

Background: Mammalian CAP1 functions in actin dynamics, with elusive mechanisms.
Results: Knockdown of CAP1 in HeLa cells leads to alterations in the actin cytoskeleton, cofilin, and FAK phosphorylation and increased cell adhesion and motility.
Conclusion: Mammalian CAP1 regulates actin cytoskeleton, cofilin, and FAK phosphorylation as well as cell adhesion.
Significance: This work presents a novel function for CAP1 in cell adhesion and insights into the CAP1/cofilin interactions.
Keywords: Actin, Cell Motility, Cell Signaling, Cofilin, Cytoskeleton
Abstract
CAP (adenylyl cyclase-associated protein) was first identified in yeast as a protein that regulates both the actin cytoskeleton and the Ras/cAMP pathway. Although the role in Ras signaling does not extend beyond yeast, evidence supports that CAP regulates the actin cytoskeleton in all eukaryotes including mammals. In vitro actin polymerization assays show that both mammalian and yeast CAP homologues facilitate cofilin-driven actin filament turnover. We generated HeLa cells with stable CAP1 knockdown using RNA interference. Depletion of CAP1 led to larger cell size and remarkably developed lamellipodia as well as accumulation of filamentous actin (F-actin). Moreover, we found that CAP1 depletion also led to changes in cofilin phosphorylation and localization as well as activation of focal adhesion kinase (FAK) and enhanced cell spreading. CAP1 forms complexes with the adhesion molecules FAK and Talin, which likely underlie the cell adhesion phenotypes through inside-out activation of integrin signaling. CAP1-depleted HeLa cells also had substantially elevated cell motility as well as invasion through Matrigel. In summary, in addition to generating in vitro and in vivo evidence further establishing the role of mammalian CAP1 in actin dynamics, we identified a novel cellular function for CAP1 in regulating cell adhesion.
Introduction
The actin cytoskeleton is essential for many cellular functions such as morphogenesis, cytokinesis, and endocytosis and cell migration. Consistently, an aberrant actin cytoskeleton underlies a variety of human diseases, such as neurodegenerative diseases and cancer metastasis (1–4). Modulation of the dynamic balance between filamentous actin (F-actin) and globular actin (G-actin)4 is a central mechanism of the regulation of the actin cytoskeleton, and one of the major proteins that control actin dynamics is CAP (adenylyl cyclase-associated protein).
CAP was first identified as a component of the yeast adenylyl cyclase complex (5) and independently in genetic screens to identify components of the yeast Ras/cAMP signaling pathway (5–7). Subsequent studies revealed that CAP homologues are conserved in all eukaryotes studied, including yeast, fungus, plants, and mammals (8). Although the role in Ras signaling does not extend beyond yeast, all CAP homologues appear to function as an actin-regulating protein (8). Not surprisingly, mounting evidence supports roles for CAP deregulation in human cancers; overexpression of both CAP isoforms is found in liver and pancreatic cancers, where they likely promote cancer cell invasion and cancer metastasis (9–13).
CAP homologues share three well conserved structural domains in the N terminus, C terminus, and proline-rich SH3 (Src homology 3) binding middle domain (8). In yeast, the N terminus of CAP binds adenylyl cyclase and is sufficient for mediating Ras signaling (8, 14–16). In contrast, the regulation of actin dynamics (and the actin cytoskeleton) is mediated by two independent mechanisms mediated by both the N termini and the C termini; yeast CAP was first shown to bind and sequester monomer actin (G-actin) through its C terminus to maintain a readily available pool of G-actin essential for dynamic actin cytoskeletal reorganization (14). The C terminus is also the most highly conserved domain among CAP homologues, and morphological defects in yeast from CAP deletion were rescued by expression of the C terminus alone of the protein or a mouse CAP1 (14, 17). In contrast, the later identified second mechanism goes through the N terminus, which cooperates with cofilin, a key actin depolymerization factor (ADF) family member, to facilitate cofilin-driven actin filament turnover (18, 19). Recently, the WH2 (Wasp homology 2) domain, which localizes between proline-rich stretches of the middle domain, has also been shown to play an important role in recharging actin monomers to facilitate actin turnover (20). Therefore, CAP is a key actin-regulating protein that controls actin dynamics through multiple mechanisms including functioning in the cofilin-mediated depolymerization cycle (21).
Mammals have two CAP isoforms, CAP1 and CAP2, and the same isoforms from different mammalian species share extremely high homology, whereas the homology between two isoforms from the same species is relatively low (8, 22). CAP1 is ubiquitously expressed in almost all tissues and cells, whereas CAP2 has a more restricted expression pattern and is found predominantly in skeletal muscle, cardiac muscle, brain, and skin (22, 23). Taken together, CAP1 appears to be required in most cells, yet CAP2 appears to have unique roles required for specific cells or tissues. Most studies so far have been on CAP1, although recent studies have shed light on the role of CAP2 in the cardiovascular system (23, 24).
Using a stable knockdown (KD) paradigm along with a rescue strategy, we efficiently knocked down CAP1 in HeLa cells. CAP1 KD led to increased actin filaments, similar to previous observations in fibroblasts (25). Unexpectedly, however, we found that CAP1-depleted HeLa cells also had greatly elevated cell motility, invasion, and adhesion. The role of CAP in cell adhesion appears to be through its interactions with the adhesion molecules focal adhesion kinase (FAK) and Talin. Moreover, CAP1 KD cells reduced cofilin phosphorylation. This study also identifies a novel cellular function for CAP1 in regulating cell adhesion, a cellular process that closely cooperates with actin dynamics to drive cell motility (26, 27).
EXPERIMENTAL PROCEDURES
Miscellaneous Reagents, Cell Culture, and Transfection
The monoclonal antibody against human CAP1 has been previously described (28). Antibodies against LIM kinase, phospho-LIM kinase, phospho-cofilin Ser-3, and phospho-FAK Tyr-397 were from Cell Signaling Technology (Danvers, MA). The cofilin antibody was from Cytoskeleton Inc. (Denver, CO). The GFP antibody, the vinculin antibody, and latrunculin A (LA) were from Sigma-Aldrich. Protein A/G beads, goat anti-Talin (C20), goat anti-actin, mouse anti-GAPDH, mouse anti-FAK, tubulin antibody, emerin antibody were all from Santa Cruz Biotechnology Inc. (Dallas, TX). Alexa Fluor 488 phalloidin, Alexa Fluor 488 goat anti-rabbit IgG (H+L), fibronectin, and PDGF were from Invitrogen. The subcellular fractionation kit was from Thermo Scientific, and both Slingshot and phospho-Slingshot antibodies were from ECM Biosciences (Versailles, KY). The Matrigel was from BD Biosciences, the tissue culture plates for fluorescence imaging were from MatTek Corp. (Ashland, MA), and the FBS was from HyClone Laboratories Inc. (Logan, UT). Cells were maintained at 37 °C supplemented with 5% CO2, as described previously (29) along with preparation of cell lysates. Transfection reagent FuGENE 6 was from Roche Diagnostics and was used following the manufacturer's instructions.
Expression Plasmids, shRNA Constructs, and Generation of CAP1 Re-expression Cells
The mouse CAP1 GFP fusion protein and deletion mutants have been described previously (30). To generate constructs expressing GST-cofilin (wild type and S3D), wild type (WT) human cofilin and the S3D mutant were subcloned into the BamHI and NotI sites of the pGEX4T-2 vector. The following primers were used in PCR amplifications: forward primer for WT, 5′-GGATCCATGGCCTCCGGTGTGGCT-3′; forward primer for S3D, 5′-GGATCCATGGCCGCCGGTGTGGCT-3′; and the reverse primer for both, 5′-GCGGCCGCTCACAAAGGCTTGCCCTC-3′. To express His6-tagged mouse CAP1 in mammalian cells, mouse Cap1 was cloned into the BamHI and NotI sites of the pcDNA4 vector using a forward primer GGATCCATTATGGCTGACATG and a reverse primer GCGGCCGCTTATCCAGCAATT. shRNA constructs targeting human CAP1 on a pRNA-U6.1/Neo vector and establishment of CAP1 KD HeLa stable cells have been described previously (30). The target sequences for two shRNA constructs are 5′-AGATGTGGATAAGAAGCAT-3′ (S2, nucleotides 519–537) and 5′-CACGACATTGCAAATCAAG-3′ (S3, nucleotides 1074–1092). Cells harboring the empty vector or a scrambled S2 that has the same composition to S2 but does not match any human mRNA were used as controls. To generate a CAP1 plasmid that harbors mismatches (pmtCAP1) allowing re-expression of CAP1 in cells harboring S3 shRNA, mismatches were introduced into the S3 target sequence of the mouse Cap1 gene with the following primers: GTCAACACAACCCTCCAGATAAAAGGCAAAATTAACTCCATTAC (forward, nucleotides shown in italic are those in the shRNA target sequence, and underlined nucleotides indicate introduced mismatches) and ATTTTGCCTTTTATCTGGAGGGTTGTGTTGACACACTTGTAGA (reverse) on a pEGFP-based plasmid and then subcloned into the BamHI and EcoRI sites of pcDNA4(A) vector using the following primers: CGCGGATCCAGATGGCTGACATGCAAAATC (forward) and CCGGAATTCTTATCCAGCGATTTCTGTC (reverse). The mismatches did not change any amino acid sequence. Stable cells re-expressing mtCAP1 were generated by transfection of S3-2 cells with pmtCAP1 followed by selection with Zeocin at 500 μg/ml for 2 weeks.
