

Background: Osteosarcoma (OS) is associated with loss of tumor suppressor p53 and increased Runx2.
Results: Runx2 and p53 levels are inversely correlated in OS. miR-34c, which targets Runx2, is absent in OS and elevated by p53.
Conclusion: p53, miR-34c, and Runx2 form a regulatory loop that is compromised in OS.
Significance: RUNX2 could be targeted by miR-34c to prevent OS growth.
Keywords: MicroRNA, Osteosarcoma, p53, Transcription Factors, Tumor Suppressor Gene
Abstract
Osteosarcoma (OS) is a primary bone tumor that is most prevalent during adolescence. RUNX2, which stimulates differentiation and suppresses proliferation of osteoblasts, is deregulated in OS. Here, we define pathological roles of RUNX2 in the etiology of OS and mechanisms by which RUNX2 expression is stimulated. RUNX2 is often highly expressed in human OS biopsies and cell lines. Small interference RNA-mediated depletion of RUNX2 inhibits growth of U2OS OS cells. RUNX2 levels are inversely linked to loss of p53 (which predisposes to OS) in distinct OS cell lines and osteoblasts. RUNX2 protein levels decrease upon stabilization of p53 with the MDM2 inhibitor Nutlin-3. Elevated RUNX2 protein expression is post-transcriptionally regulated and directly linked to diminished expression of several validated RUNX2 targeting microRNAs in human OS cells compared with mesenchymal progenitor cells. The p53-dependent miR-34c is the most significantly down-regulated RUNX2 targeting microRNAs in OS. Exogenous supplementation of miR-34c markedly decreases RUNX2 protein levels, whereas 3′-UTR reporter assays establish RUNX2 as a direct target of miR-34c in OS cells. Importantly, Nutlin-3-mediated stabilization of p53 increases expression of miR-34c and decreases RUNX2. Thus, a novel p53-miR-34c-RUNX2 network controls cell growth of osseous cells and is compromised in OS.
Introduction
Osteosarcoma (OS)5 is linked to the deregulation of osteoblast growth and differentiation. Development of OS is most prominent during adolescent bone growth and is frequently characterized by morphologically abnormal osteoblasts that directly produce defective immature bone (1). A large number of transcription factors have been shown to control osteogenic lineage progression, including RUNX2, Osx/Sp7, Dlx5, Dlx3, and Atf4 (2, 3). Consequently, deregulation of transcription factor-mediated mechanisms that promote osteoblast differentiation from mesenchymal progenitors may be critical for the onset or progression of OS formation. Furthermore, genetic alterations, including mutations in tumor suppressor and oncogenes, chromosomal amplifications, deletions, and rearrangements are associated with OS (1, 4). Inactivation of the ubiquitous tumor suppressor proteins p53 and pRB is evident in ∼50–70% of OS (5, 6). Patients with Li-Fraumeni syndrome, which is associated with germ line p53 mutations, have a higher incidence of sarcomas including osteosarcoma (5, 6). In addition, bone-specific loss of the tumor suppressors p53 and pRB in mice increases the incidence of OS (7, 8), although loss of p53 alone suffices (9). One important question is how the tumor suppressor p53 compromises the activities of osteoblast lineage- specific transcription factors to promote bone tumors.
The normal biological functions of the osteogenic master regulator RUNX2 are to promote cellular quiescence and to sustain osteogenic lineage commitment (10). To support these functions, RUNX2 is expressed at low levels during active proliferation of non-tumorigenic immortalized human and mouse calvarial osteoblasts (e.g. human fetal osteoblasts and MC3T3-E1) (11, 12). Osteoprogenitor cells with a Runx2 null mutation exhibit increased cell growth (13). Forced expression of Runx2 inhibits proliferation in several osteoblastic cell lines (e.g. MC3T3-E1, C2C12, Runx2 null osteoprogenitor cells) (11, 14). These results together clearly indicate that RUNX2 functions as a suppressor of cell proliferation in non-tumorigenic osteogenic cells. Therefore, it is necessary to resolve the apparent contradiction in the molecular etiology of bone-related cancers that the levels of RUNX2 are enhanced in a subset of OS (4, 12, 15–17). Understanding the molecular basis for this RUNX2 paradox will not only provide insight into the pathophysiology of osteosarcomas but also that of non-osseous cancer cell types in which RUNX2 is ectopically expressed (18).
Normal RUNX2 functions in bone are linked to the MDM2-p53 pathway, and RUNX2 controls expression of the p53-responsive p21 gene (9, 19, 20). Furthermore, bone-specific knock-out of p53 is dominant over loss of pRB in the predisposition to OS in mouse models (7, 8). RUNX2-dependent osteoblastic differentiation is compromised when the p53-MDM2 pathway is genetically perturbed, and genetic loss of p53 increases the differentiation-related accumulation of RUNX2 in mouse calvarial osteoblasts (9). Hence, it is critical to examine how changes in the activities of p53 and RUNX2 are interrelated. In this study, we show that p53 is an upstream post-transcriptional regulator of RUNX2 that attenuates RUNX2 levels through activation of miR-34c. The results show that loss of p53 function relieves post- transcriptional repression of RUNX2 while altering RUNX2-dependent control of osteoblast growth.
EXPERIMENTAL PROCEDURES
Tissue Analysis
Primary tissue biopsies derived from osteosarcoma patients were obtained from the archives of the National University Hospital, Singapore, the University Hospital Hamburg-Eppendorf, Hamburg, Germany, and the Medical Care Unit for Histology, Cytology, and Molecular Diagnostics, Trier, Germany following strict institutional ethical guidelines and approvals. Tissue samples were fixed, dehydrated, and embedded in paraffin. Several consecutive 4-μm sections were cut and analyzed immunohistochemically with antibodies for RUNX2 (mouse monoclonal) and Ki-67 (mouse monoclonal, Dako) according to established and previously published protocols (21–23). Adequate positive and negative controls were performed.
Cell Culture
SAOS-2 and U2OS osteosarcoma cells as well as NARF U2OS cells were cultured in McCoy's medium (Invitrogen) supplemented with 15 and 10% FBS (Atlanta), respectively, 2 mm l-glutamine (Invitrogen), penicillin/streptomycin (Invitrogen). Human fetal osteoblasts were cultured in DMEM/F-12 without phenol red (Invitrogen), 10% FBS (HyClone), penicillin/streptomycin, and human mesenchymal stem cells in α-MEM (Invitrogen) + 10% FBS and 1% penicillin/streptomycin. Mouse calvarial osteoblasts were isolated from wild-type (wt) and p53−/− mice and cultured as previously described (9).
Transfections
Cells were transfected at 30–40% confluence in 6-well plates with oligonucleotides using OligofectamineTM reagent (Invitrogen) at a final concentration of 50 nm in 1 ml of Opti-MEM (Invitrogen) according to the manufacturer's instructions. Two different small interfering RNAs (siRNAs) were used to transiently silence RUNX2, indicated as siRX2-a (ON-TARGET plus SMARTpool siRUNX2 L-012665-00 (Dharmacon)) and siRX2-b (target sequence, AAGGTTTCAACGATCTGAGATT, purchased from Qiagen). siRNAs against p53 (ON-TARGET plus SMARTpool siTP53, L-003329-00) and p21 (ON-TARGET plus SMARTpool siCDKN1A L-000389-00) were purchased from Dharmacon. Non-silencing (NS) oligos (target sequence 5′-AAT TCT CCG AAC GTG TCA CGT-3′) or Dharmacon ON-TARGET plus siControl non-targeting pool D-001810-10 were used as negative controls. SAOS-2 cells were transfected with HA-p53 plasmid as previously described (24). Cells were harvested after 48 h (unless otherwise indicated) for Western blot or gene expression analyses.
For miRNA studies, miR-34c precursors, inhibitors, Universal Negative Control #1 precursor (miR-C and antimiR-C) were purchased from Ambion and transfected with Oligofectamine at a final concentration of 50 nm according to the manufacturer's instructions. Cells were harvested after 48 h and subjected to Western blot or quantitative real time PCR (qRT-PCR) analysis.
