

Background: ApoA1, a component of HDL, promotes anti-inflammatory, immunomodulatory, and cardioprotective functions.
Results: ApoA1 suppresses tumor growth and metastasis, primarily via modulation of innate and adaptive immune responses.
Conclusion: ApoA1 impacts tumor biology at multiple levels, which appear to be linked to immunomodulatory function.
Significance: ApoA1 redirects elicited immune cells toward tumor suppression and rejection and may hold benefit as a cancer therapeutic.
Keywords: Cancer, Cancer Biology, Cancer Therapy, High Density Lipoprotein (HDL), Innate Immunity, Melanoma, Adaptive Immunity, Apolipoprotein A1 Therapy, Matrix Metalloproteinase 9 (MMP-9), Tumor-suppressive Infiltrating Leukocytes
Abstract
Here, we show that apolipoprotein A1 (apoA1), the major protein component of high density lipoprotein (HDL), through both innate and adaptive immune processes, potently suppresses tumor growth and metastasis in multiple animal tumor models, including the aggressive B16F10L murine malignant melanoma model. Mice expressing the human apoA1 transgene (A1Tg) exhibited increased infiltration of CD11b+ F4/80+ macrophages with M1, anti-tumor phenotype, reduced tumor burden and metastasis, and enhanced survival. In contrast, apoA1-deficient (A1KO) mice showed markedly heightened tumor growth and reduced survival. Injection of human apoA1 into A1KO mice inoculated with tumor cells remarkably reduced both tumor growth and metastasis, enhanced survival, and promoted regression of both tumor and metastasis burden when administered following palpable tumor formation and metastasis development. Studies with apolipoprotein A2 revealed the anti-cancer therapeutic effect was specific to apoA1. In vitro studies ruled out substantial direct suppressive effects by apoA1 or HDL on tumor cells. Animal models defective in different aspects of immunity revealed both innate and adaptive arms of immunity contribute to complete apoA1 anti-tumor activity. This study reveals a potent immunomodulatory role for apoA1 in the tumor microenvironment, altering tumor-associated macrophages from a pro-tumor M2 to an anti-tumor M1 phenotype. Use of apoA1 to redirect in vivo elicited tumor-infiltrating macrophages toward tumor rejection may hold benefit as a potential cancer therapeutic.
Introduction
The incidence of malignant melanoma, a leading cancer in people under the age of 30, is rapidly rising worldwide (1, 2). This aggressive skin cancer is known to incite significant inflammation and infiltration of phagocytic innate immune cells such as monocytes, macrophages, and myeloid dendritic cells at the site of early tumor development (3). Inflammation is a central instigator of many chronic diseases, including cardiovascular disease (4, 5), as well as cancer (6, 7). A salient feature of tumor-associated inflammation is the recruitment of innate and adaptive immune cells to the tumor microenvironment via cytokines and chemoattractants produced by the tumor cell and its associated tumor stromal cells. Within the tumor microenvironment, dysregulated interactions of innate immune myeloid cells, including myeloid-derived suppressor cells (MDSCs)2 and tumor-associated macrophages (TAMs), with T and B cells of the adaptive immune system, are believed to facilitate tumor cell immune evasion, migration, tumor-associated angiogenesis, and metastasis (6–8).
HDL is a ubiquitous plasma lipoprotein with presumed atheroprotective functions mediated primarily via its cholesterol efflux activity (9, 10). However, recent Mendelian genetics studies question the widely accepted causal role of HDL in atherosclerosis protection (11). Interestingly, a number of studies of HDL and its associated proteome have suggested an expanded repertoire of activities for the lipoprotein beyond simply serving as a shuttle of cholesterol cargo during reverse cholesterol transport, including anti-inflammatory, anti-parasitic, anti-apoptotic, and innate immune activities (9, 12, 13). Specifically, HDL functions as an anti-inflammatory molecule by interaction with the vascular endothelium and through interaction with circulating inflammatory cells (9, 12–14). HDL displays anti-parasitic functions by virtue of a subset of its particles carrying a parasite-specific lysosomal pore-forming protein (15), and HDL demonstrates anti-apoptotic and anti-oxidant activities by its ability to directly influence intracellular signaling cascades or to deliver anti-oxidant proteins such as paraoxonase 1, which stimulates the ERK1/2 and PI3K signaling pathways (16–18). Furthermore, apolipoprotein A1 (apoA1), the major structural protein of HDL, when either expressed or injected in vivo, spontaneously lipidates forming HDL and demonstrates a number of innate immune activities, including the following: (i) antiviral effects for both enveloped and nonenveloped DNA and RNA viruses (19, 20); (ii) anti-parasitic activity as mentioned above; (iii) both bacteriocidal and bacteriostatic effects (21); and (iv) modulatory effects on regulatory T cells (22). Potent bacterial toxin neutralizing activities of HDL are attributed primarily to apoA1, which binds and neutralizes both bacterial endotoxin and lipoteichoic acid (23, 24). ApoA1/HDL is thus situated at the nexus of a number of physiologically significant immune, anti-inflammatory, and anti-apoptotic functions.
A role for apoA1 as a therapeutic agent for cardiovascular disease is currently under intense study (4, 9, 12, 25). Synthetic HDL nanoparticles were recently shown to inhibit B cell lymphoma xenografts in mice in a scavenger receptor class B, type I-dependent manner (26). Interestingly, these authors did not observe any anti-tumor effect by HDL from human serum. Reconstituted nascent HDL particles have been explored for the targeted systemic delivery of anti-tumor small interfering RNA into tumor cells expressing scavenger receptor class B, type I (27). Short amphipathic α-helical peptides, presumed mimetics of apoA1 with potent anti-inflammatory activity, have also been investigated and shown to modulate ovarian cancer phenotype through binding to lysophosphatidic acid (28, 29). However, the intact apoA1 protein was observed to bind poorly to lysophosphatidic acid and to be significantly less potent than the amphipathic peptides. A role for the intact apoA1 protein as a potential chemotherapeutic agent through modulation of the immune system, thereby influencing the tumor microenvironment and cancer development, has not been examined. Here, we test the hypothesis that genetic manipulation of apoA1 levels, or direct pharmacological provision of full-length apoA1, directly impacts tumor cell biology in multiple cancer models, and we address the associated mechanisms responsible for the strikingly potent anti-neoplastic properties observed for this apolipoprotein.
EXPERIMENTAL PROCEDURES
Mice
All mouse studies were performed under approved Institutional Animal Care and Use Committee protocols at the Cleveland Clinic. C57BL/6J (wild type (WT) mice), A1KO, A1Tg+/−, A1Tg+/+, and NSG (NOD-Scid-IL2r γ) mice were purchased from The Jackson Laboratory and bred at the Biological Research Unit of the Cleveland Clinic. Nude mice (nu/nu Ncr) were obtained from Athymic Animal and Tumor Core at Case Western Reserve University (Cleveland, OH).
Tumor Cell Lines
Mouse tumor cell lines B16F10 melanoma, Lewis lung, and the human melanoma A375 were obtained from American Type Culture Collection (ATCC, Manassas, VA) and cultured in DMEM supplemented with 10% heat-inactivated fetal calf serum (FCS), 2 mm l-glutamine, and antibiotic/antimycotic (Invitrogen) at 37 °C and 5% CO2 in a humidified atmosphere. B16F10L was isolated from lung metastasis in WT C57BL/6J, and a variant expressing luciferase (firefly, Photinus pyralis) was engineered by cotransfecting B16F10L cells with pKCPIRlucBGH5.3 (the kind gift from Yan Xu, Indiana University School of Medicine) and pcDNA3 (Invitrogen, 3:1 ratio) using Lipofectamine (Invitrogen) and selected with 1.1 mg/ml G418 (Invitrogen) over 3 weeks. Pools of luciferase-expressing tumor cells were used for the study.
In Vivo Tumor Studies
8–10-Week-old male and female animals (with the exception of the human melanoma A375 experiment where females were used) were inoculated subcutaneously on both flanks with 105 tumor cells per site. For histological, RNA, and Western blotting analyses of tumor, animals were inoculated at multiple sites. Tumor volume, based on caliper measurements, was calculated three times a week according to the ellipsoid volume formula, tumor volume = (the shortest diameter)2 × the largest diameter × 0.525. Tumor volume is expressed as mean value, where n = number of tumor inoculation sites (two per animal). Whole body in vivo bioluminescent imaging was performed on shaved animals using an IVIS system (Xenogen Corp., Alameda, CA) with d-luciferin (potassium salt, Gold Biotechnology, St Louis, MO) according to manufacturer's instructions. LivingImage software (Xenogen Corp.) and Igor Image analysis software (Wave Metrics, Lake Oswego, OR) were used for data analyses. Because tumors were injected on the dorsal side, photons emanating from the ventral side of the animals were used to quantify metastasis. Metastasis was routinely observed in the abdominal, thoracic, and neck region in advanced tumors. Imaging data are expressed as total flux, photons/s, where n = number of animals per genotype or treatment group.
ApoA1, ApoA2 Injection Studies
Apolipoproteins (apoA1 or apoA2; 15–20 mg/animal) purified from human plasma (see below) were injected subcutaneously in the neck region away from the tumor injection site every other day. Plasma (EDTA) or serum levels of human apoA1 were determined using an immunoassay on an Architect ci8200 (Abbott Diagnostics, Abbot Park, IL) using a standard curve validated by adding varying levels of isolated purified human apoA1 to apoA1-KO mouse plasma.
T Cell Proliferation Assays
T cell proliferation assays were performed using mixed lymphocyte reactions. CD3+ T cells isolated by negative selection (Miltenyi Biotec, Cambridge, MA) from Balb/c splenocytes were used as T effectors (1 × 105 cells), and irradiated (1 × 105 cells, 3,000 rads) WT C57BL/6J splenocytes were used as feeder cells. CD11c cells were isolated from spleens of naive or tumor-bearing mice with CD11c FITC antibody (clone N418, 11-0114, eBioscience, San Diego) and FITC beads (120-000-293, Miltenyi Biotec). Pooled cells were co-cultured at T effector to CD11c cell ratios of 1:1, 5:1, 10:1, and 20:1 in 96-well U-bottom plates. During the last 18 h of culture, cells were pulsed with [3H]thymidine (1 μCi/well) prior to harvesting and counting in a beta scintillation counter (PerkinElmer Life Sciences).
F4/80+ Isolation
Pooled tumors were minced and digested (RPMI medium with collagenase IV (1 mg/ml, Worthington), hyaluronidase V (2.5 units/ml Sigma), and DNase I (0.1 mg/ml, Sigma) by incubating for 45 min at 37 °C. Debris and undigested tumors were removed by filtration (70 and 40 μm pore size; BD Biosciences). Dissociated cells were stained with biotinylated F4/80+ monoclonal antibody (clone CI:A3-1; AbDSerotec, Raleigh, NC) and anti-biotin microbeads using a magnetic bead sorting system (Miltenyi Biotec).
