

Background: Mutations in AIPL1, a chaperone of the lipidated visual effector phosphodiesterase-6, cause severe childhood blindness.
Results: AIPL1 binds the farnesyl lipid moiety. The unique insert region of AIPL1 is critical for this interaction.
Conclusion: The AIPL1-farnesyl interaction suggests its role in the interaction with phosphodiesterase-6 and normal function of AIPL1.
Significance: This study describes a novel mechanism of AIPL1 in retina disease.
Keywords: Fluorescence Resonance Energy Transfer (FRET), Molecular Chaperone, Phosphodiesterases, Retinal Degeneration, X-ray Scattering, AIPL1, LCA, PDE6, SAXS
Abstract
Aryl hydrocarbon receptor-interacting protein-like 1 (AIPL1) is a photoreceptor specific chaperone of the visual effector enzyme phosphodiesterase-6 (PDE6). AIPL1 has been shown to bind the farnesylated PDE6A subunit. Mutations in AIPL1 are thought to destabilize PDE6 and thereby cause Leber congenital amaurosis type 4 (LCA4), a severe form of childhood blindness. Here, we examined the solution structure of AIPL1 by small angle x-ray scattering. A structural model of AIPL1 with the best fit to the scattering data features two independent FK506-binding protein (FKBP)-like and tetratricopeptide repeat domains. Guided by the model, we tested the hypothesis that AIPL1 directly binds the farnesyl moiety. Our studies revealed high affinity binding of the farnesylated-Cys probe to the FKBP-like domain of AIPL1, thus uncovering a novel function of this domain. Mutational analysis of the potential farnesyl-binding sites on AIPL1 identified two critical residues, Cys-89 and Leu-147, located in close proximity in the structure model. The L147A mutation and the LCA-linked C89R mutation prevented the binding of the farnesyl-Cys probe to AIPL1. Furthermore, Cys-89 and Leu-147 flank the unique insert region of AIPL1, deletion of which also abolished the farnesyl interaction. Our results suggest that the binding of PDE6A farnesyl is essential to normal function of AIPL1 and its disruption is one of the mechanisms underlying LCA.
Introduction
Leber congenital amaurosis (LCA)3 is a severe, early onset, inherited retinopathy that accounts for ∼5% of all inherited retinopathies and is one of the main causes of blindness in children (1, 2). To date, 14 genes have been identified as being mutated in LCA patients, including the gene Aipl1 (aryl hydrocarbon receptor-interacting protein-like 1) (3, 4). Aipl1 mutations result in one of the most clinically severe forms of LCA (LCA type 4).
AIPL1 is expressed selectively in the retina and the pineal gland (3, 5). AIPL1 shares high sequence homology (49% identity) and similar domain organization with the ubiquitously expressed aryl hydrocarbon receptor-interacting protein (AIP) (3). AIP and AIPL1 contain an N-terminal FK506-binding protein (FKBP)-like domain and a C-terminal tetratricopeptide repeat (TPR) domain with three tetratricopeptide repeats (3, 6) (Fig. 1A). These domains are often found in proteins with chaperone activity (7). In addition, in primates, AIPL1 proteins carry at the C termini a proline-rich region of poorly understood function. LCA-linked AIPL1 mutations are found in all three domains: the FKBP-like, TPR, and the proline-rich regions (Fig. 1A). These mutations include missense, nonsense, and deletion mutations (8–10). AIP is a co-chaperone with Hsp90 in the maturation of the aryl hydrocarbon receptor (11), similar to the co-chaperone function of the FKBP and TPR domain-containing immunophilins FKBP51/52 that act on steroid hormone receptors (12). Furthermore, AIPL1 itself was found to interact with Hsp90 and Hsp70 (13). Thus, the function of AIPL1 as an essential component of a retina-specific chaperone complex has been widely hypothesized (3, 7, 14, 15). AIPL1 has been shown to interact with Nedd8 ultimate buster protein 1 (NUB1), implicated in cell cycle regulation and protein degradation (16). However, studies of AIPL1-deficient mouse models provided compelling evidence for phosphodiesterase 6 (PDE6) as the critical photoreceptor partner of AIPL1 (17, 18). AIPL1 knock-out mice exhibited rapid severe degeneration of photoreceptors and had no measurable electroretinograms (17). This phenotype is consistent with the manifestation of LCA4 in humans (10, 17). Biochemical analysis revealed that these mice had markedly reduced levels and activity of PDE6 prior to degeneration (17). The drastic reduction in PDE6 was specific, as the levels of other main photoreceptor proteins remained unaffected (17). PDE6 is a central effector enzyme in the phototransduction cascade (19, 20). Rod PDE6 is a heterotetramer composed of two homologous catalytic subunits (PDE6AB) and two copies