Lentiviral Constructs and Infections
To generate cells expressing short hairpin RNA, (shRNA)-RUNX2–5′-AAGGTTCAACGATCTGATTTG-3′ sequence was cloned in the pLVTHM vector under H1 promoter, and lentiviral particles were generated using Invitrogen BLOCK-iT kit. Generation of an Runx2 point mutation (Y433A) and deletion mutant Δ361 have been described previously (25, 26). Cells were infected with lentivirus and selected by fluorescence-activated cell sorting for GFP signal to obtain shRNA-expressing cell population. RUNX2 knockdown efficiency was confirmed by Western blot analysis.
RUNX2 Promoter and 3′-UTR Analyses
For RUNX2 promoter analysis, U2OS cells were transfected with 500 ng of empty pGL3 plasmid or RUNX2 promoter constructs (27) in Opti-MEM using FuGENE transfection reagent (Roche Diagnostics) according to the manufacturer's protocol. For 3′-UTR assays, full-length RUNX2 3′-UTR and RUNX2 3′-UTR with mutated miR-34c binding sites were cloned into a pMIR-Reporter Luciferase Plasmid (Applied BioSystems). U2OS cells were transfected with Universal Control #1 (miR-C) or miR-34c oligonucleotides (Ambion) at a final concentration of 50 nm using Oligofectamine as described above. The following day cells were transfected with 500 ng of 3′-UTR plasmids along with Renilla luciferase plasmid (Promega) using Xtremegene9 (Roche Diagnostics) following the manufacturer's instructions. After 48 h, cells were harvested in Passive Lysis Buffer (Promega), and a Dual Luciferase Reporter assay was performed according to manufacturer's instructions (Promega). Values obtained from firefly luciferase signals were normalized to Renilla luciferase activity.
γ-Irradiation
U2OS cells were seeded in 6-well plates at a density of 80,000 per well. Cell were transfected with siRNA oligonucleotides for 48 h as described above, and DNA damage was achieved with γ-irradiation (2 gray; 137Cs). Cells were allowed to recover for 6 h and harvested for molecular analyses.
Nutlin Treatment
U2OS and SAOS-2 cells were plated in 6-well plates. Cells were treated with Nutlin-3 (Sigma) at various concentrations for the indicated periods of time and subjected to RNA extraction or Western blot analyses.
Cell Cycle Analysis
The distribution of cells at specific cell cycle stages was evaluated by assessment of BrdU incorporation by flow cytometry (FACS) using an APC BrdU kit (BD Biosciences) according to the manufacturer's protocol. Before analysis, cells were transfected with NS oligo or si-p21 and treated with Nutlin-3 as described above. For cell cycle analysis, cells were incubated with 10 μm BrdU for 1 h, trypsinized, washed with phosphate-buffered saline, and fixed in cytofix/permeabilization solution (APC BrdU kit). Cells were then treated with DNase I (Roche Diagnostics) for 1 h at 37 °C, stained with anti-BrdU-FITC for 20 min at room temperature, and subjected to FACS analysis. Samples (1 × 106 cells) were analyzed using the FACStar cell sorter and Consort 30 software (BD Biosciences).
RNA Extraction and Gene Expression Analysis
Total RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. DNA was digested. and RNA was purified with the Zymo RNA purification kit (Zymo). cDNA was synthesized from 1 μg of total RNA using Supercript RT kit according to protocol provided by manufacturer (Invitrogen). qRT-PCR was performed with 7300 sequence detection system (Applied Biosystems) using SYBR® Green Master Mix (Applied Biosystems) (the primers are shown in supplemental Table 1) After normalization to GAPDH mRNA, relative expression levels and -fold induction of each target gene were calculated using a comparative CT method ((1/2TΔCT formula, where ΔCT is the difference between CT target and CT reference) with Microsoft Excel 2007®.
RNA Extraction and miRNA Expression Analysis
Total RNA was isolated using TRIzol reagent (Invitrogen). DNA was digested and RNA-purified with Zymo RNA purification kit (Zymo). RNA extraction from paraffin-embedded tissue samples was performed using High Pure FFPE RNA Micro-kit (Roche Diagnostics) according to the manufacturer's instructions. Small RNAs were poly(A)-tailed, and cDNA was synthesized according to the QuantimiR-protocol (SBI System Biosciences). Relative miRNA expression was determined with SYBR Green detection (Applied Biosystems) using Universal reverse primer and a forward primer designed for miRNAs of interest (supplemental Table 1) using a 7300 sequence detection system (Applied Biosystems/Roche Diagnostics). U6 expression was used as the internal control. The relative miRNA expression was calculated with the CT method.
Western Blot Analysis
Osteosarcoma tumor tissues were obtained from patients through an Institutional Review Board-approved protocol at the Mayo Clinic. The histology was reviewed by fellowship-trained musculoskeletal pathologists to confirm proper diagnosis. The tissues were homogenized in lysis buffer, and the protein concentration was determined. Cytoplasmic extracts containing protein (60 μg of protein) were analyzed by Western blot using anti-Runx2 (MBL International), anti-p21, anti-p53, and anti- (GAPDH) (Santa Cruz Biotechnology) antibodies.
Cells were lysed in radioimmune precipitation assay buffer (Boston Bioproducts) and 2×SDS sample buffer supplemented with protease inhibitors (Complete, EDTA-free, Roche Diagnostics) and MG132 (Calbiochem). Lysates were fractionated in a 10% acrylamide gel and subjected to immunoblotting (Bio-Rad system). Immunoblots were incubated for 1 h at room temperature or overnight at +4 ºC with the following primary antibodies: anti-RUNX2 (mouse monoclonal; MBL), anti-p53 (mouse monoclonal; Cell Signaling), anti-MDM2 (mouse monoclonal; Santa Cruz Biotechnology), anti-p14/p19ARF (rabbit polyclonal; Santa Cruz Biotechnology), anti-p21 (rabbit polyclonal; Santa Cruz Biotechnology). Anti-tubulin (mouse monoclonal; Sigma), anti-actin (goat polyclonal, Santa Cruz Biotechnology), and anti-CDK2 (rabbit polyclonal clone M2; Santa Cruz) were used as loading controls. Peroxidase-labeled secondary antibodies (Santa Cruz) were used to visualize bands with enhanced chemiluminescence (ECL) kit (PerkinElmer Life Sciences) on BioMax film (Eastman Kodak Co.).
Proliferation Assays
Cells were seeded in 6-well plates at a density of 1000 cells/cm2. Cells were counted every day using a hemocytometer. For MTS assay, cells were seeded in 96-well plates, 3000 cells/well. MTS assay (Promega) was performed according to manufacturer's instructions.
Statistical Analysis
Comparisons between groups were analyzed using analysis of variance and two-tailed unpaired Student's t test.
RESULTS
Active Expression of RUNX2 and Deregulation of Its Growth Regulatory Function in Proliferating Osteosarcoma Cells
RUNX2 suppresses growth of non-tumorigenic osteogenic mesenchymal cells, and its expression is inversely correlated with cell proliferation (1, 11, 28). Yet RUNX2 levels are frequently elevated in many but not all established OS cell lines in culture (16, 17, 29). Previous studies have shown that RUNX2 expression is selectively increased in subset of osteosarcomas and particularly in chemo-resistant tumors (1, 4). To illustrate and extend this finding, we analyzed primary tissue biopsies and metastases from OS patients (n = 21 primary tumors, n = 2 metastases) derived from Singaporean and German patient populations (Fig. 1A). Biopsies were semi-quantitatively scored for in situ expression levels of RUNX2 and the proliferation marker Ki-67 (Fig. 1B). Six samples did not show significant staining for either marker, which can be explained by necrotic morphology of the tumor tissue. Four samples showed that RUNX2 and Ki-67 levels are inversely correlated, indicating that in these samples RUNX2 levels are restricted to quiescent cells as is also observed in normal osteoblasts. However, both proteins are co-expressed in the remaining 13 biopsies. The latter observations represent 57% of these samples and corroborate previous findings with a larger patient cohort showing that RUNX2 is overexpressed in osteosarcomas that respond poorly to chemotherapy (4, 30) as well as our initial findings with established OS cell lines (supplemental Fig. 1, A and B) (16, 17, 29). Together with other results reviewed elsewhere (1, 4), it is apparent that RUNX2 and cell proliferation are not inversely coupled in a subset of osteosarcoma cells.
FIGURE 1.