Tumor Cytotoxicity Assays
Tumor-bearing animals were i.p. injected with thioglycollate on day 15 and lavaged (peritoneum) 4 days later. F4/80+ cells were purified from peritoneal exudates as described above. They were then cocultured immediately with B16F10L cells in vitro, and cell viability was assessed after 3 days. Briefly, F4/80+ cells were seeded at 5:1 and 10:1 effector (E) (F4/80+ cells) to target (T) (B16F10L tumor cells) ratios with target cells at 2,000 cells/well in 96-well flat bottom tissue culture plates in RPMI with 10% FBS and penicillin/streptomycin in a final volume of 200 μl per well. Cell viability was determined using CytoTox GLO kit (Promega, Madison, WI).
RNA and Quantitative Real Time PCR (qRT-PCR)
Total RNA was prepared using the RNeasy Mini kit (Qiagen, Valencia, CA) with on-column DNase treatment (79254, Qiagen). Tissue culture cells were processed immediately, and tumor tissue was stored in RNA later for analyses at subsequent times. RNA was reverse-transcribed (high capacity cDNA reverse transcription kit, Invitrogen), and qRT-PCR was performed using gene-specific TaqMan probes on a StepOne Plus instrument (Invitrogen). The quantity of target mRNA was normalized to the level of B2m (β2-microglobulin) in each sample. The relative number of copies of mRNA (RQ) was calculated using the ΔΔCt method.
Immunohistochemistry
Fresh frozen tumors (OCT, Tissue-Tek) were acetone-fixed before immunostaining. Immunocomplexes were visualized using alkaline phosphatase red detection kit (Vector Laboratories, Burlingham, CA) and a fluorescent microscope (Nikon Instruments) with DAPI (Vectashield, Vector Laboratories) nuclear counterstaining. The antibodies were as follows: CD11b, clone M1/70 (MAB1124, R&D Systems, Minneapolis, MN), F4/80, clone BM8 (14-4801, eBioscience), GR1, clone RB6-8C5 (550291, Pharmingen). For histological evaluation of survivin (71G4B7, Cell Signaling Technology, Inc., Danvers, MA), tumors were fixed in 10% neutral-buffered formalin, embedded in paraffin, and subjected to antigen retrieval before immunostaining. Images (collected with a ×20 objective) were analyzed with Image-Pro Plus (version 7.0, Media Cybernetics, Silver Spring, MD).
Tumor Protein Extract, Western Blotting, and Enzyme Activity Assays
Two resected tumors from each mouse were pooled 7 days post tumor inoculation, snap-frozen in liquid nitrogen, and stored at −80 °C. Protein extracts were made from tumor homogenate in lysis buffer (50 mm Tris/HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 1 mm sodium orthovanadate, 20 μg/ml aprotinin, 5 μg/ml leupeptin, 10 μg/ml pepstatin A, 1 mm PMSF, 20 mm sodium fluoride, 2 mm sodium pyrophosphate, 25 mm sodium metabysulfate, 25 mm β-glycerolphosphate, 5 mm benzamidine) by centrifuging at 20,800 × g for 30 min at 4 °C and the supernatant used for protein and enzyme analysis. MMP-9 and VEGF protein were detected with goat anti-MMP-9 (AF909, R & D Systems) and goat anti-VEGF 164 (AF-493, R & D Systems), respectively. Signal intensity was quantified using the Odyssey infrared detection system (Li-COR, Lincoln, NE). Specific proteins were normalized to β-actin on the same membrane. QuickZyme mouse MMP-9 activity assay kit (QuickZyme BioSciences, Netherlands) was used to assay enzyme activity as per the manufacturer's protocol.
Flow Cytometry
Splenocytes, isolated by mechanical disruption, were lysed for RBCs (LCK lysis buffer, Invitrogen) and stained with fluorochrome-conjugated CD11b (CD11b-PE, clone M1/70, 12-0112, eBioscience), GR1 (GR1-APC, clone RB6-8C5, 17-5931, eBioscience; GR1-PE-Cy7, clone RB6-8C5, 25-5931, eBioscience), LY-6C (LY-6C-APC, clone AL-21, 560595, Pharmingen), and F4/80 (F4/80-APC, clone BM8, 17-4801, eBioscience). Tumors were digested, and after RBC lysis (described above), cells were labeled with fluorochrome-conjugated CD45 (CD45.2-APC; clone 104, 17-0454, eBioscience), CD11b (CD11b-PE, clone M1/70, 12-0112, eBioscience), GR1 (GR1-AF700, clone RB6-8C5, 557979, Pharmingen), CD3 (CD3-AF700, clone 17A2, 561388, Pharmingen), CD4 (CD4-FITC, clone RM4-5, eBioscience), and CD8a (CD8a-PerCP, clone 53-6.7, Pharmingen). Cells were acquired on a BD LSR II FACS machine and gated on Live (Live/Dead Violet, L34955, Invitrogen), and data were analyzed using FlowJo software (Treestar Inc., Ashland, OR).
Tumor Angiogenesis
Blood vessels feeding directly into day 7 B16F10L melanoma tumors in anesthetized animals were counted under a dissecting microscope at ×12.5 magnification. Every visible vessel touching the circumference of the tumor nodule was scored as a single vessel. Two measurements were taken to assess the tumor area (the largest diameter coplanar with the skin, and a second diameter perpendicular to the first). The product of these two measurements was used as an index of tumor area. Images were captured using World Precision Instruments operating microscope PSMT5 with ×12.5 objective lens. Each experimental group contained seven mice each bearing two tumors. Tumor photographs were subjected to digital analysis using Vesgen software, where the region of interest (white in Fig. 7B, middle panel) representing the tumor mass defined the perimeter of the tumor. The output was a series of color Generation Maps (colored vessels on black background) in which the largest diameter vessels were defined as G1 (red), with each subsequent smaller generation represented as G2–G6 (Fig. 7B, right). The number of blood vessels was expressed based on total vessel area, vessel length density, and vessel diameter.
FIGURE 7.

Reduced angiogenesis in primary tumors from A1Tg mice. Angiogenesis was assessed in B16F10L-luciferase melanoma bearing mice 7 days post-tumor inoculation. A and B, number of vessels feeding directly into the primary tumor was counted under a microscope as described under “Experimental Procedures.” Images of tumor were captured at ×12.5 magnification (B, left). The region of interest (B, middle; white) representing the tumor mass defined the perimeter of the tumor. The output was a series of color generation maps (colored vessels on black background) in which the largest diameter vessels were defined as G1 (red), with each subsequent smaller generation represented as G2–G6 (B, right). These images were used to quantitate total vessel area (C), vessel length density (D), and size of vessels (E), as described under “Experimental Procedures.” F, day 7 B16F10L-luciferase subcutaneous tumors were homogenized for RNA or protein as described under “Experimental Procedures.” F, left panel, TaqMan assays for murine Vegfa were normalized to B2m and expressed as relative quantification (RQ). Bar graphs represent mean of the data series. F, right panel, VEGF dimer protein as detected by Western blot and normalized to β-actin. Horizontal bars represent median of the data series. n = number of tumor inoculation sites in A–E or the number of animals in F.
Gene Expression Analysis of HDL Treated Bone Marrow-derived Macrophages
Bone marrow progenitor cells from 10- to 11-week-old WT female C57BL/6J mice were differentiated in vitro in standard 20% (v/v) L cell conditioned medium for 7–8 days. ApoA1 purified from human plasma (described below) was used to assemble reconstituted HDL generated by the cholate dialysis method (described below). Bone marrow-derived macrophages were seeded (8 × 105 cells per well; 6-well tissue culture plate) overnight in RPMI medium in the presence of 5% (v/v) lipoprotein-deficient serum from human plasma (S5519, Sigma), 50 ng/ml recombinant human macrophage colony-stimulating factor, mCSF (300-25, PeproTech), 50 μm β-mercaptoethanol, and penicillin/streptomycin. Reconstituted HDL (0.75 mg protein/ml) was added to the medium, and RNA was isolated after 24 h. Total RNA from pooled replicates was prepared, amplified, and biotinylated for hybridization to Illumina Murine BeadChips (mouse Ref-8, version 1.1, Illumina, Inc., San Diego) using Illumina TotalPrep RNA amplification kit (AMIL1791, Applied Biosystems, Invitrogen) according to the manufacturer's protocol. Arrays were scanned in an Illumina Bead Station, and the images were processed using Illumina Bead Studio software. Raw data were quantile-normalized, background-corrected, and filtered with a detection p value of <0.01 and a 2-fold change in expression between HDL treatments versus PBS control-treated cells.
HDL, ApoA1, and ApoA2 Purification
Plasma for human HDL and apoA1/2 isolation was obtained from healthy donors after obtaining informed consent. All human study protocols were approved by the Cleveland Clinic Institutional Review Board. Throughout HDL and apoA1 isolation conditions, care was taken to ensure sterility. All nondisposable glassware used was first baked at 500 °C overnight. HDL was isolated by buoyant density ultracentrifugation (KBr; 1.063–1.21 g/ml). HDL proteins were delipidated, precipitated, dried under N2, and resuspended in 20 mm Tris, pH 8.5, freshly deionized 6 m urea supplemented with 10 mm ethanolamine (30), and then immediately further purified by anion-exchange chromatography (Q-Sepharose HP HiLoad, 20 cm2 × 15 cm) (31). Isolated apoA1 and -A2 were immediately dialyzed into sterile normal saline and either concentrated for injection or dialyzed into phosphate-buffered saline for HDL assembly. Endotoxin levels were measured by Limulus amoebocyte lysate (Charles River Laboratories, Wilmington, MA) method (0.05 < × < 0.25 EU/mg/ml protein). Reconstituted nascent HDL (2 apoA1, 20 cholesterol, 200 1-palmitoyl-2-oleoyl-l-phosphatidylcholine, mol/mol/mol) was prepared by the cholate dialysis method (32). Purity (>99%) of human apoA1 and apoA2 was confirmed by SDS-PAGE.
Lysophosphatidic Acid Analyses
Following addition of internal standard, lysophosphatidic acid (LPA) was extracted from mouse plasma and quantified by HPLC with on-line electrospray ionization tandem mass spectrometry as described previously (33), using an AB SCIEX QTRAP 5500 mass spectrometer.
Statistical Analysis
All error bars represent the mean ± S.E. unless otherwise designated. Significant differences of tumor growth between two groups were assessed by Student's t test and in multiple groups by analysis of variance. A probability value of p < 0.05 was considered significant.