of an inhibitory (Pγ) subunit (20). Light stimulates phototransduction by activating PDE6AB to hydrolyze a second messenger cGMP and close cGMP-gated channels in the photoreceptor plasma membrane (19, 20). Mutations in PDE6AB are known to cause retinal degeneration in humans and animal models by elevating intracellular cGMP concentration and triggering photoreceptor cell death (21–25). Thus, defective PDE6 function due to mutations in AIPL1 readily explains LCA4. However, the mechanism of AIPL1 action remains poorly understood. Evidence suggests that AIPL1 acts on rod PDE6 posttranslationally and stabilizes the enzyme by preventing its misassembly that leads to rapid protein degradation (26). Proper assembly of PDE6 requires correct folding and prenylation of PDE6A and PDE6B, formation of the catalytic heterodimer PDE6AB, and its association with the Pγ-subunits. PDE6A and PDE6B are farnesylated and geranylgeranylated, respectively (27). AIPL1 appears to bind to the PDE6A subunit (26), and the farnesyl modification may be involved in this interaction (28). A yeast two-hybrid screen indicated that AIPL1 interacts specifically with farnesylated retinal proteins (28). Yet, other farnesylated proteins, except PDE6, were unaffected in mouse retina lacking AIPL1 (17). In this study, we investigated whether AIPL1 can directly bind the farnesyl moiety, and we examined the possibility that LCA-linked mutations in AIPL1 may disrupt this interaction. To aid our studies, we analyzed the structure of AIPL1 in solution by SAXS and generated a molecular model of AIPL1 most consistent with the scattering data. Our analysis demonstrated high affinity binding of the farnesylated-Cys probe to the FKBP-like domain of AIPL1, thereby revealing a novel function of this domain. Furthermore, an LCA4-causing mutation C89R was found to abrogate the probe binding, suggesting an essential role for the farnesyl-mediated PDE6A interaction in normal function of AIPL1.
FIGURE 1.

A, schematic representation of the domain structure of human AIPL1 protein. AIPL1 consists of an FKBP-like domain (blue), a TPR domain with three tetratricopeptide repeats (red), and a primate-specific proline-rich region (yellow). The sequence and domain organization of mouse AIPL1 are very similar to those of human, except it is lacking the C-terminal proline-rich region. Shown are the commonly reported LCA4-linked AIPL1 mutations (4, 8–10, 54). B, gel filtration profile of AIPL1 on a calibrated Sephacryl 200 column (column volume 125 ml). Arrows indicate elution volumes of 60-, 40-, and 20-kDa standards. C, Coomassie Blue-stained gel showing peak AIPL1 gel filtration fractions.
EXPERIMENTAL PROCEDURES
Materials
AMCA, SE was purchased from Invitrogen. S-Farnesyl-l-cysteine methyl ester was purchased from Enzo Life Sciences. All other chemicals were acquired from Sigma.
Cloning, Expression, and Purification of AIPL1 and Its Domains and Mutants
DNA sequences coding the full-length AIPL1, the N-terminal domain (residues 1–161), or the C-terminal domain (residues 162–328) were PCR-amplified from mouse retinal cDNA and cloned into the pET15b vector using NdeI/BamHI sites. Mutations were introduced using standard QuikChange site-directed mutagenesis protocol. The AIPL1Δ96–143 mutant was generated by replacing the residues 96–143 of AIPL1WT with two glycine residues in a two-step PCR procedure. In the first step, the AIPL1(1–95)Gly-Gly(144–155) sequence was amplified with a forward primer containing the start codon and an NdeI site and a reverse primer corresponding to AIPL1(84–95)Gly-Gly(144–155). This PCR product was used as a forward primer in the second PCR amplification with a primer containing the stop codon and a BamHI site. This resulting PCR product was then cloned into the pET15b vector using NdeI/BamHI sites.
The His6-tagged AIPL1 proteins were expressed in BL21-codon plus Escherichia coli cells by induction with 100 μm isopropyl 1-thio-β-d-galactopyranoside at 16 °C overnight. Cell pellets were sonicated on ice (five 30-s pulses) in 50 mm Tris-HCl buffer (pH 8) containing 50 mm NaCl, 50 mm l-arginine, 50 mm l-glutamic acid, 10% glycerol, 15 mm DTT (buffer A), and protease inhibitor mixture (Roche Applied Science). The His6-tagged AIPL1 proteins were purified over Ni-NTA resin (Novagen) using buffer A containing 200 mm imidazole for elution. Ni-NTA affinity-purified AIPL1 proteins were used in fluorescence binding assays directly or after additional purification by ion exchange chromatography on a Mono Q5 column (Bio-Rad). For analysis of the structure of AIPL1 in solution by SAXS, the His6-AIPL1 protein was further purified by gel filtration chromatography on a Sephacryl 200 column.