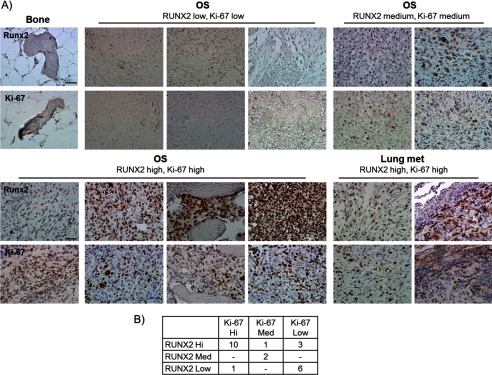
RUNX2 positively correlates with proliferation in osteosarcoma. A, tissue biopsies obtained from normal bone and primary osteosarcoma were stained for RUNX2 and proliferation marker Ki-67. B, samples were scored for staining intensities and grouped in categories with no or very low staining, medium staining, and high staining. High or medium expression of RUNX2 correlates with expression of Ki-67 in 13 of 23 samples, suggesting a loss of the reciprocal relationship between RUNX2 and cell proliferation. Scale bar, 50 μm.
Non-tumorigenic proliferating osteoblastic cells express low levels of RUNX2, which regulates a large number of target genes involved in cell proliferation and/or survival (14). Forced expression of functionally active RUNX2 proteins (i.e. WT and Y433A) but not a functionally defective RUNX2 mutant (Δ361) or GFP control protein inhibited proliferation of immortalized human O4T8 osteoblasts (Fig. 2A). These results corroborate observations in rodent mesenchymal cell types (e.g. MC3T3-E1, C2C12, and RUNX2 null osteoprogenitor cells) (11, 14). We next investigated whether RUNX2 modulates proliferation in U2OS and SAOS-2 cells. Because RUNX2 levels are elevated in OS cells, each cell type was treated with two distinct RUNX2 siRNAs, and effective knockdown of RUNX2 was confirmed by Western blot analysis (Fig. 2, B and C). Growth curves reveal that knockdown of RUNX2 using either one of the two siRNAs in U2OS cells resulted in a clear cell growth inhibition (Fig. 2B), whereas RUNX2 knockdown in SAOS-2 cells did not affect proliferation (Fig. 2C). Thus, the cell growth regulatory function of RUNX2 in OS cells was neutralized (SAOS-2) or pro-proliferative (U2OS) and in either case appeared to be fundamentally distinct from its growth-suppressive role in non- tumorigenic osteoblasts.
FIGURE 2.

RUNX2 supports cell growth in OS cell lines. A, forced expression of wild-type RUNX2 (WT) in immortalized human O48T osteoblasts inhibits cell proliferation. Similar suppression was obtained with a RUNX2 subtle point mutation (Y433A) that abrogates c-Src/Yes signaling but does not affect cell growth inhibition (30). In contrast, a truncated Runx2 protein (Δ361) that resulted from a nonsense point mutation was not able to inhibit cell growth in osteoblasts. B, growth curve analysis reveals that depletion of RUNX2 using two distinct siRNAs (siRX2-a and siRX2-b) suppresses cell growth in U2OS osteosarcoma cells. C, RUNX2 knockdown in SAOS-2 cells has no effect on cell proliferation. D, inhibition of RUNX2 with siRX2-a and siRX2-b results in activation of growth suppressive p21 pathway in U2OS cells as indicated by Western blot analysis. Effective knockdown of RUNX2 with both siRNAs does clearly increased the levels of p53 and p21. n = 3 biological replicates in 2–3 independent experiments. *, p < 0.05. #, non-specific signal.
U2OS cells express wt p53 and its classical target gene p21, whereas SAOS-2 cells have a homozygous deletion of p53 gene and are thus p53-null. We, therefore, examined whether the growth regulatory function of RUNX2 is coupled to changes in the p53-p21 pathway. Indeed, mechanistic linkage between RUNX2 and p53 was indicated by a modest increase in p53 and a more striking increase in p21 protein levels upon RUNX2 depletion in U2OS cells using two different siRNAs for Runx2 that exclude siRNA off-target effects (Fig. 2D). Taken together our data suggest that elevated levels of RUNX2 in U2OS cells, but not in SAOS-2 cells, may promote cell proliferation by attenuating the growth suppressive p53/p21 pathway.
Loss of p53 Increases RUNX2 Protein Levels
Previous studies have shown that RUNX2 is expressed at elevated levels during ex vivo differentiation of osteogenic calvarial cells isolated from conditional p53 null mutant mice in which the p53 gene was ablated by Cre recombinase-mediated excision in the osteoblast lineage (9). Therefore, we addressed whether the genetic status of p53 may influence RUNX2 gene expression in proliferating cultures. Analysis of primary calvarial osteoblasts isolated from p53 null mice show that they do not express p53, have levels of p21 below the level of detection, and yet express increased levels of the p14/p19ARF protein (Fig. 3A), which is suppressed by p53 and inhibits the p53 ubiquitin-conjugating enzyme MDM2. Strikingly, proliferating p53 null neonatal calvarial cells (Fig. 3A) and p53 null embryonic fibroblasts (data not shown) prominently express increased Runx2 protein levels. These findings establish that RUNX2 levels inversely correlate with p53 mutations in proliferating mesenchymal cells. Inactivation of the p53/p21 axis in these calvarial cells increased cell proliferation (Fig. 3B), suggesting that loss of p53 and p21 bypasses the growth suppressive potential of Runx2. In addition, Runx2 protein levels were also increased in wt calvarial osteoblasts in which p53 was depleted by RNA interference using siRNA or shRNA (Fig. 3, C and D). Thus, RUNX2 levels respond dynamically to loss of p53 function in normal murine cells rather than as a long term developmental consequence. Importantly, we observed a markedly elevated RUNX2 expression in p53-deficient SAOS-2 cells compared with U2OS cells (supplemental Fig. 1A). We also establish that knockdown of p53 increased RUNX2 levels in U2OS cells (Fig. 3E), revealing that p53 levels are rate-limiting for RUNX2 in human OS cells. Finally, we analyzed RUNX2, p53, and p21 protein expression in tumor tissues obtained from OS patients. Corroborating our in vitro findings, we find an inverse relationship between RUNX2 and p53 in human tissue samples (Fig. 3F), indicating that RUNX2 is elevated in clinical osteosarcoma upon decrease or loss of p53.
FIGURE 3.

Genetic and acute loss of p53 increases RUNX2 levels. A, primary calvarial osteoblasts were isolated from wild type (+/+), p53 null (−/−) and p53 heterozygous (+/−) mice. Markedly elevated Runx2 levels were observed in proliferating calvarial osteoblast isolated from the p53-deficient mice compared with the wt mice. Similarly, p14/p19ARF, which is normally suppressed by p53, was increased in the absence of p53. B, inactivation of p53 increased calvarial cell proliferation as demonstrated by growth curve analysis. C, acute depletion of p53 in wt calvarial cells induced by p53 siRNA (si-p53) increased Runx2 levels. D, similarly, shRNA-mediated loss of p53 in calvarial osteoblasts results in the elevation of Runx2. E, knock-down of p53 using si-p53 at various doses (10–100 nm) in U2OS cells was accompanied with dramatic down-regulation of p21 and increase of RUNX2. F, low p53 and p21 protein expression in OS samples was associated with high expression of RUNX2. *, p < 0.05.
γ-Irradiation or p14/p19ARF Induction Decreases RUNX2 Expression in a p53-dependent Manner
An intricate p53-related feedback mechanism is initiated upon DNA damage and results in a p21-mediated cell cycle block. To resolve a p53-dependent cell cycle arrest, the ubiquitin-conjugating enzyme MDM2 was activated by p53, whereas the MDM2 inhibitor p14/p19ARF was suppressed by p53 to ensure effective degradation of p53 (Fig. 4A). We, therefore, investigated whether RUNX2 protein levels are modulated by activation of p53 in response to DNA damage (Fig. 4B) or by imposing a p53 cell cycle block in response to induction of p14/p19ARF (Fig. 4, C and D). Irradiation of U2OS cells induced p53 and p21 protein accumulation as part of the DNA damage response but decreased RUNX2 levels (Fig. 4B). This reduction in RUNX2 protein levels is related to p53 expression because knockdown of p53 with siRNA prevented RUNX2 down-regulation 24 h after irradiation (Fig. 4B).
FIGURE 4.

