

Background: Human excision repair removes UV photoproducts in 30-mers in vitro, but this has not been previously observed in vivo.
Results: UV photoproducts are removed in vivo as 30-mers in complex with TFIIH both in general repair and in transcription-coupled repair.
Conclusion: Primary products of excision repair have been isolated in vivo for the first time.
Significance: The study provides novel insights into post-excision steps of human DNA repair.
Keywords: Chromatin, DNA Binding Protein, DNA Damage, DNA Nucleotide Excision Repair, DNA Repair, Dual Incision, TFIIH, Transcription-coupled Repair, Pyrimidine Dimers
Abstract
Nucleotide excision repair is the sole mechanism for removing the major UV photoproducts from genomic DNA in human cells. In vitro with human cell-free extract or purified excision repair factors, the damage is removed from naked DNA or nucleosomes in the form of 24- to 32-nucleotide-long oligomers (nominal 30-mer) by dual incisions. Whether the DNA damage is removed from chromatin in vivo in a similar manner and what the fate of the excised oligomer was has not been known previously. Here, we demonstrate that dual incisions occur in vivo identical to the in vitro reaction. Further, we show that transcription-coupled repair, which operates in the absence of the XPC protein, also generates the nominal 30-mer in UV-irradiated XP-C mutant cells. Finally, we report that the excised 30-mer is released from the chromatin in complex with the repair factors TFIIH and XPG. Taken together, our results show the congruence of in vivo and in vitro data on nucleotide excision repair in humans.
Introduction
Nucleotide excision repair (excision repair) is the sole DNA repair system for removing UV-induced DNA lesions, cyclobutane pyrimidine dimers (CPDs)2 and the (6-4) photoproducts ((6-4)PPs), as well as bulky base adducts induced by numerous chemical carcinogens and chemotherapeutic agents from the human genome (1–3). In addition, it plays a backup role in repairing oxidized and alkylated bases (3). Mutations in genes encoding excision repair proteins cause the human disease xeroderma pigmentosum (XP), which is characterized by extreme sensitivity to sunlight and a 5000-fold increased incidence of skin cancer (4, 5). There are two modes of nucleotide excision repair: general excision repair, which removes lesions from the whole genome, and transcription-coupled repair, which deals with damage in transcribed DNA strands. Human general excision repair has been reconstituted in vitro from six repair factors that include the proteins encoded by the XP genes as well as a general transcription factor and an all-purpose DNA metabolism factor: XPA, RPA, XPC, TFIIH (eight to 10 proteins, including XPB and XPD), XPG, and XPF-ERCCI (6). In contrast, there is currently no in vitro system for eukaryotic transcription-coupled repair and, hence, the mechanistic aspects of this process remain to be elucidated. In this process, RNA polymerase II stalls at the lesions to initiate repair by excision repair factors except XPC, which is not needed for transcription-coupled repair (1–3).
Experiments with the in vitro system revealed that following damage recognition by RPA, XPA, and XPC and proofreading by TFIIH, the XPG and XPF nucleases make incisions at the 6 ± 3 phosphodiester bond 3′ and the 20 ± 5 phosphodiester bond 5′, respectively, to the damaged base, releasing an oligonucleotide 24- to 32-nt in length (canonical/nominal 30-mer) carrying the lesion (7–9). The resulting gap is filled by DNA polymerases δ/ϵ and ligated to produce a 30-nt repair patch and, thus, complete the repair reaction (10). Although excision repair has been investigated in considerable detail, the following questions remain to be addressed. How is the canonical 30-mer released following the dual incisions? Do the dual incisions proceed by the same mechanism in vivo as they do in vitro? Do the concerted dual incisions occur in XP-C mutant cells, which can perform transcription-coupled repair but not general genomic repair? Finally, what happens to the excised oligonucleotides carrying the DNA damage? Recently, we addressed the question of release of the canonical 30-mer in vitro (11). Here we present data that address the other questions regarding the fundamental mechanism of human excision repair.
EXPERIMENTAL PROCEDURES
Cell Lines
A375 cells, a human melanoma cell line with high excision repair activity, were obtained as described previously (12). The following human cell lines were purchased from the NIGMS Human Genetic Cell Repository (Coriell Institute): XPA fibroblasts (XP12BE-SV, GM 04429) and XPC fibroblasts (XP4PA-SV-EB, GM15983) and its complemented cell line (XP4PA-SE2, GM 04429).
The XPA2 cell line was generated in our laboratory using the directions of the manufacturer (Invitrogen) to transfect XPA−/− cells (XP12BE-SV) with Lipofectamine 2000 and a pcDNA3 construct containing XPA with both a 5′ FLAG and a 3′ 6× His epitope. After 3–4 weeks of culturing in DMEM containing geneticin at 0.4 mg/ml, single clones were picked and further expanded in geneticin-containing medium. Expression of wild-type XPA was verified by Western blot analysis of whole cell lysates, DNA sequencing of epitope-tagged recombinant XPA in genomic DNA, and restoration of excision repair activity as assayed with a clonogenic UV survival assay (data not shown). CHO cell lines were purchased from the ATCC (WT, AA8; XPG mutant, UV135; XPF mutant, UV41) or obtained from LH Thompson, Lawrence Livermore National Laboratory (CSB mutant, UV61).
Mammalian cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum at 37 °C in a 5% CO2 humidified chamber. The XP4PA-SE2- and XPA2-transfected cells were maintained under the same conditions with the addition of 0.2 mg/ml hygromycin B or geneticin, respectively.
Antibodies
Antibodies used for immunoprecipitation (IP) included anti-mouse IgG (catalog no. sc-2025), anti-rabbit IgG (catalog no. sc-2027), anti-XPB (catalog no. sc-293), anti-XPA (catalog no. sc-28353), anti-p62 (catalog no. sc-292), and anti-XPC (catalog no.sc-74410) from Santa Cruz Biotechnology; anti-RPA34 (catalog no. NA18) from Calbiochem; anti-XPG (catalog no. A301-485A) from Bethyl; anti-XPF (catalog no. ab17798) and rabbit anti-mouse IgG (catalog no. ab46540) from Abcam; anti-CPD from Kamiya Biomedical; and anti-(6-4)PP from Cosmo Bio.
Immunoblot detection of most of the proteins involved the use of the same antibody that was used for IP. RPA and XPA were detected with antibodies from Bethyl (catalog no. A300-241A) and Santa Cruz Biotechnology (catalog no. sc-853), respectively. XPG was detected with antibodies from Santa Cruz Biotechnology (catalog no. sc-13563) for in vitro reactions and Bethyl (catalog no. A301-484A) for in vivo IP reactions.