Dynamic Light Scattering
Dynamic light scattering was used to examine the degree of monodispersity of AIPL1 samples. Purified AIPL1 was dialyzed against 50 mm Tris-HCl (pH 8.0) buffer containing 50 mm NaCl, 50 mm l-arginine, 50 mm l-glutamic acid, and 10 mm DTT (buffer B). Analyses of monodispersity of dialyzed AIPL1 samples (4 mg/ml) were performed at 22 °C using a DynaPro Nanostar instrument (Wyatt). The data were analyzed using the Dynamics 7.1.7 software.
Homology Modeling
A homology model of the N-terminal FKBP-type domain of AIPL1 (residues 1–157) was generated with Modeler 9.11 (29) using the solution NMR structure of AIP FKBP-like domain as the template (PDB ID code 2LKN) (30). The C-terminal TPR domain of AIPL1 (residues 171–328) was modeled with the hm_build macro in Yasara 12.11.25 (31) that used a number of high resolution TPR domain structures in the PDB to generate an optimized model. These domain models were then superimposed on the respective FKBP and TPR domains from the FKBP52 crystal structure (PDB ID code 1P5Q) (32) and energy-minimized using the em_run macro in Yasara to remove any steric clashes between the two domains. The resulting model with missing domain linker and the N-terminal tag was completed and refined against the SAXS data to generate the molecular model of AIPL1 as described below.
Small Angle X-ray Scattering
SAXS analysis was performed to determine the low resolution solution structure of AIPL1. SAXS data on the His6-tagged AIPL1 protein were collected at the Advanced Light Source (12.3.1 SIBYLS beamline) at the Lawrence Berkeley National Laboratories. Three different concentrations of AIPL1 (2.4, 1.6, and 0.8 mg/ml in buffer B) were exposed to the synchrotron radiation for 0.5, 1, 2, and 4 s each. Scattering from the buffer (matched against the samples by overnight dialysis) was subtracted and the sample scattering data analyzed by Primus/QT (33). AutoRg and AutoDmax were used to calculate the Rg (radius of gyration) and Dmax (maximum particle dimension from a pairwise distribution function) in Primus/QT, and ab inito envelopes were generated using the GASBOR (34), DAMMIF (35), and DAMMIN (36) programs in the ATSAS 2.4.2-1 package (37–39).
The AIPL1 homology model described above was used in the program BUNCH (40) from the ATSAS package to perform combined rigid-body/ab initio modeling against the SAXS data. The two domains were treated as rigid bodies whereas the connecting linker (residues 158–170) and the N-terminal 22 residue tag/linker were modeled as flexible dummy residues that together best fit the SAXS data. During fitting, the two domains could move independently of each other restrained by the length of the interdomain linker. The ab initio envelopes generated separately were aligned to the best-fit BUNCH model using supcomb13 (41) and visualized in PyMOL 1.5.0.4 (The PyMOL Molecular Graphics System, Schrödinger, LLC).
Preparation of the Fluorescence Probe Farnesyl-Cys-AMCA
Conjugation of AMCA-X to cysteine farnesyl methyl ester was performed at 25 °C for 1 h using, respectively, 6 mm and 2 mm solutions of the reagents in 25% acetonitrile, 25% dimethylformamide, and 100 mm HEPES (pH 7.3). The product, farnesyl-Cys-AMCA, was purified by reverse-phase HPLC. Cys-AMCA was prepared by reacting 1 mm AMCA-X with 3 mm cysteine in 50 mm HEPES (pH 7.5), 50% acetonitrile for 30 min at 25 °C.
Fluorescence Assay
Fluorescence assays were performed on a F-2500 Fluorescence Spectrophotometer (Hitachi) in 1 ml of 100 mm HEPES (pH 7.3) or 100 mm PBS (pH 7.5). Fluorescence resonance energy transfer (FRET) assay of farnesyl-Cys-AMCA binding to AIPL1 or mutants was performed with excitation at 280 nm and emission at 440 nm. Farnesyl-Cys-AMCA (150–250 nm) served as an acceptor of AIPL1 Trp fluorescence. The data were fit with equation for binding with ligand depletion,
 |
where F0 is a basal fluorescence of farnesyl-Cys-AMCA, F is the fluorescence after additions of binding protein, Fmax is the maximal fluorescence change, Bm is a concentration of farnesyl-Cys-AMCA, and X is a concentration of added protein. Concentrations of farnesyl-Cys-AMCA and AIPL1 were determined using ϵ350 = 19,000 and ϵ280 = 55,000 m−1cm−1, respectively. Fitting of the experimental data was performed with nonlinear least squares criteria using Prism software (GraphPad). The Kd values are expressed as mean ± S.E. for at least three independent measurements.