Phase Imaging and Confocal Immunofluorescence
Cells were observed and phase images were taken using a Zeiss Axiovert 200M microscope (with a 10× objective) driven by the IPLAB4 program. For staining of the actin cytoskeleton, cells were grown on MatTek plates overnight and fixed with 3.7% paraformaldehyde for 30 min before permeabilization with PBS containing 0.5% Triton for 20–30 min. The cells were then incubated with Alexa Fluor 488 phalloidin for 1 h, washed three times with PBS containing 0.1% Triton X-100, and mounted with VECTASHIELD mounting medium from Vector Laboratories Inc. (Burlingame, CA). For staining of cofilin and vinculin, permeabilized cells were blocked by incubating with 3% BSA in PBS supplemented with goat serum for 1 h at room temperature, and cells were then incubated with cofilin or vinculin antibody diluted for 50-fold for 1–2 h at room temperature. The cells were washed three times and incubated with Alexa Fluor 488 goat anti-rabbit IgG (H+L) for 1 h at room temperature. After washing for three times with PBS containing 0.1% Triton X-100, specimens were mounted, and confocal images were taken using a BD Pathway 855 imaging station with a 60× oil objective. Stacks with a 0.5-μm increment were taken for all the specimens.
Cell Spreading/Adhesion Assays and Calculation of Surface Area of Cells
For spreading assays, cells were plated on fibronectin-coated plates, and phase images were taken using a Zeiss Axiovert 200M microscope at the indicated time points. For adhesion assays, 24-well plates were coated with fibronectin for 1 h, and ∼2 × 104 control and CAP1 KD cells were plated. After 10, 20, and 30 min, cells were washed off the plate with PBS three times, and attached cells were stained with crystal violet solution and scored. For cell area measurement, cells were plated on fibronectin-coated plates overnight before phase images were captured. Cell area was measured using ImageJ software (rsb.info.nih.gov/ij). 50 cells per field were measured individually, and results from three independent experiments were analyzed using Student's t test and shown as mean ± S.E.
Cell Migration and Invasion Assays
For wound healing assays, control and CAP1 KD HeLa cells were cultured overnight on 6-well plates until confluent. A scratch (wound) was then introduced to the monolayer of cells using a pipette tip, and cells were further cultured for 16 h before images of the wound were captured using the Zeiss Axiovert 200M microscope. Transwell assays were conducted similarly to previously described (31); subconfluent cells were serum-starved overnight and then detached. ∼2 × 104 cells were plated in triplicate onto the Transwell inserts (8-μm pore size, Corning, NY), which had been placed in the wells of 12-well plates filled with medium containing 10 μg/ml PDGF. The cells were incubated overnight and stained for 15 min with a solution of 0.4% crystal violet and 10% ethanol. Non-migratory cells, which remained at the upper side of the insert, were removed by gentle wiping with a cotton swab, and cells that migrated to the other side of the membrane were counted in four random fields excluding the edge. The data were analyzed using Student's t test. For invasion assays, Transwell inserts coated with Matrigel on a membrane with 8-μm size pores were used, and the assays were conducted otherwise similarly to the Transwell assays.
Fractionation of Pellet and Supernatant Actin Fractions
The pellet and supernatant actin fractionations, which are rich in G actin and F actin, respectively, were prepared by following a recently reported protocol (32) with minor modifications. Cells were cultured to ∼80% confluence on 100-mm plates, and cell lysates were prepared by lysing cells in 2 ml of LAS buffer (50 mm PIPES, pH 6.9, 50 mm NaCl, 5 mm MgCl2, 5 mm EGTA, 5% v/v glycerol, 0.1% Nonidet P-40, 0.1% Triton X-100, 0.1% Tween, 0.1% 2-mercaptoethanol, 1 mm ATP) supplemented with protease inhibitor mixture. Cell lysates were prepared by homogenizing cells with a 25-gauge syringe, and the LA-treated control cells were harvested after treatment with 0.5 μg/ml LA for 1 h. All procedures for fractionation were performed at 37 °C because F-actin is sensitive to the temperature and depolymerizes at room temperature. The cell lysate was centrifuged at 2000 × g for 5 min first to pellet the cell debris, and then the supernatant was further centrifuged at 100,000 × g for 1 h (Beckman-Coulter, Ti 70.1 rotor). The pellet from ultracentrifugation, which contains the filament actin, was then suspended in 2 ml of LAS buffer supplemented with 1% SDS. Proportional pellet (mostly F-actin), supernatant (mostly G-actin) fractions were resolved on SDS-PAGE, and actin in each fraction was detected by Western blotting.
Subcellular Fractionation Assays
To determine cofilin localization in cells, cells were grown overnight to subconfluence and 2 × 106 Cells were fractionated into cytoplasm, nuclear, and membrane fractions using the subcellular protein fractionation kit (33) by following the manufacturer's protocol. Normalized portions of each extract from a cell lysate of 20 μg of total proteins were resolved on SDS-PAGE and analyzed by Western blotting. Tubulin was used as a marker for cytosol (34), and emerin was used as a marker for nucleus (35).
Co-immunoprecipitation and GST-Cofilin Pulldown Assays
For co-immunoprecipitation of CAP1 with FAK and Talin, 250 μl (∼300 μg of total proteins) of HeLa cell lysates was rotated/incubated with 5 μg of CAP1 antibody for 2 h at 4 °C followed by the addition of 15 μl of 50% (v/v) protein A/G beads and rotating/incubating for another 2 h. The beads were spun down and washed three times with lysis buffer and resolved on SDS-PAGE to detect co-precipitated FAK and Talin by Western blotting. For GST-cofilin pulldown of CAP1, expression of the GST fusion WT and S3D cofilin was induced in Escherichia coli strain BL21 with 1 mm isopropyl-β-d-thiogalactoside for 4 h at 37 °C. Bacteria were harvested and homogenized with sonication in PBS buffer supplemented with 1% Triton X-100 and 1 mm DTT. After centrifugation, the supernatant was collected and rotated/incubated with appropriate volumes of 50% glutathione-Sepharose 4B slurry for 2 h at 4 °C. The beads were then precipitated, washed, and resuspended in PBS buffer containing protease inhibitors until use. The amount of GST-cofilin was estimated based on Coomassie Blue staining of the proteins on gels. For pulldown of endogenous CAP1, 250 μl of HeLa cell lysate (∼200 μg of total proteins) was rotated/incubated with ∼10 μg GST fusion WT cofilin or the S3D mutant for 2 h at 4 °C. The glutathione-Sepharose beads were washed three times with lysis buffer followed by resolving on SDS-PAGE and blotting with CAP1 antibody for detecting co-precipitated CAP1. Similar procedures were followed to pull down transiently expressed GFP fusion CAP1 and deletion mutants from HEK293T lysates and for detection of the precipitated proteins in Western blotting using a GFP antibody.