RESULTS
HDL Levels Affect Tumor Development in Vivo
To initially examine the effect of apoA1 on tumor progression and metastasis, we used B16F10L melanoma tumor cells and syngeneic animal hosts either lacking apoA1 (A1KO) and having low HDL levels (19.1 ± 1.7 mg/dl HDL cholesterol), wild type (WT) C57BL/6J mice (52.0 ± 4.5 mg/dl HDL cholesterol), or mice overexpressing the human apoA1 transgene (A1Tg+/−, 110.6 ± 7.5 mg/dl HDL cholesterol) in which murine apoA1 is markedly suppressed (34). Following subcutaneous inoculation of tumor cells bilaterally on the dorsal surface of mice, tumor growth was monitored and observed to be >10-fold reduced in A1Tg+/− mice compared with A1KO (Fig. 1A, p < 0.05). In parallel studies, metastasis growth was monitored in real time by in vivo bioluminescent imaging of the ventral surface of animals inoculated instead with a B16F10L variant stably expressing Renilla luciferase. Similarly, A1Tg+/− mice exhibited a >10-fold reduction in metastasis development compared with A1KO mice (Fig. 1B, p < 0.05). To determine whether the anti-neoplastic effect of apoA1 was unique to malignant melanoma tumor growth, parallel experiments were performed inoculating Lewis lung tumor cells, another syngeneic tumor model in C57BL/6J mice. Growth of Lewis lung tumors was also significantly (p < 0.05) inhibited in A1Tg+/− mice, and with even further tumor growth inhibition in animals homozygous for the human apoA1 gene (A1Tg+/+, Fig. 1, C and D, p < 0.05). These results thus revealed dose-dependent reduction in tumor growth with increasing apoA1 levels, with A1KO mice showing the greatest rates of tumor growth, and A1Tg+/+ mice, which express human apoA1 at ∼1.5-fold higher levels relative to A1Tg+/− mice, showing minimal tumor growth (Fig. 1, C and D). Because tumor growth and metastases in A1Tg+/+ animals was so markedly inhibited and as a practical matter, in further studies outlined below, we often employed A1Tg+/− mice in experiments designed to probe mechanisms of apoA1 anti-tumor activity. A1Tg+/− mice provided sufficient amounts of tumor tissues to be harvested to obtain enough material to isolate specific cells or to determine mRNA, protein, and enzymatic activity levels.
FIGURE 1.

Enhanced tumor growth and metastasis in apoA1 null (A1KO) mice. Syngeneic C57BL/6J mice of indicated genotypes were inoculated with B16F10L melanoma (A), B16F10L-luciferase melanoma (B), or Lewis Lung (C and D) tumor cells. Tumor volume at the site of inoculation was monitored by caliper measurements (A, C, and D). D reflects Lewis lung tumor volume on day 48. Metastasis was measured by in vivo bioluminescence (B). Data points are mean values ± S.E.
Pharmacological Delivery of ApoA1 Prevents Tumor Growth and Metastasis
The above results suggested pharmacological administration of apoA1 may provide therapeutic benefit as an anti-tumor agent. To test this hypothesis, the B16F10L melanoma model was examined due to its rapid and widely metastatic disease course. A regimen of subcutaneous injection of human apoA1 on alternating days was empirically developed in A1KO animals resulting in maintenance of plasma apoA1 levels within the normal physiological range observed in humans (trough 79 ± 7 versus peak 151 ± 9 mg/dl; data not shown). In initial studies, apoA1 (15 mg/animal) was administered prior to tumor inoculation to optimize potential observed effects, and in later studies (see below), the impact of apoA1 therapy on pre-existing (palpable) tumors and metastasis was tested. Remarkably, apoA1 injections provided extraordinary protection from tumor growth and metastasis compared with the vehicle control (normal saline) (Fig. 2). Tumor growth was readily detected at the site of tumor cell inoculation in the A1KO control group within several days, whereas the lack of luminescence in the apoA1-treated group indicated a complete failure of tumor growth and development. Quantitative analyses of the bioluminescence measurements indicated apoA1 treatment inhibited both tumor growth and metastasis by over 100-fold (Fig. 2, B and C, p < 0.05 each). Further illustration of the dramatic reduction in tumor growth afforded by apoA1 injection is illustrated in Fig. 2D, in which a representative animal (with fur shaved for bioluminescence analyses) from each treatment group is shown on day 21 post-tumor inoculation. Survival analyses similarly showed dramatic protection afforded by apoA1 injections, with 100% survival in the apoA1-treated animals throughout the duration of the study (terminated on day 35). This contrasted with a 50% reduction in survival in the normal saline-treated group by day 21, and with all animals in this arm dead by day 29 (Fig. 2E).
FIGURE 2.

ApoA1 therapy confers resistance to melanoma development and improves survival in A1KO mice. Human apoA1 (15 mg per animal) or normal saline were administered in A1KO mice starting 3 weeks prior to tumor inoculation (B16F10L-luciferase melanoma) and continued for the duration of the experiment. Tumor progression was monitored by live (bioluminescent) imaging. A, representative image of the dorsal view taken on day 16 post-tumor inoculation. Tumor burden (B) reflects the sum of luminescence from both dorsal and ventral views, although only the ventral view was used to quantify metastasis (C). D, representative animals from each treatment arm are shown 21 days post-tumor inoculation. Survival plot is shown in E. Data points are mean ± S.E.
To examine the impact of apoA1 on existing tumor and metastasis, A1KO animals were inoculated with B16F10L-luciferase tumor cells, and once both tumors were palpable and metastasis was detected (day 6), subcutaneous apoA1 injections versus normal saline treatments were initiated. Within 1 week, a significant 2-fold reduction in peak tumor and metastasis burden in the apoA1-treated group was observed (Fig. 3, A and B (inset); p < 0.05 for each). With longer duration of therapy, the apoA1-treated animals showed nominal further tumor growth and metastasis, whereas the normal saline-injected group succumbed to progressive melanoma tumor development (Fig. 3, A and B). These data indicate that apoA1 not only inhibits or delays the development of tumor progression (Fig. 2) but, more importantly, apoA1 therapy can shrink and retard further development of both established tumor and metastasis (Fig. 3, A and B).
FIGURE 3.

ApoA1 but not apoA2 therapy promotes regression of established melanoma tumors and metastasis. A1KO mice were inoculated with B16F10L-luciferase melanoma. Six (A and B) or seven (C and D) days post-tumor inoculation when tumor was palpable and in vivo imaging showed metastasis, injection of human apoA1 (20 mg per animal); A–D, apoA2 (20 mg per animal); C and D, or normal saline (A–D) was initiated and continued every other day for the duration of the experiment. Tumor burden (sum of luminescence from dorsal and ventral sides) is shown in A and C, and metastasis (luminescence from ventral side) is shown in B and D. Insets (A and B) show data for the apoA1 treatment arm on an expanded scale to better illustrate tumor progression/regression before and after apoA1 treatment. Data points are mean ± S.E.
The specificity of the anti-neoplastic biological activity of apoA1 was next examined by comparing the effect of subcutaneous injections with either apoA1 or the second most abundant HDL-associated protein, apolipoprotein A2 (apoA2). A1KO mice were again first inoculated with B16F10L-luciferase tumor cells, and once tumors were both palpable and metastasis observed, subcutaneous injections of equivalent amounts (by mass) of either apoA1 or apoA2 were initiated. Remarkably, although apoA1 again promoted tumor and metastasis regression, apoA2 showed no beneficial effect on either tumor growth or metastasis (Fig. 3, C and D). Interestingly, apoA2-treated animals showed a tendency toward enhanced tumor burden and metastasis relative to the normal saline arm, but the differences did not reach statistical significance (Fig. 3C, p = 0.36, day 19; Fig. 3D, p = 0.15, day 19).
Anti-tumor Effects of ApoA1 Are Indirect and Not Associated with Altered Antigen Presentation or Systemic Lysophosphatidic Acid Levels
Efforts to reveal the mechanism(s) through which apoA1 mediates its anti-neoplastic activity initially focused on studies examining whether the lipoprotein exhibits a direct effect on cancer cells, such as reductions in proliferation or apoptosis rates. However, in vitro treatment of B16F10L tumor cells with physiological levels of either apoA1 or reconstituted nascent HDL demonstrated no significant alterations in cellular proliferation rate, various cell cycle parameters, or apoptosis rate, indicating there is no major direct inhibitory effect on the tumor cells with either apoA1 or the lipid-associated lipoprotein (data not shown).
Recent studies suggest lipid accumulation in the form of triglycerides within dendritic cells (DCs) can significantly inhibit their ability to present tumor antigen and activate T cells (35). We therefore sought to determine whether DCs in A1KO mice might be functionally defective leading to accelerated tumor growth and whether, alternatively, DCs from A1Tg mice showed enhanced capacity to stimulate allogeneic T cells. CD11c+ DCs were isolated from spleens of naive or B16F10L-bearing A1KO and A1Tg mice, and their capacity to promote allogeneic T cell proliferation was determined in mixed lymphocyte proliferation assays. DCs recovered from tumor-bearing A1KO and A1Tg+/+ mice both stimulated allogeneic T cells to an equivalent extent (p = 0.99 for comparison between CD11c+ cells from A1KO versus A1Tg mice), each mediating a 3-fold increase in T cell proliferation as compared with DCs isolated from naive A1KO and A1Tg+/+ mice (Fig. 4). The increase in T cell proliferation observed in the tumor-bearing mice is consistent with tumor-induced mobilization of DCs (36). However, the lack of demonstrable differences between DCs recovered from A1KO and A1Tg mice argues against an in vivo role for apoA1 in modulating tumor antigen presentation by DCs and subsequent T cell activation.
FIGURE 4.
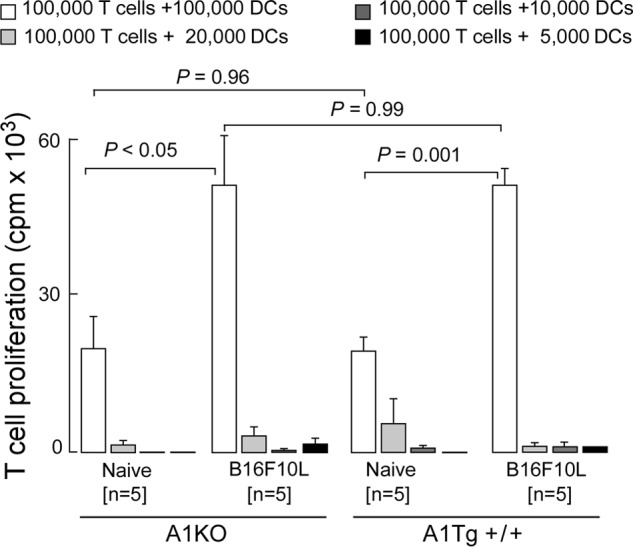
ApoA1 anti-tumor activity is independent of DC function. Allogeneic T cell proliferation induced by splenic DCs. Immunopurified CD11c+ DCs were isolated from spleens of naive and tumor-bearing A1KO or A1Tg+/+ animals 14 days post-tumor (B16F10L-luciferase) inoculation. Increasing numbers of irradiated pooled DCs were mixed with a fixed number of T cells (CD3+) isolated from naive wild type BALB/c splenocytes, and T cell proliferation was assessed by [3H]thymidine uptake as described under “Experimental Procedures.”
Studies by Farias-Eisner and co-workers (29) recently reported that injection of short anti-inflammatory amphipathic helical peptides, initially developed as apoA1 mimetics for cardiovascular disease, can inhibit ovarian cancer, presumably through binding of the pro-inflammatory lipid, LPA. We therefore quantified plasma levels of multiple distinct molecular species of lysophosphatidic acid in tumor-bearing A1KO, WT, and apoA1Tg mice using stable isotope dilution LC-MS/MS analyses. In contrast to results reported with the amphipathic peptides, no significant reductions in any LPA species were noted (Table 1). The above authors also demonstrated a significantly higher LPA binding capacity of the amphipathic peptides as compared with full-length apoA1 protein and a correlation between plasma LPA levels and tumor progression (29). The absence of any correlation between plasma levels of LPA molecular species and tumor growth in our present studies, along with the poor LPA binding activity previously noted with full-length apoA1 (29), indicates a distinct mechanism of anti-tumor activity is present with full-length apoA1 protein (this study) as compared with the short amphipathic peptides previously reported.