Purification of In Vivo-excised Oligonucleotides Containing UV Photoproducts
UV irradiation was performed as described previously (12). Briefly, culture medium was removed, and then cells were washed once with PBS before placing them under a GE germicidal lamp emitting primarily 254-nm UV light (UV-C) connected with a digital timer. Following irradiation, fresh culture medium was added to the cells, which were further incubated for the indicated length of time. The XP-C fibroblasts and their complemented derivative were irradiated with 20 J/m2 of UV-C, and other cells were exposed to 10 J/m2 of UV-C. At the indicated time points, the cells were washed with ice-cold PBS, harvested with a cell scraper in ice-cold PBS, and collected by centrifugation. Low molecular weight DNA was isolated by a modified Hirt procedure (13). Cell pellets from one 150-mm tissue culture dish (∼100 μl of packed cell volume) were resuspended in 400 μl of buffer P1 (10 mm Tris-Cl (pH8.0), 1 mm EDTA, and 100 μg/ml RNase A), lysed by adding 55 μl of 10% SDS, and incubated for 15 min at room temperature. Then, 140 μl of 5 m NaCl was added, and the tubes were inverted gently ∼10 times and stored for at least 8 h at 4 °C. Genomic DNA was removed by centrifugation in an Eppendorf 5418 microcentrifuge at maximum speed (16,873 × g) for 1 h at 4 °C. The supernatant (∼ 550 μl) was treated with 20 μg of proteinase K for 15 min at 55 °C, extracted with phenol/chloroform twice, and then precipitated with ethanol. The pellet was washed with 500 μl of 70% ethanol and resuspended in 10 μl of buffer EB (10 mm Tris-Cl (pH8.5)).
The extracted low molecular weight DNA was subjected to immunoprecipitation with anti-CPD or anti-(6-4)PP antibodies as follows: For each reaction, 5 μl of protein G Dynabeads (Invitrogen, catalog no. 10003D) slurry and 5 μl of anti-rabbit Dynabeads (Invitrogen, catalog no. 11203D) slurry were washed three times with 50 μl of wash buffer I (20 mm Tris-Cl (pH 8.0), 2 mm EDTA, 150 mm NaCl, 1% Triton X-100, and 0.1% SDS) and then incubated with 1 μl of rabbit anti-mouse IgG and 1 μl of anti-CPD or anti-(6-4)PP antibody in 20 μl of IP buffer (20 mm Tris-Cl (pH 8.0), 2 mm EDTA, 150 mm NaCl, 1% Triton X-100, and 0.5% sodium deoxycholate) for 3 h at 4 °C. After incubation, beads were separated from the liquid with a magnet and then mixed with 100 μl of IP buffer and 10 μl of DNA. The mixtures were rotated at 4 °C overnight. The beads were then washed sequentially with 200 μl each of wash buffer I, wash buffer II (20 mm Tris-Cl (pH 8.0), 2 mm EDTA, 500 mm NaCl, 1% Triton X-100, and 0.1% SDS), wash buffer III (10 mm Tris-Cl (pH 8.0), 1 mm EDTA, 150 mm LiCl, 1% Nonidet P-40, and 1% sodium deoxycholate), wash buffer IV (100 mm Tris-Cl (pH 8.0), 1 mm EDTA, 500 mm LiCl, 1% Nonidet P-40, and 1% sodium deoxycholate) and finally twice with TE (10 mm Tris-Cl (pH 8.0) and 1 mm EDTA). The oligonucleotides containing UV photoproducts were eluted by incubation with 100 μl of elution buffer (50 mm NaHCO3, 1% SDS, and 20 μg/ml glycogen) at 65 °C for 15 min. The eluted DNA was then isolated by phenol/chloroform extraction and followed by ethanol precipitation.
Immunoprecipitation of In Vivo Excision Products
Cell pellets from one 150-mm tissue culture dish were resuspended in 800 μl of ice cold buffer A (25 mm Hepes (pH 7.9), 100 mm KCl, 12 mm MgCl2, 0.5 mm EDTA, 2 mm DTT, 12.5% glycerol, 0.5% Nonidet P-40) and incubated for 10 min on ice. Resuspended cells were transferred to an ice-cold Dounce homogenizer/tissue grinder and lysed on ice with 15 strokes using a tight plunger. The chromatin fraction was then pelleted by centrifugation for 30 min at 16,873 × g at 4 °C. The supernatants were harvested, and appropriate antibodies against repair proteins (typically 2–5 μg), as described above, were added. The reactions were rotated for 3–5 h at 4 °C and then incubated overnight with 10 μl of recombinant protein A/G PLUS-agarose (Santa Cruz Biotechnology). After washing three times with 1 ml of buffer A and once with 1 ml of buffer B (25 mm Hepes (pH 7.9), 100 mm KCl, 12 mm MgCl2, 0.5 mm EDTA, 2 mm DTT, 12.5% glycerol, 1% Nonidet P-40), immunoprecipitates were eluted with 400 μl of buffer C (10 mm Tris-Cl (pH 7.5), 1 mm EDTA, and 1% SDS) at 65 °C for 15 min. The low molecular weight DNA from input, flowthrough, and immunoprecipitates were extracted by the modified Hirt procedure as described above. These DNAs were either radiolabeled directly or subjected to a second round of IP with anti-CPD or anti-(6-4)PP antibodies prior to labeling.
DNA Labeling
For 3′-end labeling, the immunoprecipitated DNA was incubated with 20 units of terminal deoxynucleotidyl transferase (NEB) and 1 μCi of [α-32P]3′-deoxyadenosine 5′-triphosphate (cordycepin 5′-triphosphate) (PerkinElmer Life Sciences) for 1 h at 37 °C in 50 μl of total reaction buffer according to the protocol of the manufacturer. For 5′ end labeling, the immunopurified DNA was treated with 1 unit of FastAP thermosensitive alkaline phosphatase (Thermo) in 45 μl of 1× T4 polynucleotide kinase buffer (NEB) for 20 min at 37 °C. After heat inactivation (75 °C, 5 min), the reaction was incubated with 10 units of T4 polynucleotide kinase (NEB) and 3 μCi of [γ-32P]ATP (MP Biomedicals) for 1 h at 37 °C. The labeling reaction was heat-inactivated (65 °C for 20 min) and then incubated with 1 μl of RNaseA/T1 mixture (Thermo) for 30 min at 37 °C to remove contaminating RNAs. After phenol-chloroform extraction and ethanol precipitation, labeled DNAs were resolved in 11% denaturing sequencing gels. For exonuclease digestion assays, the labeled DNA was purified through G25 gel filtration columns (GE Healthcare) before ethanol precipitation.