RESULTS
Knockdown of CAP1 Led to Morphological and Actin Cytoskeletal Changes in HeLa Cells
We first tested whether mammalian CAP1, like yeast CAP, binds actin in vivo by performing co-precipitation experiments. His6-tagged mouse CAP1 transiently expressed in HEK293T cells was precipitated with nickel-nitrilotriacetic acid beads from cell lysate, and a protein that is ∼46 kDa on Coomassie Blue-stained gel, which is speculated to be actin, co-precipitated with it (supplemental Fig. 1A). Consequent Western blotting with an actin antibody confirmed that the protein that co-precipitated from HeLa cell lysate was indeed actin (supplemental Fig. 1B).
To directly address cellular functions of CAP1, we used vector-based shRNA to stably knock down CAP1 in HeLa cells, which express high levels of CAP1 (Fig. 1A). Two stable knockdown clones, S2-2 and S3-2, derived from independent shRNA constructs S2 and S3, respectively, were generated (30). Both clones have efficient CAP1 knockdown as detected by Western blotting (Fig. 1A) and immunofluorescence (not shown), although S3-2 had relatively more complete CAP1 depletion than S2-2 (Fig. 1A). The quantified results of CAP1 knockdown are shown in Fig. 1B. We did not observe any elevation of CAP2 expression in CAP1 KD cells, which may serve to compensate for the loss of CAP1 function (data not shown).
FIGURE 1.

Knockdown of CAP1 in HeLa cells led to larger cell size and enhanced lamellipodia. A, KD of CAP1 in HeLa cells. S2 and S3 shRNA constructs that target independent target sequences were used to transfect HeLa cells, and the stable clones S2-2 and S3-2 were established through neomycin selection. Cell lysate was blotted for CAP1, with GAPDH blotting serving as a loading control. Vec, vector; Scr, scrambled S2. B, quantified efficiency of CAP1 KD from three independent experiments measured by densitometry, analyzed with Student's t test, and shown in the graph with error bars representing S.E. (** indicates p < 0.01). Signals were normalized to that of the cells harboring the vector (assigned a value of 1.0). C, phase images showing that CAP1 KD led to enhanced lamellipodia and increased cell size. Cells were cultured on fibronectin-coated plates overnight, and arrowheads indicate the larger cells with enhanced lamellipodia as compared with typical control cells (indicated with arrows). D, quantified results showing the average cell area by measuring an area of 50 cells per field (with three replications) using National Institutes of Health ImageJ. The results were analyzed and shown similarly to B. E, quantified results showing the percentage of cells with enhanced lamellipodia by counting 100 cells per field and three fields from each experiment (from three experiments) and analyzed and shown similarly to B (* indicates p < 0.05). F, re-expression of mtCAP1 in CAP1 KD S3-2 cells. Re-expressed CAP1 was confirmed by using CAP1 antibody as well as an antibody against His6 (not shown) in Western blotting. S3-2 cells were transfected with a pcDNA4-based CAP1 expression plasmid with additional mismatch introduced to the S3-2 target region to allow its expression; the stable clones were established by selection with Zeocin. G, phase imaging shows rescue of morphological phenotypes by re-expression of mtCAP1. Arrows indicate the larger cells with lamellipodia in cells harboring the vector, and arrowheads indicate typical cells in cells re-expressing mtCAP1. H, quantified results showing the rescue of cell area by re-expression of mtCAP1; the experiment and data analysis were conducted and shown similarly to D.
Because CAP1 is a protein that regulates actin dynamics and the actin cytoskeleton and loss of CAP caused morphological changes in yeast and Dictyostelium (5, 36, 37), we first examined whether CAP1 KD also leads to similar morphological alterations in HeLa cells. Phase imaging revealed a number of alterations in the CAP1 KD cells (Fig. 1C). First, the CAP1 KD cells (both S2-2 and S3-2) are larger than the control cells harboring either an empty vector or a scrambled S2 (indicated with arrows), with quantified cell areas shown in Fig. 1D. Second, many CAP1 KD cells have enhanced lamellipodia (peripheral ruffles), a structure that promotes directional cell migration, as indicated with arrowheads in Fig. 1C, with the percentage of cells with developed lamellipodia quantified and shown in Fig. 1E. We next designed experiments to confirm that the morphological phenotypes were truly derived from CAP1 KD by re-expressing CAP1 in S3-2 cells, which have more complete CAP1 depletion and stronger phenotypes. To do this, we modified the S3 target sequence on mouse CAP1 by introducing seven additional mismatches so that the shRNA in S3-2 cells will not recognize the expressed mutant CAP1 (mtCAP1). We confirmed expression of His6-tagged CAP1 in the stable cells by Western blotting (Fig. 1F), and the results show that the morphological alterations, including the cell size and lamellipodia phenotypes, were rescued (Fig. 1, G and H).
The morphological phenotypes suggest alterations in the actin cytoskeleton, so we next examined it by staining with fluorescent phalloidin, an actin filament-specific probe. We first observed robustly increased stress fibers and lamellipodia using wide field fluorescence imaging (not shown), and we then conducted confocal imaging to look into more details. As shown in Fig. 2A, the CAP1 KD cells (both S3-2 and S2-2 cells; cells indicated with arrows are typical ones) had enhanced stress fibers as compared with the control cells. Consistent with observations from the phase imaging (Fig. 1, C and D), phalloidin staining also shows larger cell size in CAP1 KD cells. The stress fiber phenotype is similar to that observed in NIH3T3 fibroblasts with transient CAP1 KD (25) and is consistent with the role of CAP1 as a G-actin sequestering protein; loss of CAP1 would be expected to cause more actin to be polymerized into filaments (14). Similar to the morphological changes, the actin stress fiber phenotype was rescued by re-expression of CAP1 in the rescue cells (Fig. 2B).
FIGURE 2.

Knockdown of CAP1 in HeLa cells led to actin cytoskeletal changes. A, phalloidin staining shows enhanced stress fibers in CAP1 KD cells. KD and control cells were grown on fibronectin-coated MetTak plates overnight, fixed, permeabilized, and stained with Alexa Fluor 488 phalloidin. Confocal images were taken. Arrowheads indicate cells with enhanced stress fibers in KD cells, and arrows indicate the actin cytoskeleton in typical control cells. B, re-expression of mtCAP1 in S3-2 CAP1 KD cells rescued the stress fiber phenotype. Arrowheads indicate cells with reduced stress fibers in the rescued cells, and arrows indicate cells with enhanced stress fibers (and the larger cell size) in the S3-2 cells harboring an empty pcDNA4 vector (Vec). C, fractionation of pellet (P) and supernatant (S) actin, which are rich in F-actin and G-actin respectively, shows increased F-actin in the CAP1 KD cells. Lysates from CAP1 KD HeLa cells (S2-2 and S3-2) along with cells harboring a scrambled S2 (control) were spun in ultracentrifugation at 100,000 × g for 1 h to fractionate F-actin and G-actin. The supernatant (G-actin-rich) and pellet (F-actin-rich) fractions were resolved on SDS-PAGE and blotted with an actin antibody. Scr, scrambled S2. D, the actin signals from three Western blots were measured by densitometry, and the ratio of pellet versus supernatant from three blots was calculated. The data were analyzed using Student's t test and plotted in the graph with the error bars representing S.E. (* indicates p < 0.05).
To gain further evidence that the loss of CAP1 affected the balance between F-actin and G-actin, we prepared supernatant and pellet fractions from high speed centrifugation for control and CAP1 KD cells followed by Western blotting with actin. The actin fraction in the pellet is primarily F-actin, whereas the actin fraction in the supernatant is primarily G-actin. As shown in Fig. 2C, supernatant actin and pellet actin exist at similar levels in the control cells (cells harboring scrambled S2). However, both S2-2 and S3-2 KD cells had elevated levels of pellet actin as compared with supernatant actin, thus confirming the results from fluorescence imaging that CAP1 KD led to accumulation of actin filaments. As a control for the fractionation experiment, we treated cells with LA, a toxin that inhibits actin polymerization, which caused nearly all of the actin to move to the supernatant, as expected from F-actin depolymerization by LA treatment. The quantified ratios of pellet actin versus supernatant actin are shown in Fig. 2D. Taken together, loss of CAP1 leads to significant changes in the actin cytoskeleton and cell morphology in HeLa cells characterized by increases in F-actin, stress fibers, and lamellipodia. Thus, we provide further in vivo evidence establishing roles for mammalian CAP1 in regulating actin dynamics and cell morphology.