TABLE 1.
Plasma concentration of lysophosphatidic acid species (nanomolar) in tumor-bearing mice
The indicated numbers of C57BL/6J mice were inoculated with 105 B16F10L-luciferase melanoma tumor cells per flank. Plasma was recovered from tumor-bearing animals 17 days post-tumor inoculation. Individual molecular species of LPA were quantified by LC-MS/MS as described under “Experimental Procedures.” Data are presented as mean ± S.E. p values were calculated by two-tailed Student's t test. Note that no significant difference between any of the groups for any of the LPA molecular species or the total of all LPA species was observed.
| LPA species | WT (n = 12) | A1KO (n = 8) | A1Tg+/− (n = 12) |
p value |
||
|---|---|---|---|---|---|---|
| WT versus A1KO | WT versus A1Tg+/− | A1KO versusA1Tg +/− | ||||
| C14:0LPA | 0.32 ± 0.10 | 0.34 ± 0.20 | 0.21 ± 0.08 | 0.93 | 0.42 | 0.56 |
| C16:0LPA | 25.84 ± 3.51 | 27.83 ± 3.69 | 43.79 ± 7.72 | 0.70 | 0.05 | 0.08 |
| C18:0LPA | 10.46 ± 1.17 | 12.64 ± 3.11 | 11.94 ± 1.16 | 0.53 | 0.38 | 0.84 |
| C18:1LPA | 25.26 ± 4.25 | 21.51 ± 1.91 | 34.97 ± 8.22 | 0.43 | 0.31 | 0.14 |
| C18:2LPA | 233.55 ± 22.41 | 249.01 ± 31.46 | 271.76 ± 43.31 | 0.70 | 0.44 | 0.68 |
| C18:3LPA | 2.73 ± 0.67 | 3.68 ± 0.75 | 6.03 ± 1.73 | 0.36 | 0.10 | 0.23 |
| C20:0LPA | 0.13 ± 0.08 | 0.12 ± 0.12 | 0.34 ± 0.19 | 0.97 | 0.33 | 0.35 |
| C20:4LPA | 63.59 ± 8.71 | 65.88 ± 4.91 | 88.60 ± 14.12 | 0.82 | 0.15 | 0.15 |
| C20:5LPA | 0.89 ± 0.20 | 1.29 ± 0.33 | 2.14 ± 0.55 | 0.33 | 0.05 | 0.21 |
| C22:0LPA | 0.44 ± 0.19 | 0.51 ± 0.19 | 0.45 ± 0.20 | 0.79 | 0.96 | 0.84 |
| C22:5LPA | 25.94 ± 1.29 | 29.59 ± 1.17 | 28.64 ± 3.71 | 0.05 | 0.50 | 0.81 |
| LPA (total) | 389.13 ± 36.92 | 412.39 ± 38.11 | 488.88 ± 78.37 | 0.67 | 0.27 | 0.39 |
Anti-tumor Effects of ApoA1 Require an Immunologically Intact Host
In the absence of any apparent direct effect on tumor cells, changes in antigen presentation and T cell proliferative responses in vitro, or involvement of LPA sequestration as a potential mechanism, we investigated whether the anti-tumor effect of apoA1 was mediated via other changes in the host immune response to tumor. To test this hypothesis, we examined whether apoA1 was inhibitory to melanoma progression in mice deficient in various aspects of the immune system. B16F10L melanoma cells were inoculated into the severely immunodeficient NSG mice, which lack mature lymphocytes (B and T cells), natural killer cells, and fully functional DCs (37). ApoA1 injections resulted in an approximate 5-fold reduction in tumor growth in the NSG mice (Fig. 5A). Because tumor development was still significantly repressed in response to apoA1 therapy in these immunocompromised mice, these data suggest that an appreciable portion of the anti-neoplastic activity observed with apoA1 is independent of adaptive immunity, as the scid defect within NSG mice effectively eliminates adaptive immunity. In contrast, apoA1-induced B16F10L tumor repression in the fully immunocompetent A1KO mice (Fig. 5B) was nearly complete compared with that observed in the NSG mice (Fig. 5A), indicating that elements of both innate and adaptive immunity are required for full anti-neoplastic activity by apoA1. To examine if apoA1 therapy can be extended to human tumor xenografts, nude mice, which lack T cells and show both a partial defect in B cell development and a lack of cell-mediated immunity, were inoculated with human melanoma (A375) cells, followed by either apoA1 or normal saline injections. Interestingly, tumor development was significantly retarded by nearly 2-fold in the apoA1-treated animals (p = 0.02); however, tumor growth still proceeded within these apoA1-treated nude mice (Fig. 5C). These results indicate that although a defect in T cell immunity (Fig. 5C) partially attenuates the anti-neoplastic activity of apoA1 (Fig. 5B), a significant portion of the biological effect of apoA1 is mediated by T cell-independent mechanisms. Importantly, the results in Fig. 5C also suggest apoA1 anti-tumor activity extends to human tumors. We conclude that elements of both innate and adaptive immunity are required for full anti-neoplastic activity by apoA1. Nude and NSG mice have normal levels of murine apoA1. Inhibition of tumor growth in these mice is supportive of the relevance of the apoA1 effect under normal physiological conditions where apoA1 levels are intact.
FIGURE 5.
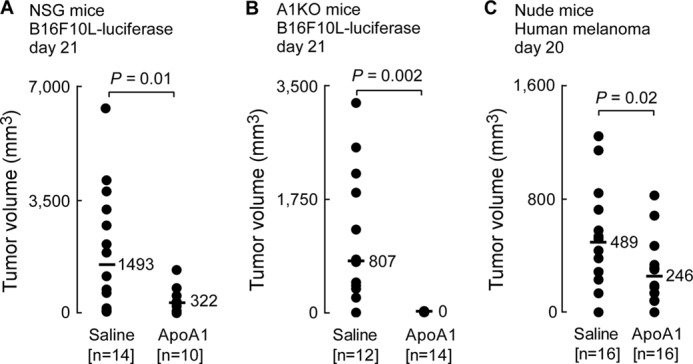
ApoA1 anti-neoplastic activity involves both innate and adaptive immunity. Human apoA1 injections (20 mg, s.c., every other day) were initiated on day 1 in NSG mice (A), A1KO mice (B), and Nude mice (C). Mean tumor volumes in mice inoculated with B16F10L-luciferase melanoma cells (A and B, day 21) or human melanoma A375 cells (C, day 20) are shown. Horizontal bars represent median tumor volumes of the data series.
ApoA1 Inhibits MDSC Expansion and Recruitment into the Tumor Environment
We next explored a potential role for MDSCs as a possible target for apoA1 actions. MDSCs play a pivotal role in tumor-induced immune suppression and constitute a major component of the leukocyte infiltrate in the tumor (38). MDSCs are a heterogeneous population of immature myeloid cells that have the properties of macrophages, granulocytes, and DCs and whose numbers normally expand in response to tumor-driven factors, including prostaglandins, CXCR2, VEGF, and GM-CSF (38, 39). In initial studies we examined the impact of reconstituted nascent HDL on cultured bone marrow-derived macrophages. Of note, in the presence of HDL, expression levels of Ptgs1 (COX-1) and Cxcl2, the ligand for CXCR2, were significantly (p < 0.01 each) reduced (supplemental Table 1). The in vivo relevance of an apoA1 effect on MDSC was therefore next examined by testing whether A1Tg mice might accumulate fewer peripheral (splenic) MDSCs in response to tumor challenge. Flow cytometry analyses of splenic cells harvested 14 days after either vehicle (i.e. naive) or tumor cell inoculation revealed that in naive tumor-free mice, CD11b+GR-1+ (MDSC) cells showed only small numbers in the spleen (Fig. 6A). Myeloid differentiation antigen GR-1 consists of two epitopes, LY-6G and LY-6C, and MDSCs consist of two major subsets, the granulocytic CD11b+LY-6GhighLY-6Clow (CD11b+GR-1high) and the monocytic CD11b+LY-6GlowLY-6Chigh (CD11b+GR-1low) populations. Further flow cytometry analyses revealed a significant (p = 0.03) 2-fold tumor-induced increase in the percentage of splenic MDSCs recovered from A1KO mice, whereas no significant increase was observed in splenic MDSCs recovered from A1Tg+/+ mice (Fig. 6A). These data suggest expansion of splenic MDSC in response to the B16F10L tumor is attenuated in A1Tg+/+ mice. Confirmation of this was observed in a subsequent experiment in which tumor-bearing spleens from A1KO and A1Tg+/− mice were examined 12 days post-tumor cell inoculation. Decreased expansion of CD11b+GR-1+ MDSCs was noted in the A1Tg+/− versus A1KO tumor-bearing mice (3.9% versus 5.9%, respectively; p = 0.02, Fig. 6B). Of note, mature macrophages (indicated by F4/80+ staining) were also significantly decreased in the spleens of the A1Tg+/− versus A1KO tumor-bearing mice (9.1% versus 11.8%; p = 0.01, Fig. 6B).
FIGURE 6.

ApoA1 therapy inhibits accumulation of MDSCs in tumor bed. A and B, flow cytometry was performed on splenocytes from indicated genotypes inoculated with normal saline (naive) or B16F10L-luciferase melanoma cells (two sites, 1 × 105 cells per site) (A) or inoculated with B16F10L-luciferase melanoma cells at four separate sites (B, 1 × 105 cells per site) and sacrificed on day 14 (A) or day 12 (B) post-tumor inoculation. Data points are mean ± S.E. C, representative day 7, B16F10L-luciferase tumor staining from A1KO and A1Tg+/− mice for GR1+ cells. Main image is at ×10 and the inset is at ×40 magnification with scale bars representing 20 μm. D, subcutaneous day 15 B16F10L-luciferase tumors were excised from A1KO tumor-bearing mice treated with apoA1 therapy (20 mg/day/animal on days 10–14 post-tumor inoculation; 105 tumor cells/site; six sites per animal) and pooled (n = number of animals/treatment arm). Tumors were digested, and single cell suspensions were surface-stained for MDSCs (CD11b+GR1+) and processed by flow cytometry as described under “Experimental Procedures.” Immune cells are expressed as frequency of live CD45.2+ cells (tumor-infiltrating leukocytes; TILs).
Current tumor immunobiology paradigms imply that MDSCs in tumor-bearing animals contribute to tumor progression, increased angiogenesis, and decreased immune surveillance, with MDSC levels being elevated not only in the circulation and peripheral immune organs but also in and around the tumor itself (38, 40). Consistent with our observation of decreased CD11b+GR-1+ cells in the spleens of tumor-bearing A1Tg mice, examination of B16F10L primary tumor tissue for the presence of CD11b+GR-1+ cells revealed that these cells were easily detected in primary tumors in A1KO mice but were significantly less abundant in A1Tg+/− mice (Fig. 6C shows a representative example). Importantly, apoA1 therapy (days 10–14 post-tumor inoculation) in A1KO mice reduced (by 50% compared with control saline-injected) the accumulation of CD11b+GR-1+ in primary tumors (Fig. 6D). Together, the above results indicate an inhibitory role for apoA1 in MDSC expansion and recruitment to the tumor microenvironment.