Circular Dichroism (CD) Analysis
CD analysis was performed to verify the folding and secondary structure of AIPL1 mutants. AIPL1 or mutants were dialyzed against 5 mm sodium phosphate buffer (pH 7.9) containing 0.1 mm DTT. CD spectra were collected on 7 μm AIPL1 protein samples in a 1-mm cuvette at 25 °C over the range of 300–180 nm with an interval of 1 nm and scan speed of 100 nm/min using a Jasco J-815 CD spectrophotometer in the Biochemistry Department (University of Iowa) or at the Keck Biophysics Facility (Northwestern University).
RESULTS
Analysis of AIPL1 Structure by SAXS
To guide our analysis of the interaction of AIPL1 with the farnesyl moiety, we examined the structure of mouse AIPL1 in solution by SAXS and generated a molecular model of the protein that agrees the best with the scattering data. With samples that are monodisperse and homogeneous, SAXS data can be used to determine three-dimensional envelopes and model the protein using existing high resolution structures of similar proteins in the PDB database (34, 42, 43). The three-step purification procedure (Ni-affinity, ion exchange, and gel filtration) yielded AIPL1 preparation of >95% purity (Fig. 1, B and C) and excellent monodispersity as measured by dynamic light-scattering (polydispersity 15%). All SAXS scattering curves superimposed quite well, reflecting no concentration dependence. However, scattering curves for the high exposures at the highest protein concentration were excluded from analysis due to observable radiation damage. An Rg value of 28.91 Å was calculated from the Guinier region (q·Rg < 1.3) of the scattering curve (Fig. 2A) (42). A bell-shaped curve in the low q region of the Kratky plot increasing somewhat in the high q region indicated that AIPL1 is mostly globular with minor presence of unfolded structure (Fig. 2B). Likewise, a plateau at low q in the Debye-Porod plot suggested that the AIPL1 sample has at least a folded core (Fig. 2C). A Dmax value of 90.81 Å was obtained from the pairwise distribution function, the shape of which also indicated a multidomain protein (Fig. 2D).
FIGURE 2.
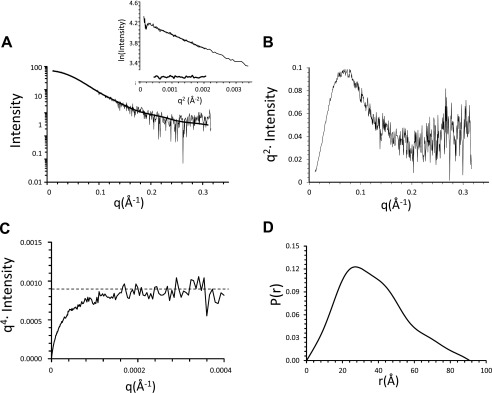
SAXS curves of mouse AIPL1. A, representative buffer-subtracted scattering curve for 0.8 mg/ml AIPL1 sample exposed for 0.5 s. Superimposed on the experimental SAXS data is the theoretical scattering curve calculated for the AIPL1 model. The linear portion of the Guinier region and the residuals after fit to the experimental data in the low q region (0.558–1.231 Å−1) such that q·Rg < 1.3 are shown in the inset. B, scattering data replotted as a Kratky plot indicating that the sample is mostly globular with some unfolded region. C, scattering data replotted as a Debye-Porod plot approaching the Porod plateau (dotted line) at low angles, indicating that the protein has a folded core. D, pairwise distance distribution function P(r), indicating a multidomain protein with a maximum dimension of approximately 91 Å.
Model of AIPL1 Consistent with SAXS Analysis
To reduce modeling bias for AIPL1 from full-length templates (such as FKBP51, PDB ID code 1KT1) (44), we generated homology models for the relatively rigid FKBP and TPR domains individually without the N-terminal and domain connecting flexible linkers. We then used a rigid body fit of these domains against the SAXS data along with ab inito modeling of the flexible linkers to guide us to the relative orientation of these fragments that best explained the experimental x-ray scattering data. This led to a remarkably good model for AIPL1 that fit the SAXS data with a χ value of 1.17 (Fig. 2A) and aligns well with the ab initio envelopes generated from the SAXS data alone (Fig. 3A). An overlay of the SAXS-based AIPL1 model with FKBP51 (PDB ID code 1KT1) (44) and FKBP52 (PDB ID code 1P5Q) (32) crystal structures using the FKBP domains for superposition highlights the heterogeneity in relative orientation between the FKBP and TPR domains possible in this class of proteins (Fig. 3B).
FIGURE 3.