CAP1 Depletion Stimulates HeLa Cell Motility and Invasion
Because actin dynamics are a key driving force of cell motility, and CAP1 facilitates actin dynamics, it is expected that CAP1 knockdown will probably reduce cell motility. However, CAP1 KD cells also have enhanced peripheral ruffles (Fig. 1, C and E), which are expected to stimulate cell motility, so we assessed cell motility in the CAP1 KD HeLa cells. We first conducted wound healing assays to assess the effects of CAP1 KD on HeLa cell motility and unexpectedly found that CAP1 KD cells had substantially increased cell motility. As shown in Fig. 3A, the wounds in the CAP1 KD cells (both S3-2 and S2-2) healed almost completely at 16 h after they were introduced, whereas those in the control cells healed only marginally after the same time. Importantly, rescue experiments also show that re-expression of mtCAP1 in KD cells reduced the cell motility phenotype (Fig. 3E). To further establish the effects on cell motility, we also conducted Transwell assays (31); the results were quantified and shown in Fig. 3B. Consistent with the results from wound healing assays, CAP1 KD cells have remarkably increased cell migration as compared with control cells. We further examined whether increased cell invasion accompanies the elevated cell motility in CAP1 KD cells and conducted Matrigel invasion assays. The invasion assays assess the capacity of cells in penetrating Matrigel, which mimics tissue invasion. As shown in the images of invaded cells in Fig. 3C, quantified in Fig. 3D, about twice the number of CAP1 KD cells invaded through the Matrigel as compared with control cells. Taken together, depletion of CAP1 led to substantially increased motility as well as invasion in HeLa cells.
FIGURE 3.
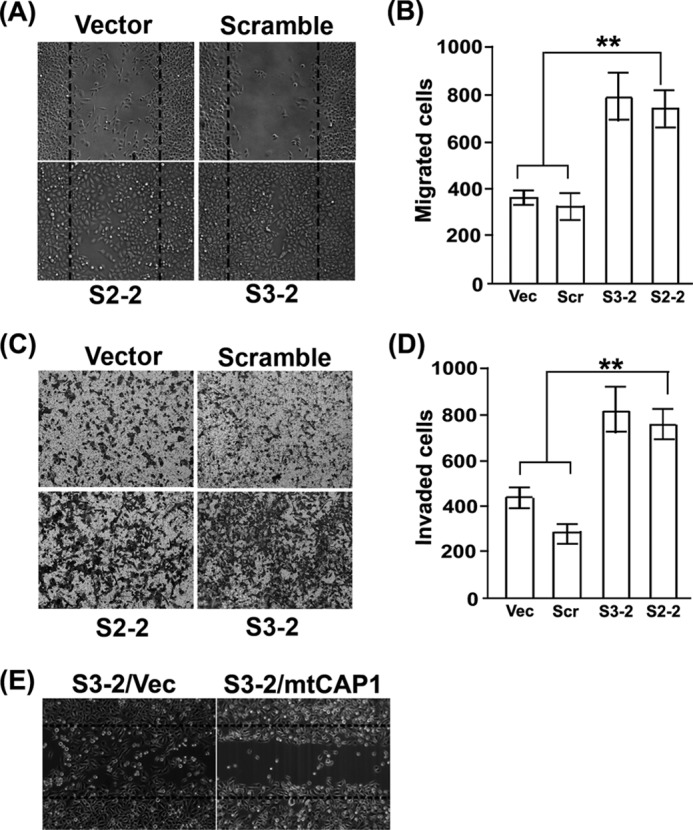
CAP1 depletion led to increased migration and invasion in HeLa cells. A, wound healing assays comparing motility of CAP1 KD and control cells. Wounds were introduced into a monolayer of confluent cells using a pipette tip, and cells were cultured for another 16 h before phase images were taken to assess the healing of the wounds. B, quantified results comparing cell motility from Transwell cell migration assays. ∼2 × 104 cells serum-starved overnight were loaded to each insert, and the inserts were placed in wells loaded with medium containing 10 μg/ml PDGF. After 16 h, cells staying on the upper side of the membrane were wiped off with a cotton swab, and the remaining (migrated) cells were scored for four representative fields. The experiment was performed three times, and the data were analyzed using Student's t test and plotted as bar graphs showing mean ± S.E. (** indicates p < 0.01). C, images of invaded cells comparing the capability of KD and control cells in penetrating Matrigel matrixes in invasion assays. D, quantified results from three independent Matrigel invasion assays. The data were collected, analyzed, and shown similarly to B. E, re-expression of mtCAP1 rescued the cell motility phenotype as assessed in the wound healing assay. Vec, vector.
Knockdown of CAP1 Leads to FAK Activation and Enhanced Cell Adhesion
During cell culture, we observed that CAP1 KD cells attached and spread more rapidly on cell culture plates than control cells (not shown), which, together with the motivation of identifying the underlying mechanism driving the motility of the CAP1 KD cells, prompted us to directly test cell adhesion and spreading. We first compared cell spreading on fibronectin-coated tissue culture plates. By 15 min after plating, many of the KD (S3-2) cells had already spread out (Fig. 4A, with arrowheads), as indicated by the formation of peripheral protrusions (S2-2 cells showed similar phenotypes; data not shown). In contrast, only very few control cells spread out. By 2 h after plating, most of the CAP1 KD cells formed multiple large protrusions, whereas the control cells had fewer and smaller protrusions (Fig. 4A, indicated with arrowheads). We next conducted detailed, quantitative adhesion assays with a time course by washing off unattached cells and scoring attached cells to assess the rate of cell adhesion. Both KD and control cells were plated on fibronectin-coated plates, and the unattached cells were washed off with PBS buffer at 10, 20, and 30 min time points; the attached cells that stayed on the plate were stained and scored. As shown in the graph with quantified results (Fig. 4B), KD cells have significantly increased numbers of attached cells than the control cells; thus, depletion of CAP1 enhances cell adhesion and spreading. Because focal adhesions are critical for cell adhesion (38) and vinculin plays a key role in focal adhesions (39), we stained vinculin to visualize focal adhesions. As in Fig. 4C, the confocal images show increased size and area of focal adhesions in CAP1 KD cells. We measured areas of focal adhesions of 20 cells each for KD and control cells, and quantified results of area of focal adhesions per cell are shown in Fig. 4D; the KD cells have significantly increased focal adhesion areas.
FIGURE 4.

CAP1 knockdown stimulates cell adhesion and spreading. A, phase imaging of cells plated on fibronectin-coated plates shows enhanced spreading of CAP1 KD (S3-2) cells as compared with the control cells (scrambled S2 (Scr)). Images were taken at 15-min and 2-h time points after the cells were plated onto the fibronectin-coated plates. The arrowheads show enhanced spreading of the CAP1 KD cells. B, cell adhesion assays show increased numbers of attached cells in KD cells at different time points (10, 20, and 30 min) after 2 × 104 cells had been plated on fibronectin-coated plates. Unattached cells were washed off the plate, and attached cells were then stained and scored. Three fields were counted for each experiment, and the experiment was performed three times. The data were analyzed using Student's t test and plotted in the graph with error bars representing S.E. (* indicates p < 0.05). C, vinculin staining showing enhanced focal adhesions in the CAP1 KD cells. Cells were cultured on fibronectin-coated MatTek plates for 5 h, fixed, permeabilized, and stained with a vinculin antibody before the confocal images were taken. The arrowheads show cells with enhanced focal adhesions in the S3-2 and S2-2 CAP1 KD cells as compared with that in the control cells (cells harboring vector and the scrambled S2, indicated with arrows). D, quantified results showing focal adhesion areas per cell in KD and control cells. The areas were measured using the National Institutes of Health ImageJ software; areas of focal adhesions (FA) in 20 cells were measured for both KD and control cells, and the data were analyzed using Student's t test, and shown as mean ± S.D. (* indicates p < 0.05). Vec, vector; Sq. Pixels, square pixels.