Tumor-associated Angiogenesis and Tumor Invasion Are Inhibited by ApoA1
Tumor neoangiogenesis is required for tumor growth and metastasis. The dramatic inhibitory effect of apoA1 on tumor growth and metastasis suggested another potential mechanism through which apoA1 may exert its anti-neoplastic effect(s) may be via inhibition of angiogenesis. Quantitative analyses of blood vessels directly feeding into day 7 primary mouse melanoma tumors revealed a significant (p < 0.005) >2-fold decrease in the number of vessels in tumors from A1Tg+/+ mice compared with A1KO hosts (Fig. 7A), consistent with apoA1 having an inhibitory effect on neoangiogenesis in the tumor bed. More detailed comparisons in A1Tg+/+ versus A1KO mice revealed marked reduction in multiple key properties of the vascular network feeding the tumor within the A1Tg+/+ mice, including reduction in vessel area, vessel length density, and size of vessels (p < 0.005 for each; Fig. 7, C–E). Thus, primary tumors from animals with the same initial tumor burden exhibited decreased angiogenesis in the presence of apoA1.
Angiogenesis can be vascular endothelial growth factor (VEGF-A)-dependent or -independent (41, 42). Examination of Vegfa mRNA expression and VEGF protein levels from tumor tissue recovered 7 days post-cancer cell inoculation in A1Tg animals exhibited a modest increase relative to that observed in A1KO animals (Fig. 7F). Thus, the reduction in angiogenesis observed within the A1Tg animals does not appear to be related to VEGF. This result contrasts with the recent finding that in an ovarian cancer model short amphipathic peptide mimetics of apoA1 directly reduced the viability of tumor cells in tissue culture and inhibited production of VEGF (28).
ApoA1 Inhibits MMP-9
Matrix metalloproteinases are zinc-dependent extracellular matrix-degrading enzymes critical for tumor angiogenesis, invasion, and metastasis in many cancers. Higher levels of active MMP-9 have been observed in more invasive and metastatic melanoma tumors (43). Expression of latent MMP-9 is regulated at the level of transcription, whereas its enzymatic activity is controlled by specific proteolytic cleavage of pro-MMP-9 (43). We therefore investigated whether apoA1 or reconstituted nascent HDL could lead to decreased levels of MMP-9 that would negatively affect cell invasion through a basement membrane. Initially, we tested this idea directly on B16F10L tumor cells in vitro using a cell invasion assay. Exposure of cells to plasma levels of either HDL or apoA1 resulted in modest (∼10%) but statistically significant impairment in tumor cell invasion across the basement membrane (data not shown). Given this small direct effect of the lipoprotein on B16F10L cell invasion in vitro, we next examined if this effect might be amplified in vivo and result as a consequence of decreased MMP-9 levels in tumors. Although no significant differences in Mmp9 mRNA level from day 7 melanoma tumors harvested from A1KO and A1Tg hosts were observed (p = 0.53, n = 8 per group, data not shown), MMP-9 protein levels were a significant 1.8-fold lower (p < 0.05) in tumors from A1Tg mice (Fig. 8A) and displayed decreased levels (>2-fold, p < 0.05) of enzymatic activity (Fig. 8B). ApoA1 thus appears to inhibit the level and activation of MMP-9 in the tumor microenvironment. In support of this, HDL inhibited the expression of Mmp9 in bone marrow-derived macrophages from WT mice (supplemental Table 1).
FIGURE 8.

Reduced MMP-9 activity in primary tumors from A1Tg mice. Day 7 B16F10L-luciferase subcutaneous tumors were homogenized for whole cell protein extract as described under “Experimental Procedures.” A, levels of MMP-9 protein detected by Western blot with β-actin as loading control. B, MMP-9 enzyme activity in tumor extracts was assayed as described under “Experimental Procedures.” Horizontal bars in A and B represent the median of the data series. n = number of animals.
ApoA1 Suppresses Expression of Survivin in the Tumor Bed
Tumors develop mechanisms to overcome death signals initiated by host cytotoxic immune cells (44). Survivin is a member of the inhibitor of apoptosis protein family of proteins with both anti-apoptotic and cell cycle promoting activities (44). In melanoma, higher cellular levels of survivin, and in particular nuclear localization of the protein, are associated with increased tumor aggressiveness and poorer patient outcome (45). Conversely, survivin down-regulation sensitizes melanoma cells to apoptosis (45). Examination of survivin protein levels and intracellular localization within day 7 primary melanoma tumors from A1Tg and A1KO mice showed survivin was expressed at a 2.7-fold lower level in A1Tg mice (Fig. 9A) with ∼24% of nuclei staining positive, as compared with ∼50% of the nuclei in the A1KO group (Fig. 9B). Tumors from A1KO mice showed higher levels of survivin in both non-nuclear and nuclear compartments (3- and 2.4-fold, respectively, p < 0.05 for each; data not shown). The higher protein levels of survivin in tumors from A1KO relative to A1Tg animals paralleled a higher survivin mRNA level observed in this group (1.4-fold, p = 0.006, data not shown). These results were also consistent with observed inhibitory effects of HDL on Birc5 (survivin) in bone marrow-derived macrophages in culture (supplemental Table 1).
FIGURE 9.
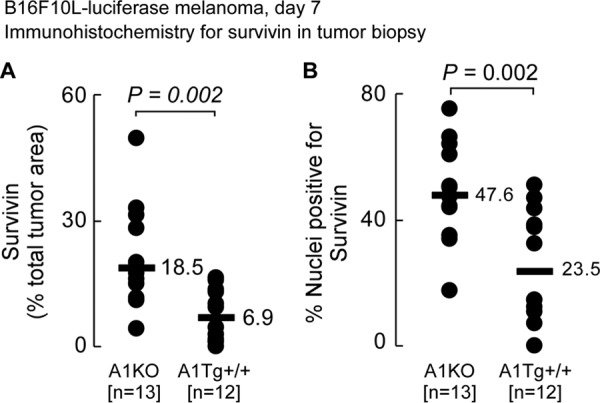
Reduced survivin expression in primary tumors from A1Tg mice. A, primary day 7 B16F10L-luciferase tumors were subjected to immunohistochemistry for survivin as described under “Experimental Procedures.” Images were captured and quantified for positive survivin staining (A) and number of nuclei positive for survivin (B) as described under “Experimental Procedures.”
ApoA1 Promotes Anti-tumor Effects in Vivo through Tumor-associated Macrophages
Many tumor-infiltrating leukocytes with tumoricidal activity are of myeloid origin and characterized by surface expression of CD11b+. We therefore next explored whether an additional potential contributory mechanism for the anti-neoplastic activity of apoA1 might occur via apoA1 influencing the extent of tumor invasion by CD11b+ monocyte-derived macrophages. This was of interest because Tall and co-workers (46) recently demonstrated mice deficient in the adenosine triphosphate-binding cassette (ABC) transporters ABCA1 and ABCG1, which are participants in HDL metabolism, display a peripheral monocytosis as a result of a myeloproliferative disorder, suggesting that HDL may inhibit the proliferation of hematopoietic stem cells linked to the observed monocytosis in the ABCA1- and ABCG1-deficient mice. We therefore first examined whether differences in levels of circulating monocytes were observed among A1KO, WT, and A1Tg mice. Hematologic analyses revealed a significant 2.5-fold higher level of circulating monocytes in A1Tg mice compared with A1KO (p < 0.001, supplemental Table 2) and a near significant increase in absolute monocyte count in WT versus A1KO mice (p = 0.06, supplemental Table 2). We therefore next examined whether there was differential recruitment of CD11b+ cells within tumor beds in our mouse model for melanoma. CD11b is an adhesion surface molecule that plays a role in adherence of immune effector cytotoxic cells, such as natural killer cells, to target cells (e.g. tumors) and is present on numerous monocyte-derived cell types, including MDSCs, macrophages, and DCs. Whether these infiltrating leukocytes fight or promote the tumor depends on the nature of the tumor microenvironment (47). Tumors from A1Tg mice (compared with those from A1KO mice) exhibited increased numbers of CD11b+ cells both within the primary tumor bed and in the surrounding stromal region (Fig. 10). Immunostaining of tumors with anti-F4/80+ antibody demonstrated colocalization with the majority of the CD11b+ staining, indicating that the CD11b+ cells are primarily macrophages (Fig. 10A, lower right panel). Double staining of the same tumor section followed by confocal microscopy proved these cells to be primarily Cd11b+F480+ (supplemental Fig. 1).
FIGURE 10.

ApoA1 promotes the infiltration of F4/80+ macrophages into tumor bed. Primary day 7 B16F10L-luciferase tumors were subjected to immunohistochemistry for CD11b+ or F4/80+ (red) and nuclei stained with DAPI (blue). The entire tumor area was captured with a ×20 lens, and digitized images were assessed for positive staining as described under “Experimental Procedures.” Representative images for anti CD11b+ or F4/80+ staining of tumors are shown in A. Top panels were stained for CD11b+, and lower panels were stained for F4/80+. Scale bars represent 500 μm. Insets from A are shown at a higher magnification in B where scale bars represent 75 μm. CD11b protein expression was quantified as described under “Experimental Procedures.” C, data indicate the numbers and genotypes of mice.
To determine whether these TAMs might fight or promote tumor growth, tumors were harvested, digested, and TAMS immunopurified. Targeted expression profiling of the recovered TAMS for 13 distinct genes associated with either M1 or M2 phenotypes revealed M1 classically activated phenotypes for TAMs recovered from primary tumors isolated from A1Tg mice compared with A1KO mice (Table 2). In further studies, F4/80+ cells were recovered from tumor-bearing A1Tg and A1KO mice and examined for tumoricidal activity at different effector (F4/80+ cells) to target (B16F10L tumor cells) ratios. F4/80+ cells recovered from the tumor-bearing A1Tg mice, but not A1KO mice, exhibited direct anti-tumor activity in vitro (Fig. 11), consistent with the expression profile observed of a classically activated M1 phenotype for macrophages in the apoA1-expressing hosts (Table 2).
TABLE 2.
B16F10L-luciferase day 17
Shown are quantitative RT-PCR results of candidate genes expressed in F4/80+ (TAMS) immunopurified from day 17 primary subcutaneous melanoma tumors, as described under “Experimental Procedures.”
| M1 immune stimulatory | M2 immune suppressive | F4/80+ (TAMs) A1Tg+/+/A1KO RQ (95% CI) |
|---|---|---|
| ↑ Ifng | 24.3 (22.1–26.7) | |
| ↑ Ccl22 | 5.44 (5.08–5.83) | |
| ↑ Il6 | 2.82 (2.68–2.96) | |
| ↑ Nos2 | 2.16 (2.01–2.33) | |
| ↑ Cx3cl1 | 2.12 (1.97–2.29) | |
| ↑ Il12b | 1.85 (1.76–1.94) | |
| ↑ Il1b | 1.80 (1.72–1.89) | |
| ↑ Cc17 | 1.36 (1.36–1.37) | |
| ↑ Cxcl10 | 1.27 (1.25–1.28) | |
| ↓ Pparg | 0.57 (0.56–0.58) | |
| ↓ Il10 | 0.66 (0.61–0.73) | |
| ↓ Chi313 | 0.78 (0.76–0.80) | |
| ↑ Arg1 | 2.31 (2.26–2.37) |
FIGURE 11.