DNA damage and p14/p19ARF induction decrease RUNX2 in a p53 dependent manner. A, DNA damage induces p53. Accumulation of p53 results in suppression of p14/p19ARF and increased MDM2, which is an inhibitor of p53. Furthermore, p53 increases p21 directly and indirectly through decreasing the levels of RUNX2, which functions as a repressor of p21. Accumulation of p21 suppresses Cyclin E/CDK2 and results in cell cycle arrest. B, γ-irradiation of U2OS induces accumulation of p53 and its target protein p21 as part of the DNA damage response as assessed by Western blot. In contrast, RUNX2 protein levels are markedly reduced as a consequence of irradiation. As shown by Western blot, the irradiation-induced down-regulation of RUNX2 is prevented by siRNA-mediated inhibition of p53, indicating that the reduction of RUNX2 is directly dependent of p53. C, the potential role of p14/p19ARF-induced p53 cell cycle block in RUNX2 levels was investigated by using a U2OS-derived NARF6 cell line that contains an IPTG-inducible p14/p19ARF gene. Induction of p14/p19ARF mRNA was analyzed by qRT-PCR after treating the cells with 1 mm IPTG for 6 and 48 h. D, induction of p14/p19ARF by 48 h IPTG treatment leads to linear activation of the MDM2-p53-p21 signaling pathway as assessed by Western blot. Simultaneously, RUNX2 protein levels are reduced. E, ectopic expression of p53 in SAOS-2 cells suppresses RUNX2. C, control. *, p < 0.05. # denotes a nonspecific signal.
We then used a U2OS-derived cell line (NARF6) that contains an IPTG-inducible p14/p19ARF gene and permits the evaluation of p53-dependent cell growth control in U2OS cells (31). IPTG induced p14/p19ARF mRNA levels (Fig. 4C), consistent with the linear activation of the p14/p19ARF-MDM2-p53-p21 signaling axis (Fig. 4A). The resulting elevation of the CDK inhibitor p21 resulted in a cell cycle block in the G1 phase as determined by FACS analysis (supplemental Fig. 2). Importantly, although the protein levels of the downstream p53 targets MDM2 and p21 were induced, RUNX2 levels decreased after 48 h of IPTG treatment (Fig. 4D). Hence, induction of endogenous p53 protein by γ-irradiation or p14/p19ARF stimulation decreased RUNX2 protein levels (Fig. 4), in contrast to the increased RUNX2 level that was apparent upon p53 depletion (Fig. 3). Because p53 is absent is SAOS-2 cells and cannot be induced by chemical inhibitors of MDM2 (Fig. 5A, supplemental Fig. 3), we ectopically expressed p53 in SAOS-2 (Fig. 4E). Reconstitution of p53 resulted in decreased RUNX2 expression in SAOS-2 cells, suggesting that RUNX2 is downstream of p53.
FIGURE 5.

Stabilization of p53 reduces RUNX2 protein and mRNA levels. A, stabilization of p53 by MDM inhibitor Nutlin-3 (1.25, 2.5, 5, and 10 μm) in U2OS cells for 48 h dose dependently increases protein levels of p53 and p21 but results in marked decrease in RUNX2 levels. In SAOS-2 cells, which are p53-negative, p21 is not detected, and Nutlin-3 treatment has no effect on RUNX2 levels at any concentration. B, the decreased RUNX2 expression upon p53 induction could result from increased RUNX2 protein degradation and/or decreased mRNA levels. To analyze the effect of p53 on mRNA levels, U2OS cells were treated with Nutlin-3 for 48 h, and p21 and RUNX2 mRNA levels were measured with RT-quantitative PCR. Stabilization of p53 by Nutlin-3 results in a dose-dependent increase in p21 mRNA levels. Low doses (1.25 and 2.5 μm) of Nutlin-3 do not drastically change RUNX2 mRNA levels. Administration of 5 or 10 μm Nutlin-3 decreases RUNX2 mRNA levels by 2-fold. C, to address whether induction of p53 alters RUNX2 gene promoter activity, U2OS cells were transfected with RUNX2 promoter-luciferase constructs for 18 h and were subsequently treated with Nutlin-3 for 32 h. Nutlin-3 treatment decreases RUNX2 transcription as measured by luciferase activity by ∼30%. This reduction in promoter activity was observed for both the full-length 0.6-kb promoter and a promoter deletion mutant. *, p < 0.05. # denotes a nonspecific signal.
Stabilization of p53 Decreases RUNX2 Gene Expression
We next established that p53 acts directly upstream of RUNX2 using the MDM2 inhibitor Nutlin-3, which blocks MDM2-dependent destabilization of p53 (32). Administration of Nutlin-3 to p53-positive U2OS cells increased protein expression of p53 and its target p21 but drastically decreased RUNX2 protein levels (Fig. 5A). The modulation in RUNX2 levels was readily apparent after 48 h of Nutlin-3 exposure (Fig. 5A) but not at 1, 2, 4, 8, or 24 h of treatment (supplemental Figs. 3 and 4). Yet protein levels of p53, p21, and Mdm2 clearly increased after both short and long treatment times in U2OS cells (Fig. 4A and supplemental Figs. 3 and 4). For comparison, Nutlin-3 treatment in p53-negative SAOS-2 cells did not appreciably modulate RUNX2 protein levels at any time point (Fig. 5A and supplemental Figs. 3 and 4). Because the effects of Nutlin-3 on RUNX2 protein levels (>24 h) were considerably delayed relative to p53, p21, and MDM2 activation (<8 h), accumulation of RUNX2 mRNA and/or protein may be indirect and perhaps proceed via a p53-dependent proxy factor (e.g. protein or small regulatory RNA).
We addressed whether changes in p53 activity alter RUNX2 mRNA levels by RT-quantitative PCR. Nutlin-3 promptly induced p21 mRNA levels by several-fold (>4) (Fig. 5B and supplemental Fig. 5). RUNX2 mRNA expression was decreased by about 2-fold after 48 h of Nutlin-3 treatment but not at earlier time points (Fig. 5B and supplemental Fig. 5). Reporter gene transcription assays were performed with vectors in which the luciferase (Luc) gene was fused to different segments (up to −391 or −160 bp) of the human RUNX2 promoter, which lacks known p53 binding sites. Nutlin-3 decreased RUNX2 transcription by 30% or less for either the −391/Luc or −160/Luc construct (Fig. 5C). Thus, the modest decreases in RUNX2 mRNA levels and in RUNX2 promoter activity (<30%) did not fully explain the strong reduction of RUNX2 protein expression after 48 h of Nutlin-3 exposure. The combined results suggest that the Nutlin-3-dependent decline in RUNX2 protein level is controlled by both transcriptional and post- transcriptional mechanisms.
RUNX2 Down-regulation upon p53 Induction Is Independent of p21-induced Cell Cycle Inhibition
Because Nutlin-3-dependent stabilization of p53 induces p21 and thus inhibits cell cycle progression, we assessed whether RUNX2 down-regulation after Nutlin-3 treatment may be related to p21-induced cell cycle arrest. We treated U2OS cells with p21 siRNA for 24 h before Nutlin-3 exposure. Treatment with p21 siRNA permitted cell growth of U2OS cells in the presence of three different doses of Nutlin-3 that decreased cell proliferation when administered to cells pretreated with non-silencing RNA (Fig. 6A). The fraction of U2OS cells that are in S phase was low after 48 h of Nutlin-3 treatment based on flow cytometry but siRNA-mediated knockdown of p21 prevents Nutlin-3-induced cell cycle inhibition (Fig. 6B). Importantly, regardless of p21-dependent effects on cell cycle progression, RUNX2 protein expression still decreased upon Nutlin-3-induced p53 accumulation (Fig. 6C). These data suggest that p53-induced RUNX2 reduction is independent of cell cycle inhibition.
FIGURE 6.