FAK is a key molecule that regulates cell adhesion (40). We next looked into whether FAK expression or activity was altered in CAP1 KD cells. For Western blotting, we used a phospho-specific antibody against FAK Tyr-397 to test potential elevations in FAK activity. Tyr-397 is an autophosphorylation site whose phosphorylation is required for focal adhesion dynamics (41), which is critical for cell motility. Phosphorylation at Tyr-397 promotes FAK association with Src (27, 42, 43) and subsequent phosphorylation of additional sites. There was no detectable alteration in FAK expression; however, we observed remarkably elevated FAK phosphorylation at Tyr-397 in both S2-2 and S3-2 CAP1 KD cells as compared with that in control cells (Fig. 5A), which is quantified and shown in Fig. 5B. FAK is phosphorylated in focal adhesions (41); larger focal adhesions in KD cells likely caused more FAK localization to the focal adhesions and contributed to the activation (phosphorylation) of FAK. Importantly, the FAK activation phenotype was also rescued by re-expression of mtCAP1, as shown in Fig. 5C. Thus, CAP1 KD leads to FAK activation.
FIGURE 5.

CAP1 depletion led to increased FAK phosphorylation (activation) and CAP1 co-IP with FAK and Talin. A, increased FAK phosphorylation at Tyr-397 (p-Y397) in CAP1 KD cells (S2-2 and S3-2) as compared with the control cells (scrambled S2 (Scr)) as detected by Western blotting. Cells were cultured to ∼80% confluence, and cell lysates were prepared and used. GAPDH serves as a loading control. B, quantified FAK phosphorylation results. Signals from three independent blots were measured by densitometry, analyzed with Student's t test, and shown in the graph with error bars representing S.E. (** indicates p < 0.01). Signals were normalized to that of the cells harboring the scrambled S2 (assigned a value of 1.0). C, re-expression of mtCAP1 in S3-2 cells rescued the elevated FAK phosphorylation as compared with cells harboring the pcDNA4 empty vector. D, FAK and Talin co-precipitates with CAP1. HeLa cells were cultured under various conditions and used for the IP experiments. NT, cells were cultured without treatment; Sus, cells were cultured in suspension for 2 h; FN, cells were cultured on fibronectin-coated plates for 1 h. Cell lysate incubated with mouse IgG in place of CAP1 antibody serves as a negative control. E, CAP1 KD, but not disruption of the F-actin by treatment with LA, abolishes co-IP of FAK with CAP1. Cells were cultured on fibronectin-coated plates, and treatment of cells with LA was for 1 h at 0.5 μg/ml before harvesting the cells. Two irrelevant lanes were cut out from the gel, and an additional image with lowered contrast was included to provide a better image of the IgG lane of the FAK co-IP.
Because KD of CAP1 in HeLa cells led to activation of FAK, we speculated that there was a physical interaction between CAP1 and FAK, either direct one-on-one binding or indirect association by forming a complex. We thus tested association of CAP1 with FAK, as well as Talin, a cytoskeletal protein (44), which is also a component of focal adhesions that provides a link between integrin and the actin cytoskeleton (26). An interaction between Talin and CAP1 has been observed previously in the yeast two-hybrid system (45). We tested for association of CAP1 with FAK and Talin through immunoprecipitation (IP) assays and found that both FAK and Talin co-precipitated with CAP1 in IP assays (Fig. 5D). We further addressed the specificity of the CAP1/FAK association by including lysates from CAP1 KD (S3-2) cells and cells treated with LA in the IP experiments. As shown in Fig. 5E, CAP1 IP did not precipitate FAK from the CAP1 KD cell lysates. In contrast, depolymerization of the F-actin by LA treatment of cells did not abolish co-IP of FAK with CAP1 (Fig. 5D), suggesting that it is unlikely that F-actin may serve to bridge FAK with CAP1. Thus, CAP1/FAK association is specific.
Biochemical and Functional Interactions between CAP1 and Cofilin
Moriyama and Yahara (18) first demonstrated that mammalian CAP1 cooperates with cofilin to facilitate cofilin-driven actin filament turnover. However, the effects of this interaction on cofilin regulation are not known, so we examined the potential effects of CAP1 KD on cofilin phosphorylation. When we examined cofilin phosphorylation at Ser-3, we found that it is dephosphorylated in CAP1 KD cells as compared with controls (Fig. 6A), with the quantified results shown in Fig. 6B. Again, re-expression of CAP1 rescued the cofilin dephosphorylation phenotype (Fig. 6E). Phosphorylation of Ser-3 by LIM kinase is pivotal in regulating cofilin activity as an ADF (46), where LIM kinase is activated by upstream signals from Rho-associated protein kinase (ROCK) (47) and p21-activated kinase (PAK) (48). Upon activation, LIM kinase phosphorylates cofilin at Ser-3, which prevents actin binding to cofilin, hence turning off its activity as an ADF (46). We tested whether CAP1 KD down-regulated LIM kinase by blotting for LIM kinase and phosphorylated LIM kinase (at Ser-506/Ser-508). Neither LIM kinase level nor the phosphorylation of the protein was decreased (Fig. 6A), suggesting that phosphatases of cofilin such as Slingshot-1L may be activated (49). We then conducted Western blotting to measure phosphorylation of Slingshot-1L and found it had slightly reduced activity (elevated phosphorylation at Ser-978) in CAP1 KD cells (Fig. 6C). The quantified results are shown in Fig. 6D. Specificity was confirmed by showing that re-expression of mtCAP1 also reduced Slingshot phosphorylation (Fig. 6E). Thus, neither LIM kinase nor Slingshot appeared to be responsible for the cofilin dephosphorylation in KD cells. We next looked into potential alterations in cofilin localization by conducting confocal imaging and found that cofilin accumulated into cytoplasmic aggregates in CAP1 KD cells (both S3-2 and S2-2 cells; confocal images are shown in Fig. 6F), whereas in control cells, cofilin appears to localize diffusely to the cytosol and nucleus, but no cofilin was found in aggregates (Fig. 6F). To further determine cofilin localization and alterations caused by CAP1 KD, we fractionated cells into nucleus and cytosolic fractions. As shown in Fig. 6G, we found that cofilin mostly localizes to the cytosol with limited localization to the nucleus in HeLa cells, which is not changed by CAP1 KD. Interestingly, we also found that nucleus cofilin is dephosphorylated. Proper preparation of the nucleus and cytosolic fractionations was confirmed by the blotting of the respective markers (tubulin for cytosol and emerin for nucleus).
FIGURE 6.

Depletion of CAP1 activates Slingshot, reduces cofilin phosphorylation, and leads to accumulation of cofilin into cytoplasmic aggregates. A, Western blotting of phospho-Ser-3 cofilin shows reduced cofilin phosphorylation in the CAP1 KD cells. The expression or phosphorylation (activity) of the cofilin kinase-LIM kinase (indicated by p-cofilin and p-LIMK) was not changed. GAPDH blotting serves as a loading control. Vec, vector. Scr, scrambled S2. B, quantified results of cofilin phosphorylation from three independent experiments analyzed using Student's t test and plotted with error bars representing S.E. (* indicates p < 0.05). Signals were normalized to that of the cells harboring the vector (assigned a value of 1.0). C, phosphorylation at Ser-978 (activity) of the cofilin phosphatase, Slingshot-1L (indicated by p-Slingshot), was elevated in CAP1 KD cells. GAPDH blotting serves as a loading control. D, quantified results of Slingshot phosphorylation from three independent experiments analyzed using Student's t test, and plotted with error bars representing S.E. (* indicates p < 0.05). Signals were normalized to that of the cells harboring the vector (assigned a value of 1.0). E, elevated phosphorylation (activation) of both cofilin and Slingshot was rescued by re-expression of mtCAP1. F, accumulation of cofilin into cytoplasmic aggregates in CAP1 KD cells as shown in confocal images. CAP1 KD and control cells were cultured overnight, fixed, permeabilized, and stained with a cofilin antibody before the confocal images were taken. The arrows indicate the cytoplasmic cofilin aggregates in both S2-2 and S3-2 CAP1 KD cells. G, cell fractionation assays show that cofilin mainly localizes to the cytosol, whereas a small amount localizes to the nucleus. Cells were grown overnight to subconfluence and fractionated into cytoplasmic and nuclear fractions using the subcellular protein fractionation kit following the manufacturer's protocol. Proportional amounts of each subcellular fraction were resolved on SDS-PAGE followed by Western blotting to detect cofilin and phospho-cofilin (p-cofilin). The samples were also blotted with emerin (nucleus marker) and tubulin (cytosol marker) to verify that the fractionation was well controlled.