Model of mouse AIPL1. A, homology models of the N-terminal FKBP (blue) and the C-terminal TPR (red) domains with relative orientation that best explains the SAXS data. The interdomain connecting loop and the flexible N-terminal tag (black) were modeled ab initio, again to be consistent with the SAXS data. A representative low resolution envelope (gray) derived from the SAXS data is overlaid with the homology model of AIPL1. B, overlay of the AIPL1 model (slate) with FKBP51 (green) and FKBP52 (pink). The FK2 domains of FKBP51/52 were aligned with the AIPL1 FKBP domain to visualize easily the range of orientations that the TPR domain can adopt. The insert region in AIPL1 (indicated by arrow) may prevent the TPR domain from adopting the conformation seen in FKBP52.
The FKBP-like Domain of AIPL1 Directly Binds Farnesylated-Cys Probe
Following S-farnesylation at a protein C-terminal “CAAX box,” the three terminal residues are cleaved, and the extreme Cys residue is carboxymethylated (27, 45, 46). Consequently, S-farnesyl-l-cysteine methyl ester modified at the amino group with AMCA-X represents an appropriate mimetic of the processed C-terminal Cys of farnesylated proteins. AIPL1 contains seven Trp residues, three in each FKBP-like domain and TPR domain and one in the linking region. AMCA-X with a peak absorbance at 350 nm is an excellent acceptor of Trp fluorescence. Thus, in the instance that AIPL1 binds farnesyl-Cys-AMCA, FRET is likely to occur from AIPL1 Trp residues to the fluorescence probe. Indeed, the Trp fluorescence of AIPL1 (excitation at 280 nm) decreased in the presence of farnesyl-Cys-AMCA concomitant with the appearance of peak AMCA fluorescence at 440 nm (Fig. 4A). The binding of farnesyl-Cys-AMCA to AIPL1 was strong (Kd = 110 nm) and specific (Fig. 4B). No FRET between AIPL1 and farnesyl-Cys-AMCA was observed in the presence of excess S-farnesyl-l-cysteine methyl ester (data not shown). In addition, using the FRET assay, AIPL1 did not bind Cys-AMCA. Thus, the assay reported primarily, if not exclusively, the interaction of AIPL1 with the farnesyl moiety.
FIGURE 4.

Binding of AIPL1 and the FKBP-domain AIPL11–161 to farnesyl-Cys-AMCA probe. A, emission spectra of farnesyl-Cys-AMCA (0.17 μm) (1), AIPL1 (0.1 μm) (2), or AIPL1 (0.1 μm) mixed with farnesyl-Cys-AMCA (0.17 μm) (3) collected with excitation at 280 nm. Inset, Ni-NTA-purified AIPL1 stained with Coomassie Blue. B, FRET assay of [farnesyl-Cys-AMCA] binding to AIPL1 (λex 280 nm, λem 440 nm). Changes in farnesyl-Cys-AMCA fluorescence F − F0 (mean ± S.E. (error bars), n = 3) in the presence of increasing concentrations of AIPL1 were fit with an equation for binding with ligand depletion. C, emission spectra of AIPL11–161 (0.2 μm) alone (1) or mixed with farnesyl-Cys-AMCA (0.25 μm) (2) (λex 280 nm). Inset, Ni-NTA-purified AIPL11–161 and AIPL1162–328 stained with Coomassie Blue. D, changes in farnesyl-Cys-AMCA fluorescence F − F0 (mean ± S.E., n = 3) in the presence of increasing concentrations of AIPL11–161 fit with an equation for binding with ligand depletion.
The model of AIPL1 in Fig. 3 shows that the N-terminal FKBP-like domain (residues 1–161) and the C-terminal TPR domain (residues 162–328) are apparently independent with no obvious interdomain contacts. We expressed these domains individually to probe their role in farnesyl binding. Both domains of AIPL1 were expressed robustly in E. coli as soluble proteins (Fig. 4C, inset). Using the FRET binding assay, the FKBP-like domain of AIPL1 bound farnesyl-Cys-AMCA comparably to the full-length protein (Fig. 4, C and D), whereas the TPR domain did not interact with the probe (data not shown). Furthermore, the TPR domain did not alter the affinity of the FKBP-like domain for the probe, when both domains were mixed in the assay (data not shown). Thus, we conclude that farnesyl-Cys-AMCA binds solely to the FKBP-like domain of AIPL1.