Exonuclease Digestion Assay
For 5′ → 3′ digestion, 3′ end-labeled DNA was incubated with 22.5 units of RecJf (NEB) for 1 h at 37 °C in a 5-μl reaction according to the recommendations of the manufacturer. For 3′ → 5′ digestion, 5′ end-labeled DNA was incubated with 0.6 units of T4 DNA polymerase (NEB) for 1 h at 37 °C in a 4-μl reaction according to the recommendations of the manufacturer. Reactions were stopped by adding 10 μl of formamide loading buffer and heating for 5 min at 95 °C. Samples were separated in 11% denaturing sequencing gels.
In Vitro Excision Repair and Immunoprecipitation Assay
Internally 32P-labeled DNA substrate (140 bp) containing a single (6-4) UV photoproduct and cell-free extract (CFE) were prepared as described previously (14). Standard excision assays with XPG (UV135) or XPF (UV41) mutant CHO CFE complemented by the appropriate proteins were carried out. Immunoprecipitations with the indicated antibodies were then performed as described previously (11). A 25-μl reaction contained 75 μg of CFE, 24 ng of purified human XPG (for UV135), or 15 ng of purified human XPF-ERCCI (for UV41). Human XPG and XPF-ERCCI proteins were purified as described (14). The mutant CHO CFE and complemented human proteins were used because our antibodies did not recognize hamster XPG and XPF.
Immunoblotting
Proteins from in vitro excision reactions and from cell lysates of UV-irradiated cells were subjected to immunoprecipitation with appropriate excision repair factor antibodies, separated by SDS-PAGE, transferred to Hybond ECL membranes (GE Healthcare), probed with the indicated antibodies, and detected with ECL reagents (GE Healthcare).
RESULTS
Recent in vitro experiments on human DNA excision repair revealed that, following dual incisions, the excised oligomer is released in a complex with TFIIH (11). We wished to find out if this happens in vivo as well. However, it was necessary to first establish that the in vivo and in vitro excision repair patterns were similar.
Dual Incisions Generate Nominal 30-mers in Vivo
Even though in vitro experiments have shown that human excision repair system excises CPD and (6-4)PP by the same dual incision mechanism on naked DNA (6, 7, 15) and in nucleosomes (16–18), it was reasonable to assume that the incision pattern might be different in the higher-order structural organization of chromatin. Indeed, to date, no study has demonstrated that UV photoproducts are released as 30-nt-long oligomers in vivo. Previous studies specifically addressing this question detected CPD in 4- to 6-nt-long oligomers (19–21). However, it could not be ascertained whether these were primary or processed reaction products (21).
To detect the excised oligonucleotides containing UV photoproducts, we used a modified Hirt procedure to purify low molecular weight DNA from irradiated cells incubated for various time periods after irradiation. The DNA was then immunoprecipitated with either anti-CPD or anti-(6-4)PP antibodies, 3′-radiolabeled, and analyzed on a sequencing gel (Fig. 1 and supplemental Fig. S1). Oligonucleotides in the range of 20–30 nucleotides were detected in the A375 human cell line, leading us to conclude that the in vivo excision pattern is similar to the in vitro pattern. To ensure the specificity of the detected signal, we used an XP-A mutant cell line and its complemented derivative. Although the XP-A cell line XP12BE, which is severely defective in nucleotide excision repair because of a splice site mutation (22), showed little to no excision product formation, the complemented derivative excised oligonucleotides containing (6-4)PPs similar to the A375 cell line (Fig. 1, B and C).
FIGURE 1.
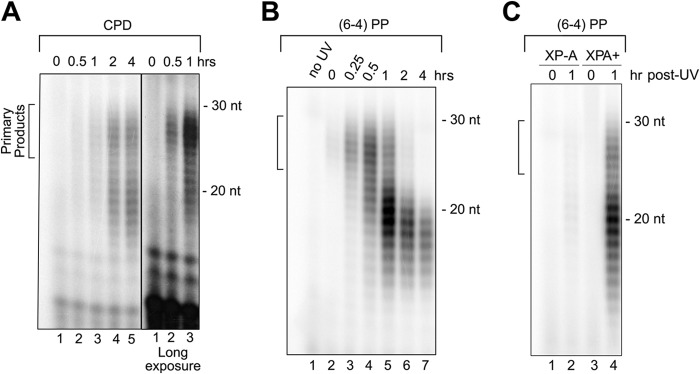
In vivo-excised oligonucleotides containing UV photoproducts. A, excised oligonucleotides containing CPDs were recovered from A375 cells at various time points after exposure to 10 J/m2 of UV-C. Because of the slow rate of excision of CPD, at later time points, the secondary excision products dominate and, therefore, longer exposure of the gel was necessary to detect the primary excision products that are present at early time points. In addition, because of the low signal-to-noise ratio for CPD at early time points, long exposure reveals small molecular weight radiolabeled species not related to repair. B, excised oligonucleotides containing (6-4)PPs were recovered from A375 cells at various time points after exposure to 10 J/m2 of UV-C. C, excised oligonucleotides containing (6-4)PPs were obtained from XP-A and its complemented (XPA+) cell lines at various time points after exposure to 10 J/m2 of UV-C. For each sample, low molecular weight DNA was extracted from one 150-mm tissue culture dish of cells by the modified Hirt procedure and then immunoprecipitated with anti-CPD or anti-(6-4)PP antibodies, respectively. Purified DNAs were 3′ end-labeled and resolved in 11% denaturing sequencing gels. Locations of primary excision products are indicated by brackets.
Two points are worth noting in Fig. 1. First, in agreement with the in vitro data and with in vivo data that measure the disappearance of UV photoproducts from genomic DNA by immunochemical methods (ELISA and slot blot), the (6-4)PPs are removed more rapidly than the CPDs (8, 12, 23). Second, although at earlier time points species in the range of 25–30 nt dominate for both photoproducts, at later time points, smaller size products become more abundant. As will be shown below, this is due to the relatively rapid degradation of the primary excision products.