CAP1 binds cofilin immobilized on an affinity column (18), and the N terminus domain of CAP1 facilitates cofilin-promoted actin dynamics. However, further molecular and biochemical mechanisms of the CAP1/cofilin interaction remained elusive. For example, it is not clear whether other domain(s) of CAP1 also plays a role in mediating their interaction. Also, it has not been reported whether Ser-3-phosphorylated cofilin, which mimics an inactive form of the ADF, still binds CAP1. Despite our considerable effort, we have been unable to co-precipitate CAP1 and cofilin in IP assays, which may be due to the low affinity of their interaction. We took an alternative approach and developed a GST-cofilin pulldown assay to precipitate CAP1 from cell lysates. As shown in Fig. 7A, GST-cofilin precipitated CAP1 from HeLa cell lysates; the GST-cofilin S3D mutant, which mimics a phosphorylated Ser-3 residue, also pulled down CAP1 but at a reduced efficiency. Therefore, the activation state of cofilin appears to partially impact its affinity with CAP1. We next mapped the CAP1 domain(s) that mediates the interaction with cofilin by GST-cofilin pulldown of GFP fusion CAP1 and deletion mutants (30) in HEK293T cells. The empty vector that expresses GFP alone was not precipitated by GST-cofilin (data not shown). We found that both the N terminus and full-length CAP1 were pulled down by GST-cofilin, whereas neither the middle domain nor the C terminus precipitated with GST-cofilin (Fig. 7B). Therefore, the N terminus of CAP1 is the primary domain that mediates the interaction between CAP1 and cofilin. These results are consistent with findings that the N terminus of CAP1 cooperates with cofilin in promoting actin dynamics (18), as well as with our previous observation that the N terminus is the sole CAP1 domain required for the mitochondrial translocation of the protein driven by a mitochondria-targeted cofilin (30).
FIGURE 7.

GST-cofilin pulldown of CAP1 and mapping of the N terminus CAP1 that interacts with cofilin. A, bacteria-purified GST-cofilin S3D (GST-cof), which harbors a mutation mimicking phosphorylated Ser-3 residue (inactive form), had reduced binding with CAP1. ∼200 μg of total protein in 250 μl of cell lysate was rotated/incubated at 4 °C with ∼10 μg of GST fusion WT cofilin or S3D mutant bound to glutathione beads for 2 h. The beads were spun briefly and washed three times followed by resolving on SDS-PAGE and Western blotting to detect co-precipitated CAP1. B, GFP fusion full-length CAP1 and derived deletion mutants (MD) were transiently expressed in HEK293T cells and used in pulldown assays. Lysates with ∼300 μg of total protein were rotated/incubated at 4 °C with ∼10 μg of GST-cofilin bound to glutathione beads for 2 h. The beads were spun briefly and washed three times with lysis buffer, and the co-precipitated CAP1 was detected with a GFP antibody in Western blotting (WB). The full-length (FL, indicated with an arrow) and N terminus domain (NT, indicated with an arrowhead) were the only ones that precipitated with GST-cofilin. CT, C terminus.
DISCUSSION
CAP has been shown as an actin-regulatory protein conserved in all eukaryotes, and evidence supports that mammalian CAP also regulates the actin cytoskeleton. The primary activity for CAP is the ability to bind and sequester actin monomers. The CAP-actin complex does not affect filament formation by itself but accelerates cofilin-dependent filament depolymerization (18). In this study, we provide both in vitro and in vivo evidence further establishing the role for mammalian CAP1 in the regulation of the actin cytoskeleton through both cofilin-dependent and cofilin-independent pathways. Depletion of CAP1 leads to accumulation of F-actin, which is likely from the reduced G-actin sequestering, as well as aberrant cofilin localization and phosphorylation. Additionally, we find that FAK phosphorylation is increased in CAP1-depleted cells, and IP experiments suggest an association of CAP1 with FAK and Talin. The CAP1 KD cells migrated more quickly; the effects on motility are likely due to increases in lamellipodia and cell spreading and adhesion.
Depletion of CAP1 reduces actin dynamics, and because actin dynamics promote cell motility and cell invasion, CAP1 KD cells would be expected to be less motile (50, 51). However, optimum motility requires a balance of actin dynamics, cell adhesion, contraction, and the generation of filopodia, lamellipodia (peripheral ruffles), and stress fibers (52). CAP1 KD HeLa cells have enhanced lamellipodia, which are likely to promote motility. The KD cells also showed increases in stress fibers, but the role of stress fibers in cell motility is somewhat complicated in comparison with universally positive roles of filopodia and lamellipodia in cell migration. Although stress fibers generate tension force essential for detaching the trailing edge of the cell during migration, cells with enhanced stress fibers such as Akt1 knock-out mouse embryonic fibroblast cells (31) often have reduced cell migration. In the case of Akt1 knockouts, the cells actually had reductions in lamellipodia, whereas the CAP1 KD cells had increases in lamellipodia. Because cell adhesion plays an important role in cell motility (26, 27, 53) and elevated activities of cell adhesion signals can also stimulate cell lamellipodia by activation of Rac/Cdc42 signaling (54, 55), we speculate that collected effects from activated cell adhesion and Rac/Cdc42 signaling probably underlie increased cell motility in the CAP1 KD cells. Thus, it is likely that in CAP1 KD cells, the activated cell adhesion signaling and increases in lamellipodia play roles in overriding negative effects on cell motility that may be caused by reduced actin turnover and additional stress fibers.
The depletion of CAP1 in fibroblasts reduced cell motility (13, 25). However, in these cells, CAP1 depletion did not enhance lamellipodia as we observed in HeLa cells. In addition to the dynamic rearrangement of the actin cytoskeleton during cell movement, cell migration also requires coordinated assembly and disassembly of cell adhesion sites (27, 52). A balance in the turnover of focal adhesions is required for optimal cell migration (27, 53, 56) as adhesion provides traction force essential for cell migration. Sufficient traction force will not be generated if there is too little adhesion for the cells to attach well; on the other hand, too much cell adhesion causes too much cell attachment, which will also hinder motility. HeLa cells are not strongly adherent and can be easily adapted to grow in suspension, so increases in adhesion are likely to promote cell motility. The changes in FAK phosphorylation and increased cell spreading show that loss of CAP1 increases cell adhesion and perhaps focal adhesion turnover as well (57). We propose that several factors may have contributed to this phenotype. The first and probably the most important one is that the depletion of CAP1 caused activation of cell adhesion signaling as indicated by FAK activation and enhanced focal adhesion, as shown in the adhesion assays and vinculin staining. Studies have shown that FAK inhibition blocks responses to cell motility cues (42). Moreover, the activated adhesion signaling has been shown to activate the Cdc42/Rac pathway (54, 55), and Cdc42/Rac are well documented drivers of cell migration. We speculate that the positive effects on protrusion and adhesion more than offset the negative effects from reduced actin dynamics. The overall effects on actin filaments appear to be similar in the HeLa cells, as was seen in fibroblasts. However, the differences in cell migration demonstrate that the net effects on cell migration are cell type-specific and not necessarily predictable by indicating the effects of CAP1 loss on a particular actin filament structure such as stress fibers.
The molecular link between CAP1 and cell adhesion may be Talin, which associates with CAP1 in yeast two-hybrid screens and co-precipitates with CAP1 (Fig. 5D). Talin is a cytoskeletal protein that controls integrin activation (44) and is also a component of focal adhesions (26). It binds to the cytoplasmic portion of integrin to induce conformational changes in integrin extracellular domains that result in increased affinity for ligands (58). In this model, depletion of CAP1 is expected to release Talin, enabling more Talin molecules to bind to the cytoplasmic portion of integrin and thus cause integrin activation through what is called the inside-out pathway (59) of integrin activation. Evidence for this model is seen in the increased spreading, adhesion, and FAK phosphorylation in the CAP1 knockdown cells.
The prevailing model for CAP function is the actin dynamics model (18). In this model, CAP actively cooperates with cofilin to depolymerize filaments during cell migration. However, the primary activity of CAP is actin monomer binding and sequestering, so depletion of CAP will reduce the pool of G-actin, making more actin molecules available to form filaments. We measured this directly by showing an alteration in the filament-monomer balance in CAP1 KD cells. Some observations in the KD cells, especially those in static cells, such as increases in stress fibers, do not necessarily require rapid changes, but probably reflect long term changes in the G-actin/F-actin balance rather than dynamic cofilin-dependent pathways.