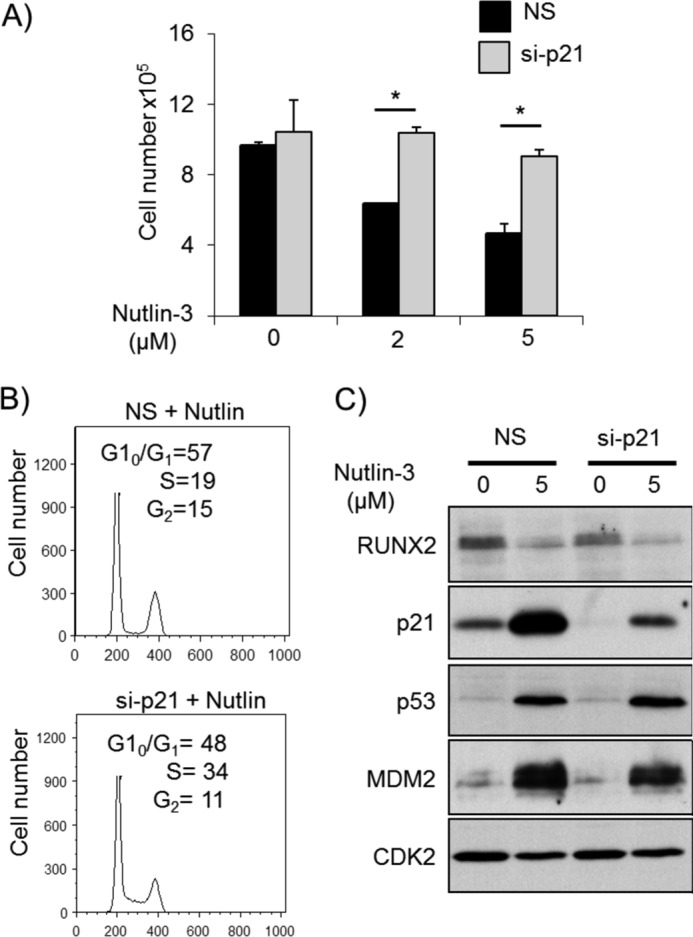
p53-induced down-regulation of RUNX2 is independent of cell cycle inhibition. A, to address whether Nutlin-3-induced RUNX2 down-regulation is a consequence of a p53-p21 pathway-induced cell cycle block, U2OS cells were transfected with non-silencing siRNA (NS) or siRNA against p21 (si-p21) for 24 h before Nutlin-3 treatment. Nutlin-3 treatment results in dose-dependent reduction in cell growth in non-silencing siRNA-treated cells. Silencing p21 by siRNA rescues Nutlin-3-induced growth defect. B, to investigate the cell cycle progression, flow cytometry analysis was performed after 48 h of Nutlin-3 treatment. Knockdown of p21 rescues the fraction of cells in S-phase compared with non-silencing siRNA treated cells (34 and 19%, respectively). C, Western blot analysis reveals a marked increase in p53, p21, and MDM2 levels and down-regulation of RUNX2 in non-silencing siRNA-transfected cells as response to Nutlin-3. Although si-p21 effectively reduces p21 protein levels, Nutlin-3 treatment results in drastic up-regulation of p53 and down-regulation of RUNX2. *, p < 0.05.
miR-34c Provides a Mechanistic Link between p53 and RUNX2
To investigate the potential contribution of microRNAs (33) to the regulation of RUNX2, we analyzed the expression of RUNX2-targeting miRNAs (34) in a panel of human cell types, including mesenchymal stromal cells, human fetal osteoblasts, and SAOS-2 and U2OS OS cell lines. Several RUNX2-targeting miRNAs were expressed at lower levels in SAOS-2 and U2OS cells compared with human mesenchymal stem cells and human fetal osteoblasts (Fig. 7A and supplemental Fig. 6). For example, miR-34c was prominently expressed in non-osteosarcoma cells but barely detected in SAOS-2 and U2OS cells (Fig. 7A). These results may reflect previous findings that miR-34 family members function as tumor suppressors in various cancers and promote cellular senescence (35, 36).
FIGURE 7.

miR-34c links p53 to RUNX2. A, to investigate whether miRNAs are involved in the regulation of RUNX2, the expression of miRNAs that target RUNX2 was measured in several human cell lines by qRT-PCR. Several miRNAs are expressed at markedly reduced levels in U2OS and SAOS-2 osteosarcoma cells compared with human mesenchymal stromal cells (hMSC) and human fetal osteoblasts (hFOB) including miR-23a, miR-30c, and miR-34c. Most strikingly, miR-34c was barely detected in osteosarcoma cell lines. B, although having no effect on miR-23a and miR-30c expression, Nutlin-3 treatment induced a 2-fold increase in the expression of miR-34c after 24 h. A more prominent dose-dependent induction was observed after 48 h of Nutlin-3 treatment. C, the direct binding of miR-34c to the RUNX2 3′-UTR was confirmed by 3′-UTR reporter assays. The entire 4-kb RUNX2 3′-UTR (wt) or RUNX2 3′-UTR in which miR-34c is mutated (mut) were cloned into the 3′-UTR of the luciferase gene in a pMIR vector. Whereas forced expression of miR-34c significantly reduced luc activity in the wt full-length RUNX2 3′ construct, it had no effect on the mutated construct, confirming that miR-34c directly targets RUNX2 in osteosarcoma cells. D, to determine whether miR-34c modulates RUNX2 protein expression, U2OS cells were transfected with 50 nm miR-34c precursor or Universal Control miRNA (miR-C). Ectopic miR-34c reduces RUNX2 protein levels after 48 h. E, U2OS cells were treated with Nutlin to induce miR-34c (Fig. 7B). Subsequent inhibition of miR-34c (antimiR-34c) partially rescued the Nutlin-induced reduction in RUNX2 expression. F, transfection of U2OS cells with miR-34c significantly reduced cell growth as measured by MTS assay. Uni-C, universal control. *, p < 0.05.
To assess whether the reciprocal relationship between RUNX2 and p53 is controlled by miRNAs, we treated U2OS cells with Nutlin-3 and measured miRNA levels in relation to RUNX2. Among the RUNX2 targeting miRNAs Nutlin-3 treatment significantly increased miR-34c expression while decreasing the levels of RUNX2 (Figs. 7B and 5A). Furthermore, ectopic expression of miR-34c in U2OS cells markedly reduced RUNX2 protein levels and suppressed luciferase activity in Luc- RUNX2–3′-UTR assays while there was no decrease in RUNX2 mRNA levels (Fig. 7, C and D, supplemental Fig. 6, B and C). Mutation of the putative miR-34c binding site in the RUNX2 3′-UTR (supplemental Fig. 6B) rescued the miR-34c-mediated decrease in luciferase activity, suggesting that miR-34c directly binds to the RUNX2 3′-UTR (Fig. 7C). Importantly, inhibition of miR-34c in the presence of Nutlin is able to partially rescue the reduction of RUNX2 expression (Fig. 7E), indicating that miR-34 is, at least in part, an important mediator of p53-dependent suppression of RUNX2. Finally, forced expression of miR-34c reduced U2OS proliferation, resembling the condition in which RUNX2 is silenced (Figs. 7F and 2C). These results demonstrate that miR-34c is Nutlin-3-dependent and that RUNX2 is a direct target of miR-34c in OS cells.
To address whether miR-34c indeed plays a role in OS, we analyzed the expression of miR-34c in OS tissue biopsies and in healthy bone (Fig. 8A). The expression level of miR-34c was found to be greatly reduced in OS tissue compared with normal bone, strongly indicating that down-regulation of miR-34c is associated with OS.
FIGURE 8.
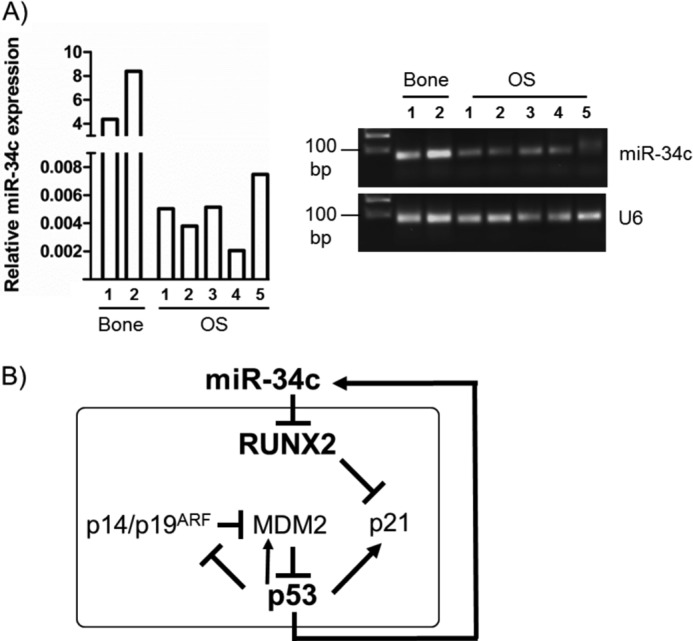
The p53/miR-34/Runx2 circuit. A, expression analysis of miR-34c in normal human bone and OS biopsies by qRT-PCR revealed significant down-regulation of miR-34c in OS compared with normal bone tissue. Values are presented as relative to U6 expression. End products of the qRT-PCR reaction were separated in 3.5% agarose gel. B, shown is a schematic of the proposed model of the p53/miR-34/Runx2 regulatory circuit.