Mode of Dual Incision in Vivo
In vitro experiments with numerous DNA lesions have shown that human excision repair removes the DNA lesion in the form of 24- to 32-nt-long oligomers by incising the 20 ± 5th phosphodiester bond 5′ and the 6 ± 3rd phosphodiester bond 3′ to the damage, with the specific incision sites being influenced by the type of DNA damage, and the surrounding sequence (1, 2, 15, 18). To find out whether this pattern, which was observed with naked DNA and DNA in nucleosomes, also holds for DNA in chromatin, we recovered the excision products from early time points after UV irradiation so that we would be working mainly with the primary excision products and then analyzed them. To determine the 3′ incision site, the excised oligonucleotides were immunoprecipitated with anti-(6-4)PP or anti-CPD antibodies and then treated with Escherichia coli RecJ exonuclease, which hydrolyzes single-stranded DNA in the 5′-to-3′ direction and stops at the photoproduct (24). The results are shown in Fig. 2A. Both (6-4)PPs and CPDs in the range of 23–29 nt before nuclease treatment (Fig. 2A, lanes 1 and 3) give rise to fragments 6–9 nt in length after RecJ digestion (lanes 2 and 4). To identify the 5′ incision site, we first immunoprecipitated with antibodies against the p62 subunit of TFIIH. Previous in vitro work (11) has shown that the primary excision products are released in a tight complex with TFIIH. Hence, by first immunoprecipitating TFIIH, we eliminate degradation products that would interfere with incision site determination. Then, the TFIIH-associated oligonucleotides were immunoprecipitated by a second round of IP with either anti-(6-4)PP or anti-CPD antibodies. The oligonucleotides were then 5′ end-labeled, treated with 3′ to 5′ exonuclease activity of T4 DNA polymerase (25), and separated on a sequencing gel. Fig. 2B shows that this treatment of 23- to 29-nt long oligomers (lanes 1 and 3) give rise to fragments 18–23 nt in length (lanes 2 and 4). The combined results from the two exonuclease digestion patterns lead us to conclude that in vivo incisions occur 17–22 phosphodiester bonds 5′ and five to eight phosphodiester bonds 3′ to the photoproducts. These values are comparable with those obtained in vitro for these photoproducts under a variety of experimental conditions (3, 7, 9). Thus, we conclude that the human excision nuclease system performs dual incisions in the same manner on naked DNA, nucleosomal DNA (16, 17), and DNA in chromatin.
FIGURE 2.
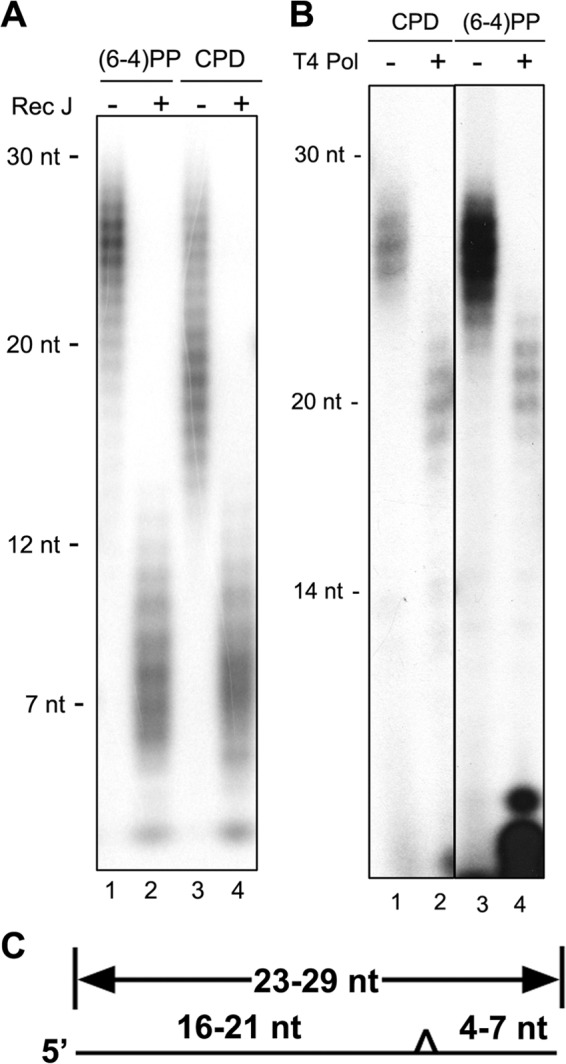
Dual incision pattern in vivo. A, the distance between the DNA lesion and the 3′ end of excised oligonucleotides. Total excised oligonucleotides containing (6-4)PPs or CPDs obtained from A375 cells either 30 min or 2 h, respectively, after 10 J/m2 of UV-C were 3′ end-labeled with 32P, treated by the 5′ to 3′ exonuclease RecJ, and analyzed in a 11% denaturing sequencing gel. B, the distance between the DNA lesion and the 5′ end of primary excised oligonucleotides. The lysates of A375 cells either 30 min or 2 h after 10 J/m2 of UV-C were immunoprecipitated with anti-p62. DNA from the immunoprecipitation reactions were then immunoprecipitated with anti-(6-4)PP or anti-CPD antibodies, respectively. The double IP-purified DNAs were then 5′ end-labeled, treated with the 3′ to 5′ exonuclease of T4 DNA polymerase, and analyzed in a 11% denaturing sequencing gel. C, schematic of dual incisions observed in vivo. The triangle depicts the UV photoproduct in the excised oligomer. Dual incision sites were mapped using Rec J (A) and the exonuclease activity of T4 polymerase (B). The numbers indicate the numbers of nucleotides 5′ and 3′ to the photoproducts, respectively.
Mode of Dual Incision in General Repair and in Transcription-coupled Repair
The incision pattern described so far is on the basis of the excision products generated by general excision repair that operates on duplex DNA in chromatin as well as by transcription-coupled nucleotide excision repair that operates on photoproducts in the transcription bubble of a stalled RNA polymerase (26, 27). We wished to analyze the incision patterns of these two repair modes separately. To this end, we used a CS-B mutant cell line that is known to have normal general repair activity but is defective in transcription-coupled repair (28) and an XP-C mutant cell line that is known to be defective in general repair but to have normal transcription-coupled repair (29, 30).
Cells were irradiated with UV-C, and then cell lysates prepared at various times after irradiation were subjected to immunoprecipitation with anti-p62 (TFIIH) antibodies. The TFIIH-bound DNA was then extracted, 3′ end-labeled, and analyzed on sequencing gels. As seen in Fig. 3A, in both XP-C and CS-B mutant cell lines, the excised oligonucleotides were in the same 25- 30-nucleotide range that is observed in wild-type cells. Thus, it appears that the dual incisions have the same pattern in transcription-coupled (XP-C mutant) and transcription-independent repair (CS-B mutant). Indeed, analysis of the excision product of the XP-C mutant by 5′ exonuclease digestion revealed the same 5′ and 3′ incision patterns as in wild-type cells (supplemental Fig. S2). We note however, that in the XP-C mutant it was necessary to use 2-fold more cells than in the wild-type control to obtain a comparable repair signal because transcription-coupled repair contributes about 20–50% of total repair at early time points after irradiation (30). In contrast, the effect of CSB mutation on the total repair rate is less drastic (28) and, therefore, with equal numbers of irradiated cells, the excision signal from the CS-B mutant was only about 20% less than the signal from the wild-type control. We also noted that the excised oligonucleotides detected in Fig. 3A include those containing CPD and those containing (6-4)PPs because they were isolated by TFIIH immunoprecipitation. The contributions of these photoproducts to the overall excision signal was analyzed by immunoprecipitation with photoproduct-specific antibodies and revealed a rather striking difference between the XP-C mutant and the complemented cell line.