Mapping the Farnesyl Binding Site of AIPL1
We first examined the possibility that the farnesyl moiety interaction site of AIPL1 corresponds to the conserved ligand-binding surface of FKPBs. A typical FKBP domain fold comprises a conical five-stranded β-sheet surrounding a short α-helix (47). Classical FKBPs, such as FKBP12, bind structurally related immunosuppressive drugs, FK506 and rapamycin, within a hydrophobic cavity between the β-sheet and the α-helix (47). Also, some members of the FKBP family are active peptidylprolylisomerases that catalyze the cis-trans conversion of peptidylprolyl bonds (48). The peptidylprolylisomerase active site overlaps with the FK506- and rapamycin-binding pocket. Although AIP and AIPL1 do not bind FK506 or possess peptidylprolylisomerase activity (30, 49), a conserved hydrophobic cavity akin to the ligand-binding site is readily identifiable in the structure of AIP FKBP domain and the model of AIPL1. This site is occluded in AIP, and apparently in AIPL1, by a long insert linking the last two β-strands (Fig. 5A) (30). However, the “insert region” appears as a flexible lid that may swing open thereby uncovering the ligand-binding site. Mutations M59A, W72S, and F149A replacing hydrophobic residues of AIPL1 corresponding to prominent ligand contacts in FKBP12 (Phe-46, Trp-59, and Phe-99) (47) were introduced into the full-length mouse AIPL1. Notably, W72S is a mutation found in LCA4 patients (9). An additional P58A mutation was generated as it is predicted to alter the position of M59 as well as the downstream hydrophobic stretch 61III63 in the vicinity of the FKBP ligand-binding site. The yields of soluble proteins for all these mutants were similar to that of the wild-type AIPL1 (data not shown). The FRET assay showed no significant differences in farnesyl-Cys-AMCA binding between these AIPL1 mutants and the parent protein (Table 1).
FIGURE 5.
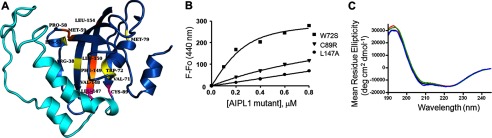
Binding of mutant AIPL1 to farnesyl-Cys-AMCA probe. A, homology model of FKBP-like domain of AIPL1 (blue) with the insert region (cyan). Residues with mutations found in LCA4 (yellow), mutated in this study as a part of the conserved FKBP ligand-binding (orange) and shown in this study to affect farnesyl-cysteine binding (magenta) are highlighted and labeled. B, changes in farnesyl-Cys-AMCA fluorescence in the presence of increasing concentrations of AIPL1 mutants W72S, C89R, and L147A recorded and fit with an equation for binding with ligand depletion. W72S is representative of the mutant with unaffected binding of farnesyl-Cys-AMCA, whereas the C89R and L147A mutations markedly impaired this interaction (Kd >2 μm). Results from one of three similar experiments for each of the mutants are shown. C, CD spectra of AIPL1 (black), C89R (red), L147A (green), and AIPL1Δ96–143 (blue) (7 μm protein each). The estimated secondary structure content of 45% helix and 13% β-strand for AIPL1, 47% helix and 12% β-strand for C89R, 46% helix and 13% β-strand for L147A, and 46% helix and 12% β-strand for AIPL1Δ96–143 is shown.
TABLE 1.
Binding of farnesyl-Cys-AMCA to AIPL1 and its mutants
| AIPL1 mutation | Kd |
|---|---|
| μm | |
| Wild type | 0.11 ± 0.01a |
| R38C | 0.15 ± 0.03 |
| K53W | 0.15 ± 0.02 |
| P58A | 0.17 ± 0.02 |
| M59A | 0.11 ± 0.01 |
| V71F | 0.19 ± 0.06 |
| W72S | 0.10 ± 0.01 |
| V96I | 0.13 ± 0.01 |
| T114I | 0.12 ± 0.01 |
| Y134F | 0.26 ± 0.01 |
| V148A | 0.21 ± 0.05 |
| F149A | 0.21 ± 0.06 |
| L150A | 0.13 ± 0.02 |
| L154A | 0.19 ± 0.01 |
a Mean ± S.E. (n ≥ 3).
Examination of the AIPL1 model reveals an unusual hydrophobic patch 147LVFL150 at the end of the insert region and the beginning of the last β-strand. According to the model, the side chains of Leu-147 and Phe-149 face the interior wall of the β-sheet, whereas Val-148 and Leu-150 together with Leu-154 are solvent-exposed on the outside of the β-sheet. The F149A mutant was already tested as a part of the FKBP ligand-binding site. Mutants with single Ala substitutions of Leu-147, Val-148, Leu-150, and Leu-154 were generated to probe the potential role of this hydrophobic site in farnesyl-Cys binding by AIPL1. V148A, L150A, and L154A mutants bound farnesyl-Cys-AMCA comparably to AIPL1 (Table 1). Interestingly, the L147A mutation markedly diminished farnesyl-Cys-AMCA binding to AIPL1 (Fig. 5). The expression level of soluble L147A was similar to that of the wild-type AIPL1, arguing against gross misfolding of the protein. The folding of L147A compared with AIPL1 was analyzed by CD spectrometry. The average CD spectra of three scans for AIPL1 and L147A are nearly superimposable (Fig. 5C). The secondary structure composition of AIPL1 and L147A was determined using Selcon 3 and Contin programs within the Dichroweb server (50–52). The estimated content of 45% helix and 13% β-strand for AIPL1 and 46% helix and 13% β-strand for L147A are in agreement with the previous CD analysis of AIPL1 (53). Thus, the L147A mutation does not appear to cause any global conformational changes or misfolding in AIPL1.