Thus, p53 may attenuate RUNX2 levels by activating miR-34c (Fig. 8B). We, therefore, propose that loss of RUNX2 targeting miRNAs, including miR-34c, in OS contributes to the elevation of RUNX2 protein in OS.
DISCUSSION
In this study we establish that the osteogenic transcription factor RUNX2 is robustly detectable in several human OS biopsies and OS cell lines in which the p53 pathway is typically compromised. In addition, RUNX2 levels are increased in mouse calvarial osteoblasts in which p53 levels are eliminated by genetic null mutation or depleted using siRNAs. Furthermore, we show that induction of p53 levels by γ-irradiation, conditional expression of the natural MDM2 inhibitor p14/p19ARF, or treatment with the pharmacological MDM2 inhibitor Nutlin-3 all decrease RUNX2 protein levels. These data together firmly demonstrate that p53, which is causally linked to OS in Li-Fraumeni patients (6), is a critical upstream regulator of RUNX2 protein expression.
The attenuation of RUNX2 expression by p53 is not directly linked to a p53-induced cell cycle arrest because increased p53 protein levels in p21-depleted cells still reduces RUNX2. The observed reductions in RUNX2 gene expression involve modest changes in RUNX2 gene transcription and mRNA levels (<30%) as well as perhaps destabilization of RUNX2 protein by ubiquitin/proteasome-dependent mechanisms similar to RUNX3, which is controlled by MDM2-p53-mediated protein degradation (37). However, the kinetics of these molecular mechanisms are typically fast (<24 h) and do not match the delayed loss of RUNX2 that is observed between 24 and 48 h of p53 stimulation by Nutlin-3. Instead, we find that several miRNAs that target RUNX2 are selectively suppressed in OS, including the p53-responsive miRNA miR-34c (35, 36), which is up-regulated 24 h after Nutlin-3 treatment and functionally suppresses RUNX2. These results are consistent with a mechanistic model in which p53 attenuates RUNX2 protein levels by increasing the levels of miR-34c and reducing the translation and/or stability of RUNX2 mRNA.
Previous studies with several non-osseous cancer cell types have shown that miR-34c is directly controlled by p53 (35, 36). Studies from our group have shown that miR-34c targets Runx2 in mouse calvarial osteoblasts (34). The novelty of the present work is that p53, miR-34c, and RUNX2 form a direct molecular axis with delayed kinetics (e.g. relative to p53-dependent induction of p21 and MDM2) in the physiological context of human osteosarcoma cells. This finding is of importance because skeletal-related regulatory mechanisms are not always conserved between species whereas miR-34c exhibits context-dependent function on its target genes and suppresses Runx2 in a cell type-specific manner (34).
Having established that there is a p53-dependent gene regulatory mechanism that controls RUNX2 levels and because loss of p53 is a major factor in the etiology of OS, the question arises, What the pathological role of RUNX2 is in OS cells? Several studies indicated that RUNX2 may promote apoptosis in OS cell cultures (37), consistent with the well established role of RUNX2 in cell growth suppression of calvarial cells. However, a RUNX2- dependent apoptotic function would be incongruent with survival of OS that express high levels of RUNX2 in patients. Results from our proliferation assays with SAOS-2 and U2OS cell cultures indicates that the cell growth-suppressive function of RUNX2 is neutralized or converted into a pro-proliferative function in these OS cells. Furthermore, genome-wide chromatin immune-precipitation experiments using microarrays (“ChIP-on-chip” analysis) for RUNX2 in SAOS-2 cells revealed that RUNX2 controls genes involved in cell motility and adhesion (38). Thus, the loss of p53 that frequently accompanies OS formation may elevate the levels of RUNX2 and increase the metastatic potential of OS.
In vivo studies using mice have indicated that loss of p53 not only promotes OS (7–9) but also positively correlates with accelerated osteoblast differentiation, which is Runx2-dependent (9). Genetic loss of the p53 antagonist MDM2 decelerates osteoblast differentiation, indicating that p53 is not just stimulatory but also required for normal osteogenesis (9). Our present work extends these previous studies by showing that RUNX2 levels respond dynamically to loss of p53 function and there is a direct mechanistic connection between p53 and RUNX2 in which p53 may attenuate RUNX2 protein levels by promoting the expression of miR-34c that directly targets the 3′-UTR of RUNX2.
If p53 has evolved to control RUNX2 levels through miR-34c in the bone-lineage, then this axis must be tightly regulated to avoid dosage insufficiency of RUNX2 during normal bone development. Hypomorphic RUNX2 alleles typically have defects in membranous but not endochondral bone (39–43). Preliminary data indicate that RUNX2 is bound to the miR-34c locus in osteogenic mesenchymal cells,6 thus potentially generating a feedback mechanism that is similar to and associated with the p14/p19ARF-MDM2-p53-miR34c pathway.
Two types of studies have produced the opposing findings that RUNX2 gene expression is either silenced (29, 44) or amplified (4, 12, 15–17) in OS. Our present data provide a plausible mechanism for the “Runx2 paradox” in which many OS cell lines and tumor biopsies express robust and sometimes elevated levels of RUNX2 as a cell growth suppressor. When p53 is rendered ineffective by null mutation, miR-34c levels decline, and RUNX2 levels rise, which suppresses p21 and enhances cell proliferation. Conversely, activation of the p53/miR-34c axis is expected to result in miR-34c-mediated depletion of RUNX2 (34) and SATB2 (45) while promoting dedifferentiation of osteoblastic cells. Either effect on p53 could be tumorigenic.
Data from patients with Li-Fraumeni syndrome and in vivo studies with p53 null mice indicate that inactivation of p53 results in an increased incidence of OS formation (1, 7, 9, 15, 34). Constitutive p53 null mice typically die due to formation of lymphomas and fibrosarcomas, whereas mice escaping death at an early age will eventually succumb to OS at later developmental stages (15). Bone-specific loss of p53 in conditional p53 null mice causes lethality due to OS formation. It is not clear why the tumor predisposing effects of p53 null mutations on tumor formation are in part bone-specific. One possible reason is that OS cell growth depends in part on the CDK inhibitor p21, which is activated by p53 and suppressed by RUNX2. This study shows that loss of p53 increases RUNX2, lowers p21 levels and should promote CDK-dependent cell cycle progression. Complicating matters is that miR-34c also attenuates osteoblast proliferation by decreasing cyclin D1, CDK4, and CDK6 (45). Because p53 suppresses RUNX2 levels by promoting expression of the miR-34c, which directly targets RUNX2, we propose that p53 null mutation perturbs the dynamic and intricate molecular equilibrium among RUNX2, p53, miR-34c, p21, and CDKs to alter cell proliferation in a bone-specific manner.
Our study also shows that the p14/p19ARF/MDM2/p53/p21 pathway and p53/miR-34c/RUNX2 axis have different kinetics; although activation of p53 results in the rapid stimulation of p21 and cell cycle arrest, the delayed reduction of RUNX2 occurs only after the subsequent activation of miR-34c. Thus, these two pathways may each serve a distinct biological purpose in osseous cells. Loss of p53 is classically coupled to increased DNA damage and immortalization. Calvarial cells in which the RUNX2 is ablated also exhibits properties of immortalized cells, but cells accumulate damaged DNA and eventually cease growth (46). Thus, both p53 and RUNX2 may protect cells against DNA damage in the osteoblast lineage. Importantly, patient data show that amplified RUNX2 gene expression correlates with resistance to chemotherapy (4, 15). We conclude that compromising p53-mediated attenuation of RUNX2 by miR-34c may favor clinically relevant and RUNX2-dependent cellular mechanisms that promote proliferative expansion or survival of OS cells.