F4/80+ cells recovered from tumor-bearing A1Tg mice are cytotoxic to tumor cells in vitro. F4/80+ effector (E) cells immunopurified from elicited peritoneal exudates of day 19 tumor-bearing animals were cocultured with B16F10L target (T) cells in vitro, and cell viability was assessed after 3 days as described under “Experimental Procedures.”
The cytokine IL-12 is known to orchestrate a Th1 anti-tumor response in major part through IFN-γ (Ifng) and downstream chemokines MIG (Cxcl9) and IP-10 (Cxcl10), which attract cytotoxic CD8 T cells into the tumor bed and thus potentiate an anti-tumor program (48, 49). The observed elevated expression levels of Ifng, Il12b, and Cxcl10 in TAMS isolated from tumor-bearing A1Tg+/+ animals (Table 2) led us to hypothesize that one mechanism through which apoA1 may promote an anti-tumor effect is via a TAM-mediated increase in accumulation of cytotoxic CD8T cells in the A1Tg+/+ tumor environment. To test this, we used flow cytometry to examine digested melanoma tumors recovered from A1Tg+/+ animals versus A1KO for CD8T cell infiltration. Consistent with this hypothesis, we observed a 4-fold increased infiltration of tumor-infiltrating leukocytes (TILs) and a 2-fold higher level of CD8 T cells in day 15 melanoma tumors from A1Tg+/+ animals compared with A1KO (Fig. 12). This observation was specific to the CD8 T subset of TILs, because CD4 T levels were similar in tumors from both genotypes (Fig. 12).
FIGURE 12.

ApoA1 promotes tumor infiltration of cytotoxic CD8 T cells. FACS analysis of pooled tumors (per genotype; six sites/animal; n = number of animals). Day 15 tumors were digested and labeled with antibodies to surface antigens as described under “Experimental Procedures.” TILs were identified as CD45.2+, and the percent of TILs that were either CD3+CD8+ or CD3+CD4+ was quantified by flow cytometry as described under “Experimental Procedures.”
DISCUSSION
This study provides unequivocal evidence for potent anti-tumorigenic activity by apoA1, the major HDL-associated protein. The scheme shown in Fig. 13 summarizes our current understanding of the net functional effects of apoA1/HDL in the tumor microenvironment. An inhibitory role for apoA1/HDL on tumor growth and metastasis is supported by numerous observations, including demonstration that mice lacking apoA1 (A1KO) develop tumors at a much more rapid pace and are at a significant survival disadvantage, whereas tumor progression is dramatically restrained, and survival is improved in a dose-dependent manner in mice expressing apoA1. Importantly, subcutaneous injection of apoA1 distant from the tumor site markedly suppressed tumor development and metastasis formation, and when provided later in the disease course it promoted existing tumor and metastasis regression.
FIGURE 13.

Net functional effects of apoA1/HDL in tumor microenvironment. Overall scheme describing role of apoA1/HDL in the tumor microenvironment in melanoma tumor biology.
Our in vitro studies indicate apoA1 and HDL do not directly inhibit tumor cell growth nor directly promote tumor cell apoptosis in culture; however, in vitro incubation studies did reveal that HDL does suppress tumor cell invasion potential in a modest but significant manner. Two prominent effects of apoA1 within the tumor bed in vivo are inhibition of tumor-associated angiogenesis and concomitant reduction in MMP-9 protein levels and activity, a critical matrix-degrading enzyme whose action culminates in the availability of pro-angiogenic factors (50). The notion that apoA1 decreases a tumor-permissive environment is based on reduced accumulation of MDSC within the tumor bed and decreased expression and nuclear localization of survivin protein in tumor tissue from apoA1 transgenic mice. This anti-apoptotic, pro-survival protein appears to be a downstream target in apoA1/HDL signaling because in separate studies we observed that direct treatment of bone marrow-derived macrophages with HDL in vitro leads to a significant (p < 0.01; 2-fold) decrease in Birc5 (survivin) gene expression (supplemental Table 1). Interestingly, HDL also significantly (p < 0.01; 4.5-fold) repressed the expression of Mmp9 in macrophages in vitro (supplemental Table 1). Thus, rather than a direct effect on the tumor cell, our data repeatedly suggest that apoA1 in vivo exerts anti-neoplastic biological effects indirectly via alterations in macrophage and other immune cell functions.
Our animal model studies demonstrated a role for apoA1 anti-tumor effect on elements of both innate and adaptive arms of immunity to realize its full anti-tumor biological activity in vivo. Thus, a prominent biological activity promoted by apoA1 appears to be suppression of peripheral expansion and tumor recruitment and accumulation of MDSCs, potent stimulators of tumor growth. The increased circulating absolute monocyte counts, and influx of CD11b+F4/80+ macrophages into tumors of A1Tg animals as compared with those from A1KO mice, suggest another biological effect of the lipoprotein is to augment the innate arm of the immune system. Importantly, in the presence of apoA1, the infiltrating macrophages exhibited an anti-tumorigenic M1 classically activated phenotype. Interestingly, HDL and apoA1 have previously been shown to inhibit phorbol myristate acetate- or LPS-induced surface expression of CD11b adhesion molecules in human monocytes (51). ApoA1 mediates this effect in vitro through the ATP-binding cassette transporter ABCA1 (51). Disturbance of lipid raft formation, presumably mediated via reductions in membrane cholesterol content by apoA1 and HDL as cholesterol acceptors via the ABCA1 or ABCG1 transporter, respectively, has been suggested to inhibit downstream signaling, altering some cellular functions in immune cells. ApoA1/HDL were reported to inhibit monocyte differentiation into functional dendritic cells (52) and were shown to inhibit the capacity of antigen-presenting cells to stimulate T cell activation (53). Although the above studies were conducted in vitro, the possibility that apoA1-dependent modulation of cholesterol content of the immune cells within the tumor bed occurs and somehow participates in the phenotype observed in the present studies remains to be explored. Previous studies have also demonstrated that mice deficient in both ABCA1 and ABCG1 (DKO) exhibit leukocytosis as a result of hyper-proliferation of hematopoietic stem and progenitor cells (46). Transplantation of DKO bone marrow hematopoietic stem and progenitor cells into apoA1 transgenic mice reversed this myeloproliferative disorder (46). These data lead to the suggestion that HDL and ABCA1/ABCG1 transporters inhibit leukocytosis in major part through disruption of cholesterol-rich lipid rafts and their associated downstream signaling. In contrast, we observed increased circulating monocytes in A1Tg, twice as much as in A1KO mice (supplemental Table 2). At this time we cannot offer any explanation for this apparent discrepancy but suggest that regulation of myeloid cell homeostasis by lipid metabolism pathways is likely to be complex.
Given that MDSCs (54, 55), as well as M2 but not M1 macrophages (56), are proangiogenic, the reduction in angiogenesis observed in the tumors of apoA1Tg mice, and apoA1-treated mice appears also to be attributable in part to a reduction in the number of functional myeloid cells with pro-angiogenic activity. Complete apoA1 anti-tumor activity required both adaptive and innate arms of immunity (Fig. 5). ApoA1 anti-tumor activity was associated with TAM elaboration of enhanced Ifng and accumulation of CD8 T lymphocytes (Fig. 12). T cell infiltration of solid tumors such as melanoma is in general considered indicative of a favorable prognosis (57, 58).
These findings have clinical relevance at multiple levels. First, our studies demonstrated that human apoA1 dramatically reduces tumor growth and metastasis in malignant melanoma and Lewis lung carcinoma and within nude mice inoculated with human malignant melanoma tumor cells, supporting the notion that apoA1 anti-neoplastic activity is a general phenomenon and not species- or cancer-specific. However, clearly more human tumor types need to be screened to more fully understand the breadth of tumor types with which apoA1-dependent anti-tumor activity is observed. Furthermore, during the conduct of these studies, a large meta analysis of randomized controlled trials of lipid-altering therapies was reported that suggested an inverse relationship between plasma HDL cholesterol levels and the incidence of cancer development during the conduct of the trials (59). Specifically, for every 10 mg/dl increase in plasma HDL cholesterol level among trial participants, a significant 36% lower risk of cancer incidence was noted over 625,000 person-years of follow-up and >8,000 incidents of cancers cumulatively among the trials included in the meta analysis (59). Although such meta analyses are hypotheses generating and cannot serve as proof for an anti-cancer effect of HDL/apoA1 in humans, they are provocative. It is thus also of interest that another recent population-based prospective cohort study (n = 17,779) in China similarly reported a 2-fold cancer risk associated with low HDL cholesterol (60). Collectively, the present studies, coupled with these epidemiological studies in humans, suggest that HDL may be linked to tumor cell biology in humans.
As noted in the Introduction, studies with a series of amphipathic helical peptides that possess potent anti-inflammatory activities were recently reported to impact ovarian cancer tumor growth (29). These amphipathic peptides were initially reported to serve as apoA1 mimetic peptides in atherosclerosis studies, but more recent experiments in a variety of cardiovascular and inflammation models suggest their biological activities are markedly distinct from apoA1. It is thus perhaps not surprising that the relatively modest anti-tumor mechanism(s) reported for the short amphipathic peptides appear markedly distinct from those observed in this study because the peptide mimetics in tissue culture directly reduced the viability of ovarian cancer cells and the amphipathic peptides inhibited production of VEGF (28, 29). In this study, we observed that intact apoA1 promoted neither of these direct activities on cultured tumor cells. Moreover, it has been suggested that the amphipathic peptides act through binding of lysophosphatidic acid (29). However, this does not appear to be the case with intact apoA1 in this study because in separate stable isotope dilution LC-MS/MS experiments we observed no significant differences in plasma levels of multiple distinct molecular species of lysophosphatidic acid of tumor-bearing A1KO, WT, and A1Tg mice (Table 1).
One interesting observation in this study is the consistent demonstration of lower levels of splenic MDSCs in A1Tg tumor-bearing mice compared with A1KO animals, as well as reduced accumulation of these immune suppressive cells with apoA1 therapy (Fig. 6D). MDSCs are the major cellular mediators of defective anti-tumor immunity, and typically expand in response to pro-inflammatory signals originating from both tumor and host cells within the tumor microenvironment. The above effect of apoA1 on MDSC numbers thus suggest that apoA1 inhibits tumor-initiated inflammatory signals that lead to expansion of these immunosuppressive cells. It is of interest to note that monocyte-derived macrophages from human subjects with low HDL were recently reported to exhibit a pro-inflammatory gene expression signature (61). Similarly, we also observed that incubation of bone marrow-derived macrophages in the presence versus the absence of reconstituted nascent HDL particles results in suppression of pro-inflammatory genes such as S100a8, S100a9, Mmp9, Ccl2 and that of the immunosuppressive cytokine, Il10, and chemokine, Ccl7 (p < 0.01 for each, supplemental Table 1). We also observed fewer GR1+ cells in the tumor microenvironment of apoA1-expressing hosts (Fig. 6C). ApoA1/HDL was recently shown to inhibit neutrophil activation, adhesion, and migration (62).
