sphingomyelin synthesis in the Golgi apparatus requires phosphatidylinositol 4-kinase IIα. Mol. Biol. Cell 21, 4141–4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Weggen S., Eriksen J. L., Das P., Sagi S. A., Wang R., Pietrzik C. U., Findlay K. A., Smith T. E., Murphy M. P., Bulter T., Kang D. E., Marquez-Sterling N., Golde T. E., Koo E. H. (2001) A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414, 212–216 [DOI] [PubMed] [Google Scholar]
- 64. Lingwood D., Simons K. (2010) Lipid rafts as a membrane-organizing principle. Science 327, 46–50 [DOI] [PubMed] [Google Scholar]
- 65. Lee S. J., Liyanage U., Bickel P. E., Xia W., Lansbury P. T., Jr., Kosik K. S. (1998) A detergent-insoluble membrane compartment contains Aβ in vivo. Nat. Med. 4, 730–734 [DOI] [PubMed] [Google Scholar]
- 66. Vetrivel K. S., Cheng H., Kim S. H., Chen Y., Barnes N. Y., Parent A. T., Sisodia S. S., Thinakaran G. (2005) Spatial segregation of γ-secretase and substrates in distinct membrane domains. J. Biol. Chem. 280, 25892–25900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ehehalt R., Keller P., Haass C., Thiele C., Simons K. (2003) Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 160, 113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li Y. M., Xu M., Lai M. T., Huang Q., Castro J. L., DiMuzio-Mower J., Harrison T., Lellis C., Nadin A., Neduvelil J. G., Register R. B., Sardana M. K., Shearman M. S., Smith A. L., Shi X. P., Yin K. C., Shafer J. A., Gardell S. J. (2000) Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405, 689–694 [DOI] [PubMed] [Google Scholar]
- 69. Bickel P. E., Scherer P. E., Schnitzer J. E., Oh P., Lisanti M. P., Lodish H. F. (1997) Flotillin and epidermal surface antigen define a new family of caveolae-associated integral membrane proteins. J. Biol. Chem. 272, 13793–13802 [DOI] [PubMed] [Google Scholar]
- 70. Holcomb L., Gordon M. N., McGowan E., Yu X., Benkovic S., Jantzen P., Wright K., Saad I., Mueller R., Morgan D., Sanders S., Zehr C., O'Campo K., Hardy J., Prada C. M., Eckman C., Younkin S., Hsiao K., Duff K. (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 4, 97–100 [DOI] [PubMed] [Google Scholar]
- 71. Hwang Y. P., Jeong H. G. (2010) Ginsenoside Rb1 protects against 6-hydroxydopamine-induced oxidative stress by increasing heme oxygenase-1 expression through an estrogen receptor-related PI3K/Akt/Nrf2-dependent pathway in human dopaminergic cells. Toxicol. Appl. Pharmacol. 242, 18–28 [DOI] [PubMed] [Google Scholar]
- 72. Hien T. T., Kim N. D., Pokharel Y. R., Oh S. J., Lee M. Y., Kang K. W. (2010) Ginsenoside Rg3 increases nitric oxide production via increases in phosphorylation and expression of endothelial nitric oxide synthase: essential roles of estrogen receptor-dependent PI3-kinase and AMP-activated protein kinase. Toxicol. Appl. Pharmacol. 246, 171–183 [DOI] [PubMed] [Google Scholar]
- 73. Kang J. H., Song K. H., Woo J. K., Park M. H., Rhee M. H., Choi C., Oh S. H. (2011) Ginsenoside Rp1 from Panax ginseng exhibits anti-cancer activity by down-regulation of the IGF-1R/Akt pathway in breast cancer cells. Plant Foods Hum. Nutr. 66, 298–305 [DOI] [PubMed] [Google Scholar]
- 74. Lan T. H., Xu Z. W., Wang Z., Wu Y. L., Wu W. K., Tan H. M. (2011) Ginsenoside Rb1 prevents homocysteine-induced endothelial dysfunction via PI3K/Akt activation and PKC inhibition. Biochem. Pharmacol. 82, 148–155 [DOI] [PubMed] [Google Scholar]
- 75. Volpicelli-Daley L. A., Lucast L., Gong L. W., Liu L., Sasaki J., Sasaki T., Abrams C. S., Kanaho Y., De Camilli P. (2010) Phosphatidylinositol-4-phosphate 5-kinases and phosphatidylinositol 4,5-bisphosphate synthesis in the brain. J. Biol. Chem. 285, 28708–28714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wenk M. R., Pellegrini L., Klenchin V. A., Di Paolo G., Chang S., Daniell L., Arioka M., Martin T. F., De Camilli P. (2001) PIP kinase Iγ is the major PI(4,5)P(2) synthesizing enzyme at the synapse. Neuron 32, 79–88 [DOI] [PubMed] [Google Scholar]
- 77. Di Paolo G., Moskowitz H. S., Gipson K., Wenk M. R., Voronov S., Obayashi M., Flavell R., Fitzsimonds R. M., Ryan T. A., De Camilli P. (2004) Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 431, 415–422 [DOI] [PubMed] [Google Scholar]
- 78. Kwik J., Boyle S., Fooksman D., Margolis L., Sheetz M. P., Edidin M. (2003) Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc. Natl. Acad. Sci. U.S.A. 100, 13964–13969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Golub T., Caroni P. (2005) PI(4,5)P2-dependent microdomain assemblies capture microtubules to promote and control leading edge motility. J. Cell Biol. 169, 151–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tong J., Nguyen L., Vidal A., Simon S. A., Skene J. H., McIntosh T. J. (2008) Role of GAP-43 in sequestering phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) to raft bilayers. Biophys. J. 94, 125–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fraering P. C., Ye W., Strub J. M., Dolios G., LaVoie M. J., Ostaszewski B. L., van Dorsselaer A., Wang R., Selkoe D. J., Wolfe M. S. (2004) Purification and characterization of the human γ-secretase complex. Biochemistry 43, 9774–9789 [DOI] [PubMed] [Google Scholar]
- 82. Winkler E., Kamp F., Scheuring J., Ebke A., Fukumori A., Steiner H. (2012) Generation of Alzheimer disease-associated amyloid β42/43 peptide by γ-secretase can be inhibited directly by modulation of membrane thickness. J. Biol. Chem. 287, 21326–21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Minogue S., Chu K. M., Westover E. J., Covey D. F., Hsuan J. J., Waugh M.G. (2010) Relationship between phosphatidylinositol 4-phosphate synthesis, membrane organization, and lateral diffusion of PI4KIIα at the trans-Golgi network. J. Lipid Res. 51, 2314–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang R., Sweeney D., Gandy S. E., Sisodia S. S. (1996) The profile of soluble amyloid β protein in cultured cell media. Detection and quantification of amyloid β protein and variants by immunoprecipitation-mass spectrometry. J. Biol. Chem. 271, 31894–31902 [DOI] [PubMed] [Google Scholar]
- 85. Qi-Takahara Y., Morishima-Kawashima M., Tanimura Y., Dolios G., Hirotani N., Horikoshi Y., Kametani F., Maeda M., Saido T. C., Wang R., Ihara Y. (2005) Longer forms of amyloid β protein: implications for the mechanism of intramembrane cleavage by γ-secretase. J. Neurosci. 25, 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Schmidt S. D., Nixon R. A., Mathews P. M. (2005) ELISA method for measurement of amyloid-β levels. Methods Mol. Biol. 299, 279–297 [DOI] [PubMed] [Google Scholar]