Acknowledgments
We thank the members of our laboratories and especially Sneha Gupta, Jitesh Pratap, Mohammed Hassan, Aveek Mazumdar, Kaleem Zaidi, Tripti Gaur, Kakoli Das, Jonathan Gordon, Shirwin Pockwinse, and Saskia Schröder for assistance with experimentation, critical comments, technical advice, and sharing of reagents and/or general support as well as Judy Rask for assistance with preparation of the manuscript. We thank Carol Prives for providing inducible U2OS cells. We also thank the past and present investigators at the National University of Singapore, including Suresh Nathan, Kakoli Das, Dominique Voon, Motomi Osato, and Victor Nurcombe, for stimulating discussions and/or sharing unpublished data. We also acknowledge the support of our colleague Barry Perreira who passed away while this work was in progress.
This work was supported, in whole or in part, by National Institutes of Health Grants AR49069 (to A. v-W.) and CA082834 (to G. S. S.). This work was also supported by grant sponsors Iniciativa Cientifico Milenio P09/016-F (to M. G.), Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) 1095234 (to M. G.), and Deutsche Forschungsgemeinschaft Grant HE 5208/2-1 (to E. H.).

This article contains supplemental Table 1 and Figs. 1–6.
M. van der Deen, H. Taipaleenmäki, Y. Zhang, N. M. Teplyuk, A. Gupta, S. Cinghu, K. Shogren, A. Maran, M. J. Yaszemski, L. Ling, S. M. Cool, D. T. Leong, C. Dierkes, J. Zustin, M. Salto-Tellez, Y. Ito, S.-C. Bae, M. Zielenska, J. A. Squire, J. B. Lian, J. L. Stein, G. P. Zambetti, S. N. Jones, M. Galindo, E. Hesse, G. S. Stein, and A. J. van Wijnen, unpublished observations.
- OS
- osteosarcoma
- NS
- non-silencing
- qRT-PCR
- quantitative real time PCR
- miRNA
- microRNA
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- CDK
- cyclin-dependent kinase
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside.
REFERENCES
- 1. Kansara M., Thomas D. M. (2007) Molecular pathogenesis of osteosarcoma. DNA Cell Biol. 26, 1–18 [DOI] [PubMed] [Google Scholar]
- 2. Jensen E. D., Gopalakrishnan R., Westendorf J. J. (2010) Regulation of gene expression in osteoblasts. Biofactors 36, 25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marie P. J. (2008) Transcription factors controlling osteoblastogenesis. Arch. Biochem. Biophys. 473, 98–105 [DOI] [PubMed] [Google Scholar]
- 4. Martin J. W., Zielenska M., Stein G. S., van Wijnen A. J., Squire J. A. (2011) The role of RUNX2 in osteosarcoma oncogenesis. Sarcoma 2011, 282745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fuchs B., Pritchard D. J. (2002) Etiology of osteosarcoma. Clin. Orthop. Relat. Res. 397, 40–52 [DOI] [PubMed] [Google Scholar]
- 6. Hansen M. F. (1991) Molecular genetic considerations in osteosarcoma. Clin. Orthop. Relat. Res. 270, 237–246 [PubMed] [Google Scholar]
- 7. Berman S. D., Calo E., Landman A. S., Danielian P. S., Miller E. S., West J. C., Fonhoue B. D., Caron A., Bronson R., Bouxsein M. L., Mukherjee S., Lees J. A. (2008) Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc. Natl. Acad. Sci. U.S.A. 105, 11851–11856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walkley C. R., Qudsi R., Sankaran V. G., Perry J. A., Gostissa M., Roth S. I., Rodda S. J., Snay E., Dunning P., Fahey F. H., Alt F. W., McMahon A. P., Orkin S. H. (2008) Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 22, 1662–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lengner C. J., Steinman H. A., Gagnon J., Smith T. W., Henderson J. E., Kream B. E., Stein G. S., Lian J. B., Jones S. N. (2006) Osteoblast differentiation and skeletal development are regulated by Mdm2-p53 signaling. J. Cell Biol. 172, 909–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lian J. B., Stein G. S., Javed A., van Wijnen A. J., Stein J. L., Montecino M., Hassan M. Q., Gaur T., Lengner C. J., Young D. W. (2006) Networks and hubs for the transcriptional control of osteoblastogenesis. Rev. Endocr. Metab. Disord. 7, 1–16 [DOI] [PubMed] [Google Scholar]
- 11. Galindo M., Pratap J., Young D. W., Hovhannisyan H., Im H. J., Choi J. Y., Lian J. B., Stein J. L., Stein G. S., van Wijnen A. J. (2005) The bone-specific expression of RUNX2 oscillates during the cell cycle to support a G1-related anti-proliferative function in osteoblasts. J. Biol. Chem. 280, 20274–20285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nathan S. S., Pereira B. P., Zhou Y. F., Gupta A., Dombrowski C., Soong R., Pho R. W., Stein G. S., Salto-Tellez M., Cool S. M., van Wijnen A. J. (2009) Elevated expression of Runx2 as a key parameter in the etiology of osteosarcoma. Mol. Biol. Rep. 36, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pratap J., Galindo M., Zaidi S. K., Vradii D., Bhat B. M., Robinson J. A., Choi J.-Y., Komori T., Stein J. L., Lian J. B., Stein G. S., van Wijnen A. J. (2003) Cell growth regulatory role of Runx2 during proliferative expansion of pre-osteoblasts. Cancer Res. 63, 5357–5362 [PubMed] [Google Scholar]
- 14. Teplyuk N. M., Galindo M., Teplyuk V. I., Pratap J., Young D. W., Lapointe D., Javed A., Stein J. L., Lian J. B., Stein G. S., van Wijnen A. J. (2008) Runx2 regulates G-protein coupled signaling pathways to control growth of osteoblast progenitors. J. Biol. Chem. 283, 27585–27597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martin J. W., Yoshimoto M., Ludkovski O., Thorner P. S., Zielenska M., Squire J. A., Nuin P. A. (2010) Analysis of segmental duplications, mouse genome synteny, and recurrent cancer-associated amplicons in human chromosome 6p21-p12. Cytogenet. Genome Res. 128, 199–213 [DOI] [PubMed] [Google Scholar]
- 16. Pereira B. P., Zhou Y., Gupta A., Leong D. T., Aung K. Z., Ling L., Pho R. W., Galindo M., Salto-Tellez M., Stein G. S., Cool S. M., van Wijnen A. J., Nathan S. S. (2009) Runx2, p53, and pRB status as diagnostic parameters for deregulation of osteoblast growth and differentiation in a new pre-chemotherapeutic osteosarcoma cell line (OS1). J. Cell Physiol. 221, 778–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. San Martin I. A., Varela N., Gaete M., Villegas K., Osorio M., Tapia J. C., Antonelli M., Mancilla E. E., Pereira B. P., Nathan S. S., Lian J. B., Stein J. L., Stein G. S., van Wijnen A. J., Galindo M. (2009) Impaired cell cycle regulation of the osteoblast-related heterodimeric transcription factor Runx2-Cbf in osteosarcoma cells. J. Cell Physiol. 221, 560–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pratap J., Lian J. B., Stein G. S. (2011) Metastatic bone disease. Role of transcription factors and future targets. Bone 48, 30–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang X., Kua H. Y., Hu Y., Guo K., Zeng Q., Wu Q., Ng H. H., Karsenty G., de Crombrugghe B., Yeh J., Li B. (2006) p53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis, and bone remodeling. J. Cell Biol. 172, 115–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Westendorf J. J., Zaidi S. K., Cascino J. E., Kahler R., van Wijnen A. J., Lian J. B., Yoshida M., Stein G. S., Li X. (2002) Runx2 (Cbfa1, AML-3) interacts with histone deacetylase 6 and represses the p21(CIP1/WAF1) promoter. Mol. Cell. Biol. 22, 7982–7992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Das K., Leong D. T., Gupta A., Shen L., Putti T., Stein G. S., van Wijnen A. J., Salto-Tellez M. (2009) Positive association between nuclear Runx2 and oestrogen-progesterone receptor gene expression characterises a biological subtype of breast cancer. Eur. J. Cancer 45, 2239–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Das K., Mohd Omar M. F., Ong C. W., Bin Abdul Rashid S., Peh B. K., Putti T. C., Tan P. H., Chia K. S., Teh M., Shah N., Soong R., Salto-Tellez M. (2008) TRARESA. A tissue microarray-based hospital system for biomarker validation and discovery. Pathology 40, 441–449 [DOI] [PubMed] [Google Scholar]