The pronounced anti-neoplastic effects observed for apoA1 in vivo in this study add to the growing list of evidence indicating that the lipoprotein has evolved to function as more than simply a shuttle of cholesterol cargo. It remains to be shown whether anti-tumor activity of apoA1/HDL arises in part through modulation of immune cell function via regulation of cholesterol content in critical subcellular compartments. Identification of potent immune modulatory effects with resultant anti-tumor biological activity of apoA1 suggests that pharmacological delivery of apoA1 may be exploited for therapeutic gain as an anticancer agent or for tumor/metastasis regression. Indeed, our studies show administration of apoA1 after palpable tumor formation and metastasis detection leads to 50% shrinkage of peak tumor and metastasis burden within a week. It is of interest that despite the failure of multiple HDL cholesterol-raising drugs, apoA1 infusions or apoA1-elevating drugs have thus far shown potential therapeutic benefit and are being developed as potential therapeutics for cardiovascular disease (25, 63–65). This study thus indicates that apoA1 also represents a viable therapeutic agent for evaluation in malignant melanoma and possibly other cancers in humans.
Acknowledgments
We thank the core services from the Case Western Reserve University Comprehensive Cancer Center, the Flow Cytometry and Imaging Core at Lerner Research Institute, and the following persons for their help in these studies: C. Tannenbaum, Ph.D. (Immunology, Lerner Research Institute), for critical discussions and review of the manuscript; M. Johansen for review of the manuscript; and A. Vasanji, Ph.D., A. Cotleur, J. Peterson, Ph.D., M. Pepoy, and B. Gopalan for technical assistance. The mass spectrometry instrumentation used is housed within the Cleveland Clinic Mass Spectrometry Facility and is supported in part through a Center of Innovation by AB SCIEX.
This work was supported, in whole or in part, by National Institutes of Health Grants P01HL098055, P01HL076491, and HL094525. This work was also supported in part by a grant from the LeDucq Fondation. Drs. Zamanian-Daryoush, DiDonato, and Hazen report being listed as co-inventors on pending and issued patents held by the Cleveland Clinic. Dr. Hazen reports having been paid as a consultant for the following companies: Abbott, Cleveland Heart Lab, Esperion, Lilly, Liposcience Inc., Merck & Co., Inc., and Pfizer Inc. and receiving research funds from Abbott, Cleveland Heart Lab, Liposcience Inc., and Pfizer Inc.
This article was selected as a Paper of the Week.

This article contains supplemental Fig. 1 and Tables 1 and 2.
- MDSC
- myeloid-derived suppressor cell
- TAM
- tumor-associated macrophage
- DC
- dendritic cell
- TIL
- tumor infiltrating leukocyte
- LPA
- lysophosphatidic acid
- NSG
- NOD Scid IL2r γ.
REFERENCES
- 1. Linos E., Swetter S. M., Cockburn M. G., Colditz G. A., Clarke C. A. (2009) Increasing burden of melanoma in the United States. J. Invest. Dermatol. 129, 1666–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Godar D. E. (2011) Worldwide increasing incidences of cutaneous malignant melanoma. J. Skin Cancer 2011:858425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Navarini-Meury A. A., Conrad C. (2009) Melanoma and innate immunity–active inflammation or just erroneous attraction? Melanoma as the source of leukocyte-attracting chemokines. Semin. Cancer Biol. 19, 84–91 [DOI] [PubMed] [Google Scholar]
- 4. Libby P., Ridker P. M., Hansson G. K. (2011) Progress and challenges in translating the biology of atherosclerosis. Nature 473, 317–325 [DOI] [PubMed] [Google Scholar]
- 5. Weber C., Noels H. (2011) Atherosclerosis: current pathogenesis and therapeutic options. Nat. Med. 17, 1410–1422 [DOI] [PubMed] [Google Scholar]
- 6. Mantovani A. (2010) Molecular pathways linking inflammation and cancer. Curr. Mol. Med. 10, 369–373 [DOI] [PubMed] [Google Scholar]
- 7. Ben-Neriah Y., Karin M. (2011) Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 12, 715–723 [DOI] [PubMed] [Google Scholar]
- 8. Colotta F., Allavena P., Sica A., Garlanda C., Mantovani A. (2009) Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30, 1073–1081 [DOI] [PubMed] [Google Scholar]
- 9. Rye K. A., Bursill C. A., Lambert G., Tabet F., Barter P. J. (2009) The metabolism and anti-atherogenic properties of HDL. J. Lipid Res. 50, S195–S200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Khera A. V., Cuchel M., de la Llera-Moya M., Rodrigues A., Burke M. F., Jafri K., French B. C., Phillips J. A., Mucksavage M. L., Wilensky R. L., Mohler E. R., Rothblat G. H., Rader D. J. (2011) Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 364, 127–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Voight B. F., Peloso G. M., Orho-Melander M., Frikke-Schmidt R., Barbalic M., Jensen M. K., Hindy G., Hólm H., Ding E. L., Johnson T., Schunkert H., Samani N. J., Clarke R., Hopewell J. C., Thompson J. F., Li M., Thorleifsson G., Newton-Cheh C., Musunuru K., Pirruccello J. P., Saleheen D., Chen L., Stewart A. F., Schillert A., Thorsteinsdottir U., Thorgeirsson G., Anand S., Engert J. C., Morgan T., Spertus J., Stoll M., Berger K., Martinelli N., Girelli D., McKeown P. P., Patterson C. C., Epstein S. E., Devaney J., Burnett M.-S., Mooser V., Ripatti S., Surakka I., Nieminen M. S., Sinisalo J., Lokki M.-L., Havulinna A., de Faire U., Gigante B., Ingelsson E., Zeller T., Wild P., de Bakker P. I., Klungel O. H., Maitland-van der Zee A.-H., Peters B. J., de Boer A., Grobbee D. E., Kamphuisen P. W., Deneer V. H., Elbers C. C., Onland-Moret N. C., Hofker M. H., Wijmenga C., Verschuren W. M., Boer J. M., van der Schouw Y. T., Rasheed A., Frossard P., Demissie S., Willer C., Do R., Ordovas J. M., Abecasis G. R., Boehnke M., Mohlke K. L., Daly M. J., Guiducci C., Burtt N. P., Surti A., Gonzalez E., Purcell S., Gabriel S., Marrugat J., Peden J., Erdmann J., Diemert P., Willenborg C., König I. R., Fischer M., Hengstenberg C., Ziegler A., Buysschaert I., Lambrechts D., Van de Werf F., Fox K. A., El Mokhtari N. E., Rubin D., Schrezenmeir J., Schreiber S., Schäfer A., Danesh J., Blankenberg S., Roberts R., McPherson R., Watkins H., Hall A. S., Overvad K., Rimm E., Boerwinkle E., Tybjaerg-Hansen A., Cupples L. A., Reilly M. P., Melander O., Mannucci P. M., Ardissino D., Siscovick D., Elosua R., Stefansson K., O'Donnell C. J., Salomaa V., Rader D. J., Peltonen L., Schwartz S. M., Altshuler D., Kathiresan S., Perola M. (2012) Plasma HDL cholesterol and risk of myocardial infarction: a Mendelian randomisation study. Lancet 380, 572–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Camont L., Chapman M. J., Kontush A. (2011) Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol. Med. 17, 594–603 [DOI] [PubMed] [Google Scholar]
- 13. Gordon S. M., Hofmann S., Askew D. S., Davidson W. S. (2011) High density lipoprotein: it's not just about lipid transport anymore. Trends Endocrinol. Metab. 22, 9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cockerill G. W., Rye K. A., Gamble J. R., Vadas M. A., Barter P. J. (1995) High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler. Thromb. Vasc. Biol. 15, 1987–1994 [DOI] [PubMed] [Google Scholar]
- 15. Vanhamme L., Paturiaux-Hanocq F., Poelvoorde P., Nolan D. P., Lins L., Van Den Abbeele J., Pays A., Tebabi P., Van Xong H., Jacquet A., Moguilevsky N., Dieu M., Kane J. P., De Baetselier P., Brasseur R., Pays E. (2003) Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature 422, 83–87 [DOI] [PubMed] [Google Scholar]
- 16. Bhattacharyya T., Nicholls S. J., Topol E. J., Zhang R., Yang X., Schmitt D., Fu X., Shao M., Brennan D. M., Ellis S. G., Brennan M. L., Allayee H., Lusis A. J., Hazen S. L. (2008) Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 299, 1265–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Souza J. A., Vindis C., Nègre-Salvayre A., Rye K. A., Couturier M., Therond P., Chantepie S., Salvayre R., Chapman M. J., Kontush A. (2010) Small, dense HDL 3 particles attenuate apoptosis in endothelial cells: Pivotal role of apolipoprotein A-I. J. Cell. Mol. Med. 14, 608–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fuhrman B., Gantman A., Aviram M. (2010) Paraoxonase 1 (PON1) deficiency in mice is associated with reduced expression of macrophage SR-BI and consequently the loss of HDL cytoprotection against apoptosis. Atherosclerosis 211, 61–68 [DOI] [PubMed] [Google Scholar]
- 19. Srinivas R. V., Venkatachalapathi Y. V., Rui Z., Owens R. J., Gupta K. B., Srinivas S. K., Anantharamaiah G. M., Segrest J. P., Compans R. W. (1991) Inhibition of virus-induced cell fusion by apolipoprotein A-I and its amphipathic peptide analogs. J. Cell. Biochem. 45, 224–237 [DOI] [PubMed] [Google Scholar]
- 20. Singh I. P., Chopra A. K., Coppenhaver D. H., Ananatharamaiah G. M., Baron S. (1999) Lipoproteins account for part of the broad nonspecific antiviral activity of human serum. Antiviral Res. 42, 211–218 [DOI] [PubMed] [Google Scholar]
- 21. Tada N., Sakamoto T., Kagami A., Mochizuki K., Kurosaka K. (1993) Antimicrobial activity of lipoprotein particles containing apolipoprotein Al. Mol. Cell. Biochem. 119, 171–178 [DOI] [PubMed] [Google Scholar]
- 22. Wilhelm A. J., Zabalawi M., Owen J. S., Shah D., Grayson J. M., Major A. S., Bhat S., Gibbs D. P., Jr., Thomas M. J., Sorci-Thomas M. G. (2010) Apolipoprotein A-I modulates regulatory T cells in autoimmune LDLr−/−, ApoA-I−/− mice. J. Biol. Chem. 285, 36158–36169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Levine D. M., Parker T. S., Donnelly T. M., Walsh A., Rubin A. L. (1993) In vivo protection against endotoxin by plasma high density lipoprotein. Proc. Natl. Acad. Sci. U.S.A. 90, 12040–12044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grunfeld C., Marshall M., Shigenaga J. K., Moser A. H., Tobias P., Feingold K. R. (1999) Lipoproteins inhibit macrophage activation by lipoteichoic acid. J. Lipid Res. 40, 245–252 [PubMed] [Google Scholar]