FIGURE 3.

Transcription-coupled excision and general excision products in vivo from XP-C and CS-B mutant cell lines. A, size of primary excised oligonucleotides by transcription-coupled repair and general repair. XP-C human fibroblast and their complemented derivative (XPC+) and CHO CS-B mutant and isogenic wild-type CHO cells were exposed to UV-C and then harvested and lysed immediately or 30 min after irradiation. Cell lysates were immunoprecipitated with anti-p62. DNA from the immunoprecipitations were purified, 3′ end-labeled, and then analyzed in a 11% denaturing sequencing gel. B, XPC-independent repair efficiency of CPDs and (6-4)PPs by transcription-coupled repair. Total excised oligonucleotides containing (6-4)PPs or CPDs obtained from an XP-C mutant and its complemented derivative that were irradiated with 20 J/m2 of UV-C and, 30 min later, were harvested for isolation of the excised oligonucleotides by IP with photoproduct-specific antibodies followed by 3′ end-labeling and analysis in a 11% sequencing gel. The whole image is from the same gel, although the order of the lanes has been changed for clarity. Excision products were quantified with ImageQuant 5.2 software (GE Healthcare) and given as numbers relative to CPD in each set. We note that because anti-CPD and anti-(6-4)PP antibodies have slightly different IP efficiencies, these values should be considered semiquantitative. Nevertheless, the comparison between the wild-type and XP-C mutant compensates for this technical limitation. In addition, because the XP-C mutant cell line has weak excision repair capability (and only transcription-coupled repair), two 150-mm tissue culture dishes of XP-C cells were used for each sample, whereas just one 150-mm tissue culture dish for each sample for other cell lines was used in A and B.
Fig. 3B shows the results of these experiments. Cyclobutane pyrimidine dimers are notoriously poor substrates for the human excision nuclease system, being excised at 10–20% the rate of excision of (6-4)PPs both in vivo (23) and in vitro (8, 31). In agreement with these findings, about 70% of the excised oligonucleotides from the complemented cells contain (6-4)PPs (Fig. 3B, lanes 5–8). Strikingly, in the XP-C mutant, the ratio is reversed. Nearly 70% of the excised oligonucleotides contain CPDs and only about 30% contain (6-4)PPs (Fig. 3B, lanes 1–4). This seemingly paradoxical finding is the result of a unique property of transcription-coupled repair. In transcription-coupled repair, RNA polymerase II makes a very stable elongation complex at the site of the UV photoproduct (27, 32), which is now in part of the transcription elongation bubble (27). Within such a structure, the rate of excision of the poorly recognized CPDs is stimulated 5–10-fold, whereas that of efficiently recognized (6-4)PPs is unaffected (31, 33, 34). As a consequence, CPDs and (6-4)PPs in transcription elongation bubbles are repaired at about the same rate (29). Because at the UV dose used in our experiments there are about 5 times more CPDs than (6-4)PPs (23), the probability of CPDs being in a transcription bubble is 5-fold higher, and because within such a structure they are repaired at the same rate as the (6-4)PPs, the excised oligonucleotides are expected to contain more CPDs than (6-4)PPs, as we observed in our experiments.
Mechanism of Release of the Dual Incision Product In Vivo
Recent in vitro experiments indicate that the excised oligomer is released from DNA in a tight complex with TFIIH (11). These experiments also revealed that secondary excision products produced by nucleolytic degradation of the primary product were bound by RPA. To find out whether, in fact, in vivo the primary excision product was released in complex with TFIIH, we carried out in vitro and in vivo repair experiments in parallel, immunoprecipitated each of the six excision nuclease factors, and analyzed the immunoprecipitates for DNA content. The results are shown in Fig. 4. In agreement with the previous report, in vitro, TFIIH is bound to the primary excision product (Fig. 4A, lane 8 in the left and right panels). Of interest, XPG (Fig. 4A, left panel, lane 9) and to a lesser extent XPF (right panel, lane 9) also bring down the primary excision product. These factors were not tested in the previous study (11). In fact, it appears that XPG strongly interacts with TFIIH (6, 35, 36) and, therefore, that the excision complex is most likely released in the form of DNA-TFIIH-XPG. There is only weak interaction between XPF and TFIIH (35), and it is reasonable to assume that some XPF remains bound to the 5′ end (which is generated by XPF) of the excised oligomer for a period of time.
FIGURE 4.

The primary excised oligonucleotides are released in complex with TFIIH-XPG and XPF both in vitro and in vivo. A, in vitro experiments with cell-free extracts. The indicated proteins were immunoprecipitated from excision repair reactions with an internally labeled 140-bp-long duplex containing (6-4)PP with specific antibodies. Left panel, XPG-mutant CHO (UV135) CFE and purified human XPG were used. Right panel, XPF-mutant CHO (UV41) CFE and purified human XPF-ERCCI were used. DNA from the immunoprecipitations were purified and then analyzed in a 11% denaturing sequencing gel. Immunoprecipitates were also analyzed by SDS-PAGE and Western blotting. B, in vivo experiments. The indicated proteins were immunoprecipitated with specific antibodies from A375 cells 2 h after exposure to 10 J/m2 of UV-C. A portion of immunoprecipitates was analyzed by SDS-PAGE and Western blotting. DNA from the remaining immunoprecipitates were then immunoprecipitated with anti-CPD antibodies, 3′ end-labeled, and analyzed in a 11% denaturing sequencing gel. The XPG immunoprecipitation reaction was performed on extracts from a separate experiment. Locations of the primary excision products are indicated by brackets. We note that the low titer of the XPG antibody necessitated the use of high amount of antibody in IP experiments that resulted in some background in Western blots. Thus, + in A, lane 9 (left panel) and B, lane 15 indicate a cross-reacting IgG light chain that migrates in the vicinity of the XPA band. Similarly, the low amount of XPG in the in vivo experiment and the low titer of the XPG antibody necessitated the use of a high amount of the antibody, which reveals a cross-reacting band (‡) not related to XPG in the flowthrough of B, lane 8.