- 23. Ong C. W., Kim L. G., Kong H. H., Low L. Y., Wang T. T., Supriya S., Kathiresan M., Soong R., Salto-Tellez M. (2010) Computer-assisted pathological immunohistochemistry scoring is more time-effective than conventional scoring, but provides no analytical advantage. Histopathology 56, 523–529 [DOI] [PubMed] [Google Scholar]
- 24. Chi X. Z., Kim J., Lee Y. H., Lee J. W., Lee K. S., Wee H., Kim W. J., Park W. Y., Oh B. C., Stein G. S., Ito Y., van Wijnen A. J., Bae S. C. (2009) Runt-related transcription factor RUNX3 is a target of MDM2-mediated ubiquitination. Cancer Res. 69, 8111–8119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choi J.-Y., Pratap J., Javed A., Zaidi S. K., Xing L., Balint E., Dalamangas S., Boyce B., van Wijnen A. J., Lian J. B., Stein J. L., Jones S. N., Stein G. S. (2001) Subnuclear targeting of Runx/Cbfa/AML factors is essential for tissue-specific differentiation during embryonic development. Proc. Natl. Acad. Sci. U.S.A. 98, 8650–8655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zaidi S. K., Sullivan A. J., Medina R., Ito Y., van Wijnen A. J., Stein J. L., Lian J. B., Stein G. S. (2004) Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 23, 790–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y., Hassan M. Q., Xie R. L., Hawse J. R., Spelsberg T. C., Montecino M., Stein J. L., Lian J. B., van Wijnen A. J., Stein G. S. (2009) Co-stimulation of the bone-related Runx2 P1 promoter in mesenchymal cells by SP1 and ETS transcription factors at polymorphic purine-rich DNA sequences (Y-repeats). J. Biol. Chem. 284, 3125–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Young D. W., Hassan M. Q., Pratap J., Galindo M., Zaidi S. K., Lee S. H., Yang X., Xie R., Javed A., Underwood J. M., Furcinitti P., Imbalzano A. N., Penman S., Nickerson J. A., Montecino M. A., Lian J. B., Stein J. L., van Wijnen A. J., Stein G. S. (2007) Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2. Nature 445, 442–446 [DOI] [PubMed] [Google Scholar]
- 29. Thomas D. M., Johnson S. A., Sims N. A., Trivett M. K., Slavin J. L., Rubin B. P., Waring P., McArthur G. A., Walkley C. R., Holloway A. J., Diyagama D., Grim J. E., Clurman B. E., Bowtell D. D., Lee J. S., Gutierrez G. M., Piscopo D. M., Carty S. A., Hinds P. W. (2004) Terminal osteoblast differentiation, mediated by runx2 and p27KIP1, is disrupted in osteosarcoma. J. Cell Biol. 167, 925–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sadikovic B., Thorner P., Chilton-Macneill S., Martin J. W., Cervigne N. K., Squire J., Zielenska M. (2010) Expression analysis of genes associated with human osteosarcoma tumors shows correlation of RUNX2 overexpression with poor response to chemotherapy. BMC Cancer 10, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stott F. J., Bates S., James M. C., McConnell B. B., Starborg M., Brookes S., Palmero I., Ryan K., Hara E., Vousden K. H., Peters G. (1998) The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 17, 5001–5014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vassilev L. T., Vu B. T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., Fotouhi N., Liu E. A. (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 [DOI] [PubMed] [Google Scholar]
- 33. Jones K. B., Salah Z., Del Mare S., Galasso M., Gaudio E., Nuovo G. J., Lovat F., LeBlanc K., Palatini J., Randall R. L., Volinia S., Stein G. S., Croce C. M., Lian J. B., Aqeilan R. I. (2012) miRNA signatures associate with pathogenesis and progression of osteosarcoma. Cancer Res. 72, 1865–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y., Xie R. L., Croce C. M., Stein J. L., Lian J. B., van Wijnen A. J., Stein G. S. (2011) A program of microRNAs controls osteogenic lineage progression by targeting transcription factor Runx2. Proc. Natl. Acad. Sci. U.S.A. 108, 9863–9868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Corney D. C., Flesken-Nikitin A., Godwin A. K., Wang W., Nikitin A. Y. (2007) MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 67, 8433–8438 [DOI] [PubMed] [Google Scholar]
- 36. He L., He X., Lim L. P., de Stanchina E., Xuan Z., Liang Y., Xue W., Zender L., Magnus J., Ridzon D., Jackson A. L., Linsley P. S., Chen C., Lowe S. W., Cleary M. A., Hannon G. J. (2007) A microRNA component of the p53 tumour suppressor network. Nature 447, 1130–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eliseev R. A., Dong Y. F., Sampson E., Zuscik M. J., Schwarz E. M., O'Keefe R. J., Rosier R. N., Drissi M. H. (2008) Runx2-mediated activation of the Bax gene increases osteosarcoma cell sensitivity to apoptosis. Oncogene 27, 3605–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van der Deen M., Akech J., Lapointe D., Gupta S., Young D. W., Montecino M. A., Galindo M., Lian J. B., Stein J. L., Stein G. S., van Wijnen A. J. (2012) Genomic promoter occupancy of runt-related transcription factor RUNX2 in Osteosarcoma cells identifies genes involved in cell adhesion and motility. J. Biol. Chem. 287, 4503–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Komori T., Yagi H., Nomura S., Yamaguchi A., Sasaki K., Deguchi K., Shimizu Y., Bronson R. T., Gao Y.-H., Inada M., Sato M., Okamoto R., Kitamura Y., Yoshiki S., Kishimoto T. (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89, 755–764 [DOI] [PubMed] [Google Scholar]
- 40. Liu J. C., Lengner C. J., Gaur T., Lou Y., Hussain S., Jones M. D., Borodic B., Colby J. L., Steinman H. A., van Wijnen A. J., Stein J. L., Jones S. N., Stein G. S., Lian J. B. (2011) Runx2 protein expression utilizes the Runx2 P1 promoter to establish osteoprogenitor cell number for normal bone formation. J. Biol. Chem. 286, 30057–30070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lou Y., Javed A., Hussain S., Colby J., Frederick D., Pratap J., Xie R., Gaur T., van Wijnen A. J., Jones S. N., Stein G. S., Lian J. B., Stein J. L. (2009) A Runx2 threshold for the cleidocranial dysplasia phenotype. Hum. Mol. Genet. 18, 556–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yoshida C. A., Furuichi T., Fujita T., Fukuyama R., Kanatani N., Kobayashi S., Satake M., Takada K., Komori T. (2002) Core-binding factor β interacts with Runx2 and is required for skeletal development. Nat. Genet. 32, 633–638 [DOI] [PubMed] [Google Scholar]
- 43. Yoshida C. A., Yamamoto H., Fujita T., Furuichi T., Ito K., Inoue K., Yamana K., Zanma A., Takada K., Ito Y., Komori T. (2004) Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 18, 952–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kansara M., Tsang M., Kodjabachian L., Sims N. A., Trivett M. K., Ehrich M., Dobrovic A., Slavin J., Choong P. F., Simmons P. J., Dawid I. B., Thomas D. M. (2009) Wnt inhibitory factor 1 is epigenetically silenced in human osteosarcoma, and targeted disruption accelerates osteosarcomagenesis in mice. J. Clin. Invest. 119, 837–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wei J., Shi Y., Zheng L., Zhou B., Inose H., Wang J., Guo X. E., Grosschedl R., Karsenty G. (2012) miR-34s inhibit osteoblast proliferation and differentiation in the mouse by targeting SATB2. J. Cell Biol. 197, 509–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zaidi S. K., Pande S., Pratap J., Gaur T., Grigoriu S., Ali S. A., Stein J. L., Lian J. B., van Wijnen A. J., Stein G. S. (2007) Runx2 deficiency and defective subnuclear targeting bypass senescence to promote immortalization and tumorigenic potential. Proc. Natl. Acad. Sci. U.S.A. 104, 19861–19866 [DOI] [PMC free article] [PubMed] [Google Scholar]