We also found that depletion of CAP1 reduced phosphorylation of cofilin and caused its accumulation into cytoplasmic aggregates. Reduced cofilin phosphorylation was not associated with changes in expression or activity of LIM kinase or hyper-activation of Slingshot. We propose that cofilin aggregation prevented LIM kinase access. Also, because CAP1 KD leads to robust cofilin dephosphorylation or activation in cells, accumulation into cytoplasmic aggregates may serve as a mechanism to protect the cells by containing the abnormally high activity of cofilin. The punctate structure (aggregates) may contain cofilin and cleaved actin, which is similar to the rod-like structures seen in neurons treated with cytotoxic stimuli (60, 61). Because deregulation of the actin cytoskeleton has been observed in neurodegenerative diseases, and KD of CAP1 causes cofilin aggregates similar to those seen in Alzheimer disease (62), it would not be surprising if deregulation of CAP1 is found to play roles in neurodegenerative diseases as well.
In conclusion, in addition to providing in vitro and in vivo evidence further establishing roles of mammalian CAP1 in actin dynamics and cofilin machinery, our studies demonstrate a novel role for CAP1 in cell adhesion signaling. The observation that CAP1 KD HeLa cells have increased cell motility shows that, depending on cell context, it can either promote or inhibit cell motility.
This work was supported by startup funds from the Arkansas Biosciences Institute and Arkansas State University, as well as National Scientist Development Grant NSDG 0630394N from the American Heart Association (to G. L. Z.).

This article contains supplemental Fig. 1.
- G-actin
- globular actin
- CAP
- adenylyl cyclase-associated protein
- mtCAP1
- mutant CAP1
- ADF
- actin depolymerization factor
- KD
- knockdown
- FAK
- focal adhesion kinase
- LA
- latrunculin A
- IP
- immunoprecipitation.
REFERENCES
- 1. Bamburg J. R., Wiggan O. P. (2002) ADF/cofilin and actin dynamics in disease. Trends Cell Biol. 12, 598–605 [DOI] [PubMed] [Google Scholar]
- 2. Bamburg J. R., Bloom G. S. (2009) Cytoskeletal pathologies of Alzheimer disease. Cell Motil. Cytoskeleton 66, 635–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olson M. F., Sahai E. (2009) The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis 26, 273–287 [DOI] [PubMed] [Google Scholar]
- 4. Hall A. (2009) The cytoskeleton and cancer. Cancer Metastasis Rev. 28, 5–14 [DOI] [PubMed] [Google Scholar]
- 5. Field J., Vojtek A., Ballester R., Bolger G., Colicelli J., Ferguson K., Gerst J., Kataoka T., Michaeli T., Powers S., et al. (1990) Cloning and characterization of CAP, the S. cerevisiae gene encoding the 70 kd adenylyl cyclase-associated protein. Cell 61, 319–327 [DOI] [PubMed] [Google Scholar]
- 6. Fedor-Chaiken M., Deschenes R. J., Broach J. R. (1990) SRV2, a gene required for RAS activation of adenylate cyclase in yeast. Cell 61, 329–340 [DOI] [PubMed] [Google Scholar]
- 7. Field J., Nikawa J., Broek D., MacDonald B., Rodgers L., Wilson I. A., Lerner R. A., Wigler M. (1988) Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol. Cell. Biol. 8, 2159–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hubberstey A. V., Mottillo E. P. (2002) Cyclase-associated proteins: CAPacity for linking signal transduction and actin polymerization. FASEB J. 16, 487–499 [DOI] [PubMed] [Google Scholar]
- 9. Vezina C. M., Walker N. J., Olson J. R. (2004) Subchronic exposure to TCDD, PeCDF, PCB126, and PCB153: effect on hepatic gene expression. Environ. Health Perspect. 112, 1636–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shibata R., Mori T., Du W., Chuma M., Gotoh M., Shimazu M., Ueda M., Hirohashi S., Sakamoto M. (2006) Overexpression of cyclase-associated protein 2 in multistage hepatocarcinogenesis. Clin. Cancer Res. 12, 5363–5368 [DOI] [PubMed] [Google Scholar]
- 11. Zucchi I., Mento E., Kuznetsov V. A., Scotti M., Valsecchi V., Simionati B., Vicinanza E., Valle G., Pilotti S., Reinbold R., Vezzoni P., Albertini A., Dulbecco R. (2004) Gene expression profiles of epithelial cells microscopically isolated from a breast-invasive ductal carcinoma and a nodal metastasis. Proc. Natl. Acad. Sci. U.S.A. 101, 18147–18152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. He J., Whelan S. A., Lu M., Shen D., Chung D. U., Saxton R. E., Faull K. F., Whitelegge J. P., Chang H. R. (2011) Proteomic-based biosignatures in breast cancer classification and prediction of therapeutic response. Int. J. Proteomics 2011, 896476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yamazaki K., Takamura M., Masugi Y., Mori T., Du W., Hibi T., Hiraoka N., Ohta T., Ohki M., Hirohashi S., Sakamoto M. (2009) Adenylate cyclase-associated protein 1 overexpressed in pancreatic cancers is involved in cancer cell motility. Lab. Invest. 89, 425–432 [DOI] [PubMed] [Google Scholar]
- 14. Gerst J. E., Ferguson K., Vojtek A., Wigler M., Field J. (1991) CAP is a bifunctional component of the Saccharomyces cerevisiae adenylyl cyclase complex. Mol. Cell. Biol. 11, 1248–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mintzer K. A., Field J. (1994) Interactions between adenylyl cyclase, CAP, and RAS from Saccharomyces cerevisiae. Cell. Signal. 6, 681–694 [DOI] [PubMed] [Google Scholar]
- 16. Wang J., Suzuki N., Nishida Y., Kataoka T. (1993) Analysis of the function of the 70-kilodalton cyclase-associated protein (CAP) by using mutants of yeast adenylyl cyclase defective in CAP binding. Mol. Cell. Biol. 13, 4087–4097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vojtek A. B., Cooper J. A. (1993) Identification and characterization of a cDNA encoding mouse CAP: a homolog of the yeast adenylyl cyclase associated protein. J. Cell Sci. 105, 777–785 [DOI] [PubMed] [Google Scholar]
- 18. Moriyama K., Yahara I. (2002) Human CAP1 is a key factor in the recycling of cofilin and actin for rapid actin turnover. J. Cell Sci. 115, 1591–1601 [DOI] [PubMed] [Google Scholar]
- 19. Quintero-Monzon O., Jonasson E. M., Bertling E., Talarico L., Chaudhry F., Sihvo M., Lappalainen P., Goode B. L. (2009) Reconstitution and dissection of the 600-kDa Srv2/CAP complex: roles for oligomerization and cofilin-actin binding in driving actin turnover. J. Biol. Chem. 284, 10923–10934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chaudhry F., Little K., Talarico L., Quintero-Monzon O., Goode B. L. (2010) A central role for the WH2 domain of Srv2/CAP in recharging actin monomers to drive actin turnover in vitro and in vivo. Cytoskeleton 67, 120–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DesMarais V., Ghosh M., Eddy R., Condeelis J. (2005) Cofilin takes the lead. J. Cell Sci. 118, 19–26 [DOI] [PubMed] [Google Scholar]
- 22. Yu G., Swiston J., Young D. (1994) Comparison of human CAP and CAP2, homologs of the yeast adenylyl cyclase-associated proteins. J. Cell Sci. 107, 1671–1678 [DOI] [PubMed] [Google Scholar]
- 23. Peche V., Shekar S., Leichter M., Korte H., Schröder R., Schleicher M., Holak T. A., Clemen C. S., Ramanath-Y B., Pfitzer G., Karakesisoglou I., Noegel A. A. (2007) CAP2, cyclase-associated protein 2, is a dual compartment protein. Cell. Mol. Life Sci. 64, 2702–2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peche V. S., Holak T. A., Burgute B. D., Kosmas K., Kale S. P., Wunderlich F. T., Elhamine F., Stehle R., Pfitzer G., Nohroudi K., Addicks K., Stöckigt F., Schrickel J. W., Gallinger J., Schleicher M., Noegel A. A. (2013) Ablation of cyclase-associated protein 2 (CAP2) leads to cardiomyopathy. Cell. Mol. Life Sci. 70, 527–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bertling E., Hotulainen P., Mattila P. K., Matilainen T., Salminen M., Lappalainen P. (2004) Cyclase-associated protein 1 (CAP1) promotes cofilin-induced actin dynamics in mammalian nonmuscle cells. Mol. Biol. Cell 15, 2324–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Critchley D. R. (2000) Focal adhesions – the cytoskeletal connection. Curr. Opin. Cell Biol. 12, 133–139 [DOI] [PubMed] [Google Scholar]