Importantly, when the immunoprecipitation reactions were performed on lysates of UV-irradiated cells 2 h following irradiation, essentially identical results were obtained (Fig. 4B). TFIIH (Fig. 4B, lane 12) and XPG (lane 15) were almost exclusively bound to the primary excision product in the 24- 30-nt size, whereas RPA was mostly bound to oligonucleotides in the range of 15–20 nucleotides (lane 10). XPF, as in the in vitro reaction, was bound to a small fraction of the primary excision product (Fig. 4B, lane 14).
DISCUSSION
In this study, we have addressed several interrelated questions regarding the basic mechanism of excision repair. Is the dual incision pattern from chromatin in vivo the same as the pattern observed with naked DNA in vitro? What is the incision pattern in transcription-coupled repair observed in XP-C mutant cells? Is the excision product released from the DNA duplex in the same manner in vivo and in vitro? Finally, how stable is the excised oligomer-repair protein complex?
Dual Incision Patterns in Vivo and in Vitro
Our data show that, within the resolution of our assay, the concerted dual incision event that was first discovered in vitro with naked DNA is the manner by which damage is removed from chromatin in vivo. It is known that, during excision repair in vivo, there is a considerable amount of covalent and non-covalent structural changes that facilitate the access of the DNA repair enzymes to the damage (37). Hence, it is quite likely that the excision repair system in the course of its assembly at the damage site the DNA is essentially in the form of a conventional B-form duplex, enabling the dual incisions to proceed as it does in the in vitro reaction with cell-free extracts or the reconstituted excision repair system, that is, incisions at the 20th ± 5 phosphodiester bond 5′ and the 6th ± 3 phosphodiester bond 3′ to the damage.
Dual Incision in the XP-C Mutant
It has been reported that in XP-C mutant cells, UV photoproducts in the template strand of transcribed genes are repaired in an XPC-independent manner and with about the same efficiency as repair elsewhere with the entire set of excision repair factors (29, 30). Even though the E. coli transcription-coupled repair has been reconstituted in vitro (38), currently there is no in vitro system for eukaryotic transcription-coupled repair. However, it has been found that RNA polymerase II stalled at a CPD does not inhibit excision of the photoproduct (27) and that a CPD in a transcription bubble-like substrate can be excised by the five core repair factors in the absence of XPC (33). However, there was no information regarding the excision pattern of UV photoproducts in XP-C mutants in which the excised oligomer is exclusively generated by transcription-coupled repair. In this study, for the first time, the sizes of the excision fragments and the pattern of dual incision in an XP-C mutant have been determined. We find that the dual incision initiated by transcription-coupled repair and carried out with five core repair factors along with RNA polymerase II is essentially the same as that generated by the six core repair factors independently of transcription. It thus appears that RNA polymerase II stalled at a photoproduct functions primarily as a damage recognition factor in a manner that XPC does elsewhere, enabling the five core factors that are present in the actual dual incision complex, even in transcription-independent repair, to assemble at the site of damage with the same topography and carry out dual incision in the same manner. Moreover, as both CPD and (6-4)PPs are equally efficient in blocking RNA polymerase; the polymerase, in turn, functions as an equally efficient damage recognition factor for both lesions. This explains their equally efficient removal by transcription-coupled repair in contrast to their visibly different repair rates in transcription-independent repair, in which the damage recognition factors XPC, RPA, and XPA recognize (6-4)PPs more efficiently than the CPDs.
Release of the Excised Oligonucleotide
Recent work revealed the surprising finding that the excised oligonucleotide is released in a tight complex with TFIIH (11). Of the other repair factors tested, XPA and XPC were not associated with the excision products, and RPA was found to be bound mainly to the secondary excision products that were released from TFIIH. In this study, we have confirmed those findings and have shown that they are a faithful representation of the in vivo events, as the same results were obtained with the in vivo-generated excision products. Furthermore, in this work, we have also tested the two nucleases that are responsible for generating the dual incisions for their associations with the nuclease excision product. We have found that XPG is bound as tightly as TFIIH to the excision product, but this appears to be the consequence of tight association between XPG and TFIIH (6, 35). In contrast, XPF binds only weakly to the excised oligomer, and this binding appears to be independent of TFIIH. Taking these findings and the known protein-protein interactions among the six excision repair factors into account, we propose a modified form of the recently reported model for mammalian excision repair (Fig. 5). The primary excision product is released in the form of DNA (nominal 30-mer)-TFIIH-XPG, and TFIIH and XPG are in contact with one another and with the oligomer within the ternary complex. Some of the excised oligomer is also bound to XPF at the 5′ terminus, but this association is weaker than that of TFIIH and XPG and, as a consequence, only a fraction of excised oligomer-protein complexes contain XPF.
FIGURE 5.
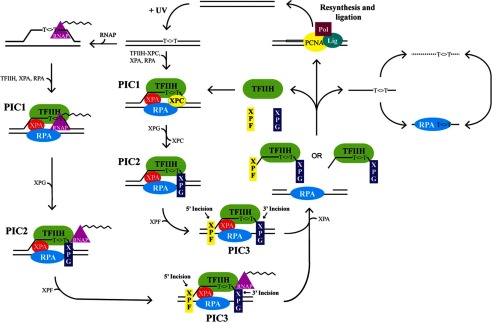
Model of human nucleotide excision repair. UV exposure generates photoproducts in DNA, as shown by the thymine dimer (T<>T). In the general repair pathway, the nucleotide excision repair factors TFIIH-XPC, XPA, and RPA assemble on the damaged DNA in a random and cooperative manner that involves the kinetic proofreading and helicase activities of TFIIH (PIC1, preincision complex 1). XPG enters and stabilizes the preincision complex, which coincides with XPC leaving the damaged DNA (PIC2, preincision complex 2). XPF associates with the other repair factors at the site of damage, followed by incision events by the XPF and XPG nucleases (PIC3, preincision complex 3). In the transcription-coupled repair pathway, transcribing RNA polymerase (RNAP) is blocked by DNA lesions. Then, repair factors (XPC is not required) assemble at the lesion site, and the two nucleases XPG and XPF enter sequentially to form the preincision complex. Because the transcription-coupled repair produces the same nominal 30-nt primary excision products as general repair, the two pathways should have the same PIC3 and the same dual incision events, although the RNAP does not dissociate from the complex. After dual incision, RPA remains on the gapped DNA to promote the subsequent repair resynthesis and ligation steps of excision repair. The excision product is released in complex with the TFIIH-XPG complex and partially with XPF after the dual incision events. Release of the excised oligonucleotides from these repair factors recycles them for new rounds of repair. The released 30-mer can then be targeted for degradation by nucleases or bound by RPA. (Modified from Ref. 11.)