- 25. Degoma E. M., Rader D. J. (2011) Novel HDL-directed pharmacotherapeutic strategies. Nat. Rev. Cardiol. 8, 266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang S., Damiano M. G., Zhang H., Tripathy S., Luthi A. J., Rink J. S., Ugolkov A. V., Singh A. T., Dave S. S., Gordon L. I., Thaxton C. S. (2013) Biomimetic, synthetic HDL nanostructures for lymphoma. Proc. Natl. Acad. Sci. U.S.A. 110, 2511–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shahzad M. M., Mangala L. S., Han H. D., Lu C., Bottsford-Miller J., Nishimura M., Mora E. M., Lee J. W., Stone R. L., Pecot C. V., Thanapprapasr D., Roh J. W., Gaur P., Nair M. P., Park Y. Y., Sabnis N., Deavers M. T., Lee J. S., Ellis L. M., Lopez-Berestein G., McConathy W. J., Prokai L., Lacko A. G., Sood A. K. (2011) Targeted delivery of small interfering RNA using reconstituted high-density lipoprotein nanoparticles. Neoplasia 13, 309–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gao F., Vasquez S. X., Su F., Roberts S., Shah N., Grijalva V., Imaizumi S., Chattopadhyay A., Ganapathy E., Meriwether D., Johnston B., Anantharamaiah G. M., Navab M., Fogelman A. M., Reddy S. T., Farias-Eisner R. (2011) L-5F, an apolipoprotein A-I mimetic, inhibits tumor angiogenesis by suppressing VEGF/basic FGF signaling pathways. Integr. Biol. 3, 479–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Su F., Kozak K. R., Imaizumi S., Gao F., Amneus M. W., Grijalva V., Ng C., Wagner A., Hough G., Farias-Eisner G., Anantharamaiah G. M., Van Lenten B. J., Navab M., Fogelman A. M., Reddy S. T., Farias-Eisner R. (2010) Apolipoprotein A-I (apoA-I) and apoA-I mimetic peptides inhibit tumor development in a mouse model of ovarian cancer. Proc. Natl. Acad. Sci. U.S.A. 107, 19997–20002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Osborne J. C., Jr. (1986) Delipidation of plasma lipoproteins. Methods Enzymol. 128, 213–222 [DOI] [PubMed] [Google Scholar]
- 31. Weisweiler P. (1987) Isolation and quantitation of apolipoproteins A-I and A-II from human high-density lipoproteins by fast-protein liquid chromatography. Clin. Chim. Acta 169, 249–254 [DOI] [PubMed] [Google Scholar]
- 32. Matz C. E., Jonas A. (1982) Micellar complexes of human apolipoprotein A-I with phosphatidylcholines and cholesterol prepared from cholate-lipid dispersions. J. Biol. Chem. 257, 4535–4540 [PubMed] [Google Scholar]
- 33. Murph M., Tanaka T., Pang J., Felix E., Liu S., Trost R., Godwin A. K., Newman R., Mills G. (2007) Liquid chromatography mass spectrometry for quantifying plasma lysophospholipids: potential biomarkers for cancer diagnosis. Methods Enzymol. 433, 1–25 [DOI] [PubMed] [Google Scholar]
- 34. Rubin E. M., Ishida B. Y., Clift S. M., Krauss R. M. (1991) Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high density lipoprotein size subclasses. Proc. Natl. Acad. Sci. U.S.A. 88, 434–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Herber D. L., Cao W., Nefedova Y., Novitskiy S. V., Nagaraj S., Tyurin V. A., Corzo A., Cho H. I., Celis E., Lennox B., Knight S. C., Padhya T., McCaffrey T. V., McCaffrey J. C., Antonia S., Fishman M., Ferris R. L., Kagan V. E., Gabrilovich D. I. (2010) Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 16, 880–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kusmartsev S., Gabrilovich D. I. (2006) Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol. Immunother. 55, 237–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shultz L. D., Lyons B. L., Burzenski L. M., Gott B., Chen X., Chaleff S., Kotb M., Gillies S. D., King M., Mangada J., Greiner D. L., Handgretinger R. (2005) Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2Rγ null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 174, 6477–6489 [DOI] [PubMed] [Google Scholar]
- 38. Gabrilovich D. I., Nagaraj S. (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Condamine T., Gabrilovich D. I. (2011) Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 32, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang L., DeBusk L. M., Fukuda K., Fingleton B., Green-Jarvis B., Shyr Y., Matrisian L. M., Carbone D. P., Lin P. C. (2004) Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 6, 409–421 [DOI] [PubMed] [Google Scholar]
- 41. West X. Z., Malinin N. L., Merkulova A. A., Tischenko M., Kerr B. A., Borden E. C., Podrez E. A., Salomon R. G., Byzova T. V. (2010) Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 467, 972–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim S., Karin M. (2011) Role of TLR2-dependent inflammation in metastatic progression. Ann. N. Y. Acad. Sci. 1217, 191–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hofmann U. B., Houben R., Bröcker E. B., Becker J. C. (2005) Role of matrix metalloproteinases in melanoma cell invasion. Biochimie 87, 307–314 [DOI] [PubMed] [Google Scholar]
- 44. McKenzie J. A., Grossman D. (2012) Role of the apoptotic and mitotic regulator survivin in melanoma. Anticancer Res. 32, 397–404 [PubMed] [Google Scholar]
- 45. Piras F., Murtas D., Minerba L., Ugalde J., Floris C., Maxia C., Colombari R., Perra M. T., Sirigu P. (2007) Nuclear survivin is associated with disease recurrence and poor survival in patients with cutaneous malignant melanoma. Histopathology 50, 835–842 [DOI] [PubMed] [Google Scholar]
- 46. Yvan-Charvet L., Pagler T., Gautier E. L., Avagyan S., Siry R. L., Han S., Welch C. L., Wang N., Randolph G. J., Snoeck H. W., Tall A. R. (2010) ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328, 1689–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grivennikov S. I., Greten F. R., Karin M. (2010) Immunity, inflammation, and cancer. Cell 140, 883–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Colombo M. P., Trinchieri G. (2002) Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 13, 155–168 [DOI] [PubMed] [Google Scholar]
- 49. Tannenbaum C. S., Tubbs R., Armstrong D., Finke J. H., Bukowski R. M., Hamilton T. A. (1998) The CXC chemokines IP-10 and Mig are necessary for IL-12-mediated regression of the mouse RENCA tumor. J. Immunol. 161, 927–932 [PubMed] [Google Scholar]
- 50. Bauvois B. (2012) New facets of matrix metalloproteinases MMP-2 and MMP-9 as cell surface transducers: outside-in signaling and relationship to tumor progression. Biochim. Biophys. Acta 1825, 29–36 [DOI] [PubMed] [Google Scholar]
- 51. Murphy A. J., Woollard K. J., Hoang A., Mukhamedova N., Stirzaker R. A., McCormick S. P., Remaley A. T., Sviridov D., Chin-Dusting J. (2008) High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler. Thromb. Vasc. Biol. 28, 2071–2077 [DOI] [PubMed] [Google Scholar]
- 52. Kim K. D., Lim H. Y., Lee H. G., Yoon D. Y., Choe Y. K., Choi I., Paik S. G., Kim Y. S., Yang Y., Lim J. S. (2005) Apolipoprotein A-I induces IL-10 and PGE2 production in human monocytes and inhibits dendritic cell differentiation and maturation. Biochem. Biophys. Res. Commun. 338, 1126–1136 [DOI] [PubMed] [Google Scholar]
- 53. Wang S. H., Yuan S. G., Peng D. Q., Zhao S. P. (2012) HDL and ApoA-I inhibit antigen presentation-mediated T cell activation by disrupting lipid rafts in antigen presenting cells. Atherosclerosis 225, 105–114 [DOI] [PubMed] [Google Scholar]
- 54. Shojaei F., Wu X., Malik A. K., Zhong C., Baldwin M. E., Schanz S., Fuh G., Gerber H. P., Ferrara N. (2007) Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat. Biotechnol. 25, 911–920 [DOI] [PubMed] [Google Scholar]
- 55. Kujawski M., Kortylewski M., Lee H., Herrmann A., Kay H., Yu H. (2008) Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Invest. 118, 3367–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lamagna C., Aurrand-Lions M., Imhof B. A. (2006) Dual role of macrophages in tumor growth and angiogenesis. J. Leukocyte Biol. 80, 705–713 [DOI] [PubMed] [Google Scholar]
- 57. Pagès F., Galon J., Dieu-Nosjean M. C., Tartour E., Sautès-Fridman C., Fridman W. H. (2010) Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene 29, 1093–1102 [DOI] [PubMed] [Google Scholar]
- 58. Clemente C. G., Mihm M. C., Jr., Bufalino R., Zurrida S., Collini P., Cascinelli N. (1996) Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 77, 1303–1310 [DOI] [PubMed] [Google Scholar]
- 59. Jafri H., Alsheikh-Ali A. A., Karas R. H. (2010) Baseline and on-treatment high-density lipoprotein cholesterol and the risk of cancer in randomized controlled trials of lipid-altering therapy. J. Am. Coll. Cardiol. 55, 2846–2854 [DOI] [PubMed] [Google Scholar]
- 60. Chen W., Lu F., Liu S. J., DU J. B., Wang J. M., Qian Y., Shen C., Jin G. F., Hu Z. B., Shen H. B. (2012) Cancer risk and key components of metabolic syndrome: a population-based prospective cohort study in Chinese. Chin. Med. J. 125, 481–485 [PubMed] [Google Scholar]
- 61. Sarov-Blat L., Kiss R. S., Haidar B., Kavaslar N., Jaye M., Bertiaux M., Steplewski K., Hurle M. R., Sprecher D., McPherson R., Marcel Y. L. (2007) Predominance of a proinflammatory phenotype in monocyte-derived macrophages from subjects with low plasma HDL cholesterol. Arterioscler. Thromb. Vasc. Biol. 27, 1115–1122 [DOI] [PubMed] [Google Scholar]
- 62. Murphy A. J., Woollard K. J., Suhartoyo A., Stirzaker R. A., Shaw J., Sviridov D., Chin-Dusting J. P. (2011) Neutrophil activation is attenuated by high-density lipoprotein and apolipoprotein A-I in in vitro and in vivo models of inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 1333–1341 [DOI] [PubMed] [Google Scholar]
- 63. Nissen S. E., Tsunoda T., Tuzcu E. M., Schoenhagen P., Cooper C. J., Yasin M., Eaton G. M., Lauer M. A., Sheldon W. S., Grines C. L., Halpern S., Crowe T., Blankenship J. C., Kerensky R. (2003) Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA 290, 2292–2300 [DOI] [PubMed] [Google Scholar]
- 64. Tardif J. C., Grégoire J., L'Allier P. L., Ibrahim R., Lespérance J., Heinonen T. M., Kouz S., Berry C., Basser R., Lavoie M. A., Guertin M. C., Rodés-Cabau J., and Effect of rHDL on Atherosclerosis-Safety and Efficacy (ERASE) Investigators (2007) Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA 297, 1675–1682 [DOI] [PubMed] [Google Scholar]
- 65. Sacks F. M., Rudel L. L., Conner A., Akeefe H., Kostner G., Baki T., Rothblat G., de la Llera-Moya M., Asztalos B., Perlman T., Zheng C., Alaupovic P., Maltais J. A., Brewer H. B. (2009) Selective delipidation of plasma HDL enhances reverse cholesterol transport in vivo. J. Lipid Res. 50, 894–907 [DOI] [PMC free article] [PubMed] [Google Scholar]