Farnesyl-Cys Binding by LCA-linked Mutants of AIPL1
Defective farnesyl binding by AIPL1 may hypothetically be the underlying mechanism of certain LCA-linked mutations in the FKBP-like domain. To investigate this hypothesis, we generated the following mutations found as homozygous or compound heterozygous alleles in LCA patients: R38C, K53W (R53W in human AIPL1), V71F, M79T, C89R, V96I, T114I, and Y134F (8–10, 16). The H82Y mutation (54) was excluded from the analysis as this position is not conserved among species and corresponds to Gly-82 in mouse AIPL1. One of the selected AIPL1 mutants, M79T, failed to express as a soluble protein, suggesting defects in the overall folding. The other AIPL1 mutants expressed well although the yields of soluble proteins varied somewhat (data not shown). The R38C, K53W, V71F, V96I, T114I, and Y134F mutations had no significant effect on the binding of farnesyl-Cys-AMCA to AIPL1 (Table 1). In contrast, the C89R mutation drastically impaired this interaction (Fig. 5). CD analysis of the C89R mutant produced the spectrum similar to that of the wild-type AIPL1 (47% helix and 12% β-strand), ruling out global conformational changes in the mutant protein (Fig. 5C). Thus, the C89R mutation locally alters the farnesyl-binding site, or Cys-89 makes direct contacts with farnesyl-Cys-AMCA.
The Insert Region of AIPL1 Is Essential to the FarnesylBinding
The two mutations disrupting the farnesyl-Cys probe binding to AIPL1, C89R and L147A, flank the insert region (Fig. 5A). To test potential role of this region in the AIPL1/farnesyl interaction, we generated the insert deletion mutant, AIPL1Δ96–143. AIPL1Δ96–143 failed to bind farnesyl-Cys-AMCA in the FRET assay. The proper folding of the mutant was confirmed by CD analysis, which indicated a secondary structure composition of 46% α-helix and 12% β-strand (Fig. 5C).
DISCUSSION
AIPL1 is a specific chaperone for the visual effector enzyme, PDE6. Mutations in AIPL1 cause destabilization of PDE6 and underlie LCA4 (3, 8, 17, 18). In photoreceptor cells, AIPL1 may function as a critical component of a multiprotein chaperone complex involving Hsp90, Hsp70, and other co-chaperones (13). Hsp90/70 proteins interact with the AIPL1 TPR domain, and several LCA-associated mutations within the TPR domain reduced this interaction (13). However, it is not known whether Hsp90/70 proteins directly assist the AIPL1-mediated folding and/or assembly of PDE6. Another well investigated partner of AIPL1 is NUB1 (7, 16), but the evidence linking this interaction to PDE6 stability is lacking. Co-expression of PDE6 with AIPL1 in COS cells failed to produce functional PDE6 (18), suggesting the existence of yet additional photoreceptor-specific chaperone(s) for the enzyme.
The mechanism of AIPL1 as a PDE6 chaperone is not fully understood. The PDE6A and PDE6B subunits are prenylated in the cytosol in the rod inner segment. The lipidation promotes association of PDE6A and PDE6B with the endoplasmic reticulum membranes, where the CAAX box is further processed via proteolysis and methylation (27, 45, 46, 55). The timing of PDE6AB heterodimerization in PDE6 biosynthesis remains unclear, but AIPL1 is capable of binding PDE6A prior to the dimer formation and binding of the Pγ-subunits (26). Consistent with the idea that AIPL1 binding to PDE6A precedes PDE6AB dimerization, PDE6 subunits appear to misassemble in the absence of AIPL1 (26). The fact that AIPL1 interacts primarily with PDE6A (26), which is farnesylated, and the earlier report of the AIPL1 interaction with farnesylated proteins (28) raised a possibility of direct binding of AIPL1 to the farnesyl moiety. Our study reveals a strong interaction of farnesyl-Cys-AMCA with AIPL1. The binding affinity of farnesyl-Cys-AMCA for AIPL1 is comparable with the probe affinity for the prenyl-binding protein PrBP/δ (56, 57). The structure of PrBP/δ complexed with a small G protein Rheb shows that the farnesylated-Cys of Rheb is entirely buried in the hydrophobic pocket of PrBP/δ (58). Hence, we surmise that the farnesyl moiety is likely to be substantially occluded in the PDE6A complex with AIPL1, and PDE6A would not appreciably interact with the membrane until AIPL1 is released from the complex. Furthermore, the main membrane binding site of PDE6AB is the geranylgeranylated C terminus of PDE6B (59). Thus, our results in the context of the previous studies (26, 59) favor the following sequence of AIPL1 interactions during the biosynthesis of PDE6: (i) AIPL1 binds PDE6A after the protein farnesylation in the cytosol, (ii) the binding of AIPL1 allows correct assembly of PDE6A with the geranylgeranylated PDE6B, (iii) assembled PDE6AB is then targeted by the prenylated C terminus of PDE6B to the ER membrane for further processing, and (iv) membrane binding of PDE6AB facilitates release of AIPL1.