Lifetime of the Excised Oligonucleotide-TFIIH Complex
UV irradiation is known to inhibit transcription, and this was ascribed to a physical barrier of UV photoproducts to the progression of RNA polymerase (2). The discovery that TFIIH is both a core excision repair factor as well as a general transcription factor (36) led to the realization that transcription initiation would also be inhibited as a result of sequestration of TFIIH in repair complexes at damage sites. The recent finding that the excised oligomer is in a tight complex with TFIIH led to the notion of a third mechanism of transcription inhibition by sequestering TFIIH in the post-excision complex (11). The finding that this complex has a half-life of 3.3 h in vitro raised the possibility that this mode of inhibition may have a serious impact on transcription initiation as well as on the rate of excision repair. However, there was the possibility that, under in vivo conditions, the lifetime of this complex might be shorter and, hence, that the rate of TFIIH release may not drastically affect transcription initiation and excision repair kinetics. It is difficult to determine the precise kinetics of release of CPD oligomers from TFIIH in vivo because CPD repair is slow and continues at essentially the same rate for 12 h in human cells, and, thus, excised oligonucleotide-TFIIH complexes persist for as long because as the old complexes disappear, new ones are generated. In contrast, most of the (6-4)PPs are excised in about 2 h in cells irradiated with 10 J/m2 (12) and, hence, any (6-4)PP-containing oligomer that remains associated with TFIIH after about 2 h could be considered to represent the stability of the TFIIH-excised oligomer complex. In fact, even though we do detect some (6-4)PP primary excision products (24–30-mers) 2 h post-irradiation, at 4 h the ∼20-mer degradation products predominate, and by 8 h, the (6-4)PP oligomers have mostly disappeared (data not shown), indicating that the excised oligonucleotides are degraded to small (< 10) nucleotides that are not detectable in our experimental system. Moreover, as apparent from the size range of the excised oligomer with both CPD and (6-4)PPs after about 2 h of UV irradiation, fragments of ∼20 nucleotides in size predominate, and these are nucleolytic degradation products of the primary excision oligomer and are no longer in complex with TFIIH. Thus, it appears that, in vivo, the excised oligomer bound to TFIIH is released from TFIIH at a faster rate than we observed previously in vitro (11). This release will enable TFIIH to enter new rounds of repair and transcription, and the contribution of TFIIH-30-mer complex to inhibition of transcription is not as severe as the effect of TFIIH sequestration in preincision repair complexes. Identifying the factors that promote the faster rate of release of excised oligomers from TFIIH in vivo compared with that in vitro will be important for developing comprehensive models of nucleotide excision repair and for our understanding of how repair efficiency impacts human disease, including skin cancer.
Association of the Excised Oligonucleotide with RPA
Our observation that excised oligomers form complexes with RPA in vivo that are unique from those formed with TFIIH is identical to our recent report using cell-free extract in vitro (11). Because the distribution of lengths of excised oligonucleotides that are associated with RPA are generally shorter than those bound to TFIIH, we assume that the binding of the oligomers to RPA occurs after release from TFIIH and after limited degradation by nucleases in the cell. As the major single-stranded DNA binding protein in humans and other eukaryotes (39, 40), it is perhaps not too surprising that RPA binds to these small, damage-containing oligomers. Whether there is physiological significance to this association remains to be determined.
In conclusion, this study confirms the validity of the in vitro data on human excision repair with regard to its relevance to in vivo events and, importantly, it shows that damage is excised from chromatin with a higher-order structure in the same way that it is excised from a DNA duplex and provides insight into damage recognition and product processing following dual incision by nucleotide excision repair. We would also note that the immunoprecipitation techniques employed here to isolate excised DNA repair intermediates following UV irradiation should be applicable to any DNA lesion that can be processed by the excision repair machinery. Furthermore, the high signal-to-noise ratio our methodology generates should be of particular utility to cell culture and animal model systems in which the biochemistry of excision repair may have been previously difficult to examine in vivo.
This work was supported, in whole or in part, by National Institutes of Health Grant GM32833.

This article contains supplemental Figs. S1 and S2.
- CPD
- cyclobutane pyrimidine dimer
- (6-4)PP
- (6-4) pyrimidine-pyrimidone UV photoproducts
- XP
- xeroderma pigmentosum
- IP
- immunoprecipitation
- nt
- nucleotide(s)
- TFIIH
- transcription factor II H
- RPA
- replication protein A
- CFE
- cell-free extract
- UV-C
- ultraviolet C.
REFERENCES
- 1. Sancar A. (1996) DNA excision repair. Annu. Rev. Biochem. 65, 43–81 [DOI] [PubMed] [Google Scholar]
- 2. Wood R. D. (1997) Nucleotide excision repair in mammalian cells. J. Biol. Chem. 272, 23465–23468 [DOI] [PubMed] [Google Scholar]
- 3. Reardon J. T., Sancar A. (2005) Nucleotide excision repair. Prog. Nucleic Acid Res. Mol. Biol. 79, 183–235 [DOI] [PubMed] [Google Scholar]
- 4. Cleaver J. E. (1968) Defective repair replication of DNA in xeroderma pigmentosum. Nature 218, 652–656 [DOI] [PubMed] [Google Scholar]
- 5. DiGiovanna J. J., Kraemer K. H. (2012) Shining a light on xeroderma pigmentosum. J. Invest. Dermatol. 132, 785–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mu D., Park C. H., Matsunaga T., Hsu D. S., Reardon J. T., Sancar A. (1995) Reconstitution of human DNA repair excision nuclease in a highly defined system. J. Biol. Chem. 270, 2415–2418 [DOI] [PubMed] [Google Scholar]
- 7. Huang J. C., Svoboda D. L., Reardon J. T., Sancar A. (1992) Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer. Proc. Natl. Acad. Sci. U.S.A. 89, 3664–3668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reardon J. T., Sancar A. (2003) Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev. 17, 2539–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Svoboda D. L., Taylor J. S., Hearst J. E., Sancar A. (1993) DNA repair by eukaryotic nucleotide excision nuclease. Removal of thymine dimer and psoralen monoadduct by HeLa cell-free extract and of thymine dimer by Xenopus laevis oocytes. J. Biol. Chem. 268, 1931–1936 [PubMed] [Google Scholar]