- 27. Mitra S. K., Hanson D. A., Schlaepfer D. D. (2005) Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68 [DOI] [PubMed] [Google Scholar]
- 28. Freeman N. L., Field J. (2000) Mammalian homolog of the yeast adenylyl cyclase associated protein, CAP/Srv2p, regulates actin filament assembly. Cell Motil. Cytoskeleton 45, 106–120 [DOI] [PubMed] [Google Scholar]
- 29. Zhou G. L., Zhuo Y., King C. C., Fryer B. H., Bokoch G. M., Field J. (2003) Akt phosphorylation of serine 21 on Pak1 modulates Nck binding and cell migration. Mol. Cell. Biol. 23, 8058–8069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang C., Zhou G. L., Vedantam S., Li P., Field J. (2008) Mitochondrial shuttling of CAP1 promotes actin- and cofilin-dependent apoptosis. J. Cell Sci. 121, 2913–2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou G. L., Tucker D. F., Bae S. S., Bhatheja K., Birnbaum M. J., Field J. (2006) Opposing roles for Akt1 and Akt2 in Rac/Pak signaling and cell migration. J. Biol. Chem. 281, 36443–36453 [DOI] [PubMed] [Google Scholar]
- 32. Ishikawa-Ankerhold H. C., Gerisch G., Müller-Taubenberger A. (2010) Genetic evidence for concerted control of actin dynamics in cytokinesis, endocytic traffic, and cell motility by coronin and Aip1. Cytoskeleton 67, 442–455 [DOI] [PubMed] [Google Scholar]
- 33. Cascio S., Zhang L., Finn O. J. (2011) MUC1 protein expression in tumor cells regulates transcription of proinflammatory cytokines by forming a complex with nuclear factor-κB p65 and binding to cytokine promoters: importance of extracellular domain. J. Biol. Chem. 286, 42248–42256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Subramanian C., Hada M., Opipari A. W., Jr., Castle V. P., Kwok R. P. (2013) CREB-binding protein regulates Ku70 acetylation in response to ionization radiation in neuroblastoma. Mol. Cancer Res. 11, 173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fairley E. A., Kendrick-Jones J., Ellis J. A. (1999) The Emery-Dreifuss muscular dystrophy phenotype arises from aberrant targeting and binding of emerin at the inner nuclear membrane. J. Cell Sci. 112, 2571–2582 [DOI] [PubMed] [Google Scholar]
- 36. Kawamukai M., Gerst J., Field J., Riggs M., Rodgers L., Wigler M., Young D. (1992) Genetic and biochemical analysis of the adenylyl cyclase-associated protein, cap, in Schizosaccharomyces pombe. Mol. Biol. Cell 3, 167–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Noegel A. A., Rivero F., Albrecht R., Janssen K. P., Köhler J., Parent C. A., Schleicher M. (1999) Assessing the role of the ASP56/CAP homologue of Dictyostelium discoideum and the requirements for subcellular localization. J. Cell Sci. 112, 3195–3203 [DOI] [PubMed] [Google Scholar]
- 38. Wehrle-Haller B. (2012) Assembly and disassembly of cell matrix adhesions. Curr. Opin. Cell Biol. 24, 569–581 [DOI] [PubMed] [Google Scholar]
- 39. Carisey A., Ballestrem C. (2011) Vinculin, an adapter protein in control of cell adhesion signalling. Eur. J. Cell Biol. 90, 157–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Michael K. E., Dumbauld D. W., Burns K. L., Hanks S. K., García A. J. (2009) Focal adhesion kinase modulates cell adhesion strengthening via integrin activation. Mol. Biol. Cell 20, 2508–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hamadi A., Bouali M., Dontenwill M., Stoeckel H., Takeda K., Rondé P. (2005) Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J. Cell Sci. 118, 4415–4425 [DOI] [PubMed] [Google Scholar]
- 42. Schlaepfer D. D., Mitra S. K., Ilic D. (2004) Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim. Biophys. Acta 1692, 77–102 [DOI] [PubMed] [Google Scholar]
- 43. Schaller M. D., Hildebrand J. D., Shannon J. D., Fox J. W., Vines R. R., Parsons J. T. (1994) Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell. Biol. 14, 1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Calderwood D. A., Ginsberg M. H. (2003) Talin forges the links between integrins and actin. Nat. Cell Biol. 5, 694–697 [DOI] [PubMed] [Google Scholar]
- 45. O'Neill K. E. (1996) An Analysis and Functional Characterization of Human and Yeast Cyclase Associated Protein. Ph.D thesis, Columbia University [Google Scholar]
- 46. Yang N., Higuchi O., Ohashi K., Nagata K., Wada A., Kangawa K., Nishida E., Mizuno K. (1998) Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393, 809–812 [DOI] [PubMed] [Google Scholar]
- 47. Ohashi K., Nagata K., Maekawa M., Ishizaki T., Narumiya S., Mizuno K. (2000) Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J. Biol. Chem. 275, 3577–3582 [DOI] [PubMed] [Google Scholar]
- 48. Edwards D. C., Sanders L. C., Bokoch G. M., Gill G. N. (1999) Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1, 253–259 [DOI] [PubMed] [Google Scholar]
- 49. Huang T. Y., DerMardirossian C., Bokoch G. M. (2006) Cofilin phosphatases and regulation of actin dynamics. Curr. Opin. Cell Biol. 18, 26–31 [DOI] [PubMed] [Google Scholar]
- 50. Carlier M. F., Pantaloni D. (2007) Control of actin assembly dynamics in cell motility. J. Biol. Chem. 282, 23005–23009 [DOI] [PubMed] [Google Scholar]
- 51. Pollard T. D., Borisy G. G. (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 [DOI] [PubMed] [Google Scholar]
- 52. Brakebusch C., Fässler R. (2003) The integrin-actin connection, an eternal love affair. EMBO J. 22, 2324–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vicente-Manzanares M., Choi C. K., Horwitz A. R. (2009) Integrins in cell migration–the actin connection. J. Cell Sci. 122, 199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Price L. S., Leng J., Schwartz M. A., Bokoch G. M. (1998) Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol. Biol. Cell 9, 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Etienne-Manneville S., Hall A. (2001) Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCζ. Cell 106, 489–498 [DOI] [PubMed] [Google Scholar]
- 56. Schlaepfer D. D., Mitra S. K. (2004) Multiple connections link FAK to cell motility and invasion. Curr. Opin. Genet. Dev. 14, 92–101 [DOI] [PubMed] [Google Scholar]
- 57. Ren X. D., Kiosses W. B., Sieg D. J., Otey C. A., Schlaepfer D. D., Schwartz M. A. (2000) Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 113, 3673–3678 [DOI] [PubMed] [Google Scholar]
- 58. Calderwood D. A. (2004) Integrin activation. J. Cell Sci. 117, 657–666 [DOI] [PubMed] [Google Scholar]
- 59. Legate K. R., Wickström S. A., Fässler R. (2009) Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 23, 397–418 [DOI] [PubMed] [Google Scholar]
- 60. Minamide L. S., Striegl A. M., Boyle J. A., Meberg P. J., Bamburg J. R. (2000) Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat. Cell Biol. 2, 628–636 [DOI] [PubMed] [Google Scholar]
- 61. Bernstein B. W., Chen H., Boyle J. A., Bamburg J. R. (2006) Formation of actin-ADF/cofilin rods transiently retards decline of mitochondrial potential and ATP in stressed neurons. Am. J. Physiol. Cell Physiol. 291, C828–C839 [DOI] [PubMed] [Google Scholar]
- 62. Mitake S., Ojika K., Hirano A. (1997) Hirano bodies and Alzheimer's disease. Kaohsiung J. Med. Sci. 13, 10–18 [PubMed] [Google Scholar]