- 10. Reardon J. T., Thompson L. H., Sancar A. (1997) Rodent UV-sensitive mutant cell lines in complementation groups 6–10 have normal general excision repair activity. Nucleic Acids Res. 25, 1015–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kemp M. G., Reardon J. T., Lindsey-Boltz L. A., Sancar A. (2012) Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J. Biol. Chem. 287, 22889–22899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gaddameedhi S., Kemp M. G., Reardon J. T., Shields J. M., Smith-Roe S. L., Kaufmann W. K., Sancar A. (2010) Similar nucleotide excision repair capacity in melanocytes and melanoma cells. Cancer Res. 70, 4922–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hirt B. (1967) Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26, 365–369 [DOI] [PubMed] [Google Scholar]
- 14. Reardon J. T., Sancar A. (2006) Purification and characterization of Escherichia coli and human nucleotide excision repair enzyme systems. Methods Enzymol. 408, 189–213 [DOI] [PubMed] [Google Scholar]
- 15. Mu D., Hsu D. S., Sancar A. (1996) Reaction mechanism of human DNA repair excision nuclease. J. Biol. Chem. 271, 8285–8294 [DOI] [PubMed] [Google Scholar]
- 16. Hara R., Mo J., Sancar A. (2000) DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol. Cell. Biol. 20, 9173–9181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hara R., Sancar A. (2002) The SWI/SNF chromatin-remodeling factor stimulates repair by human excision nuclease in the mononucleosome core particle. Mol. Cell. Biol. 22, 6779–6787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang D., Hara R., Singh G., Sancar A., Lippard S. J. (2003) Nucleotide excision repair from site-specifically platinum-modified nucleosomes. Biochemistry 42, 6747–6753 [DOI] [PubMed] [Google Scholar]
- 19. Boling M. E., Setlow J. K. (1966) The resistance of Micrococcus radiodurans to ultraviolet radiation. 3. A repair mechanism. Biochim. Biophys. Acta 123, 26–33 [DOI] [PubMed] [Google Scholar]
- 20. La Belle M., Linn S. (1982) In vivo excision of pyrimidine dimers is mediated by a DNA N-glycosylase in Micrococcus luteus but not in human fibroblasts. Photochem. Photobiol. 36, 319–324 [DOI] [PubMed] [Google Scholar]
- 21. Weinfeld M., Gentner N. E., Johnson L. D., Paterson M. C. (1986) Photoreversal-dependent release of thymidine and thymidine monophosphate from pyrimidine dimer-containing DNA excision fragments isolated from ultraviolet-damaged human fibroblasts. Biochemistry 25, 2656–2664 [DOI] [PubMed] [Google Scholar]
- 22. Cleaver J. E., Thompson L. H., Richardson A. S., States J. C. (1999) A summary of mutations in the UV-sensitive disorders. Xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Hum. Mutat. 14, 9–22 [DOI] [PubMed] [Google Scholar]
- 23. Mitchell D. L. (1988) The relative cytotoxicity of (6-4) photoproducts and cyclobutane dimers in mammalian cells. Photochem. Photobiol. 48, 51–57 [DOI] [PubMed] [Google Scholar]
- 24. Burdett V., Baitinger C., Viswanathan M., Lovett S. T., Modrich P. (2001) In vivo requirement for RecJ, ExoVII, ExoI, and ExoX in methyl-directed mismatch repair. Proc. Natl. Acad. Sci. U.S.A. 98, 6765–6770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doetsch P. W., Chan G. L., Haseltine W. A. (1985) T4 DNA polymerase (3′-5′) exonuclease, an enzyme for the detection and quantitation of stable DNA lesions. The ultraviolet light example. Nucleic Acids Res. 13, 3285–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hanawalt P. C., Spivak G. (2008) Transcription-coupled DNA repair. Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 9, 958–970 [DOI] [PubMed] [Google Scholar]
- 27. Selby C. P., Drapkin R., Reinberg D., Sancar A. (1997) RNA polymerase II stalled at a thymine dimer. Footprint and effect on excision repair. Nucleic Acids Res. 25, 787–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Venema J., Mullenders L. H., Natarajan A. T., van Zeeland A. A., Mayne L. V. (1990) The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc. Natl. Acad. Sci. U.S.A. 87, 4707–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Hoffen A., Venema J., Meschini R., van Zeeland A. A., Mullenders L. H. (1995) Transcription-coupled repair removes both cyclobutane pyrimidine dimers and 6-4 photoproducts with equal efficiency and in a sequential way from transcribed DNA in xeroderma pigmentosum group C fibroblasts. EMBO J. 14, 360–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Venema J., van Hoffen A., Karcagi V., Natarajan A. T., van Zeeland A. A., Mullenders L. H. (1991) Xeroderma pigmentosum complementation group C cells remove pyrimidine dimers selectively from the transcribed strand of active genes. Mol. Cell. Biol. 11, 4128–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mu D., Tursun M., Duckett D. R., Drummond J. T., Modrich P., Sancar A. (1997) Recognition and repair of compound DNA lesions (base damage and mismatch) by human mismatch repair and excision repair systems. Mol. Cell. Biol. 17, 760–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lindsey-Boltz L. A., Sancar A. (2007) RNA polymerase. The most specific damage recognition protein in cellular responses to DNA damage? Proc. Natl. Acad. Sci. U.S.A. 104, 13213–13214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mu D., Sancar A. (1997) Model for XPC-independent transcription-coupled repair of pyrimidine dimers in humans. J. Biol. Chem. 272, 7570–7573 [DOI] [PubMed] [Google Scholar]
- 34. Mu D., Wakasugi M., Hsu D. S., Sancar A. (1997) Characterization of reaction intermediates of human excision repair nuclease. J. Biol. Chem. 272, 28971–28979 [DOI] [PubMed] [Google Scholar]
- 35. Araújo S. J., Nigg E. A., Wood R. D. (2001) Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol. Cell. Biol. 21, 2281–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Drapkin R., Reardon J. T., Ansari A., Huang J. C., Zawel L., Ahn K., Sancar A., Reinberg D. (1994) Dual role of TFIIH in DNA excision repair and in transcription by RNA polymerase II. Nature 368, 769–772 [DOI] [PubMed] [Google Scholar]
- 37. Gong F., Kwon Y., Smerdon M. J. (2005) Nucleotide excision repair in chromatin and the right of entry. DNA Repair 4, 884–896 [DOI] [PubMed] [Google Scholar]
- 38. Selby C. P., Sancar A. (1993) Molecular mechanism of transcription-repair coupling. Science 260, 53–58 [DOI] [PubMed] [Google Scholar]
- 39. Wold M. S. (1997) Replication protein A. A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 66, 61–92 [DOI] [PubMed] [Google Scholar]
- 40. Fanning E., Klimovich V., Nager A. R. (2006) A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 34, 4126–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]