Unlike prototypical immunophilin FKBP domains, the FKBP domain of AIPL1 does not bind FK506 or exhibit peptidylprolylisomerase activity (30, 49). The FKBP domain may contribute to the AIPL1/NUB1 interaction, but the sites of this interaction are not well defined (16). The role of the AIPL1 FKBP domain, particularly with respect to the function as PDE6 chaperone, remained obscure. A further main finding of our study is the demonstration that farnesylated-Cys binds exclusively to the FKBP domain of AIPL1. The TPR domain of AIPL1 neither promoted nor inhibited the binding of farnesylated-Cys. To map the AIPL1 farnesyl-Cys binding site, we performed mutational analysis of selected residues from candidate hydrophobic areas. Several residues from the hydrophobic pocket corresponding to the conserved FKBP ligand-binding site were found not to be involved in the interaction with the farnesyl-Cys. However, Leu-147 proximal to the FKBP ligand pocket is shown to be critical for the farnesyl-Cys binding. Additional clues to the farnesyl-Cys binding site were provided by the analysis of the LCA-linked mutations of in the AIPL1 FKBP domain. Similar to the L147A mutation, the C89R mutation markedly diminished the interaction of farnesyl-Cys-AMCA with AIPL1. CD analysis of the C89R and L147A mutants rules out protein misfolding or global conformational changes. Cys-89 and Leu-147 are situated in close proximity in the AIPL1 model in a hydrophobic pocket (Fig. 5A). More intriguingly, these residues mark the start and the end of an extensive insert region unique to the FKBP domains of AIP and AIPL1 (30). This indicated a potential role of the insert in farnesyl binding. Indeed, deletion of this region in the AIPL1Δ96–143 mutant completely abolished farnesyl-Cys-AMCA binding.
The model of AIPL1 (Figs. 3, A and B, and 5A) suggests two interesting features of the insert region that may be important for AIPL1 function. First, this region may influence the relative orientation between the FKBP and TPR domains. The domain orientation in the AIPL1 model is similar to that in FKBP51, and it is rather different in FKBP52 (Fig. 3B). The insert region in AIPL1 would engage in steric clashes with the TPR domain preventing it from adopting the domain orientation seen in FKBP52. Second, this region serves a lid over the wider side of the conical β-sheet. Local conformational changes in the hinge sites of the insert region would affect the opening/closing of this lid. Cys-89 and Leu-147 may be essential to the conformation of the insert region, which either provides access to or itself is a part of the lipid-binding site. Our results support the latter model, where the insert region plays a direct and critical role in farnesyl binding. A full understanding of the AIPL1 farnesyl-binding site and the selectivity of AIPL1 for the farnesylated PDE6A protein awaits future structural studies.
Acknowledgments
We thank the staff at the SIBYLS beamline at the Advanced Light Source, Lawrence Berkeley National Laboratory, for SAXS data collection. The SIBYLS beamline at the Advanced Light Source, Lawrence Berkeley National Laboratory, is supported in part by the Department of Energy program Integrated Diffraction Analysis Technologies (IDAT) under Contract DE-AC02-05CH11231.
This work was supported, in whole or in part, by National Institutes of Health Grants EY-10843 and EY-12682 (to N. O. A.).
- LCA
- Leber congenital amaurosis
- AIP
- aryl hydrocarbon receptor-interacting protein
- AIPL1
- aryl hydrocarbon receptor-interacting protein-like 1
- AMCA-X, SE
- 6-((7-amino-4-methylcoumarin-3-acetyl)amino)hexanoic acid, succinimidyl ester
- FKBP
- FK506-binding proteins
- Ni-NTA
- nickel-nitrilotriacetic acid
- NUB1
- Nedd8 ultimate buster protein 1
- PDB
- Protein Data Bank
- PDE6
- photoreceptor phosphodiesterase-6
- SAXS
- small angle x-ray scattering
- TPR
- tetratricopeptide repeat.
REFERENCES
- 1. Koenekoop R. K. (2004) An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv. Ophthalmol. 49, 379–398 [DOI] [PubMed] [Google Scholar]
- 2. Kaplan J., Bonneau D., Frézal J., Munnich A., Dufier J. L. (1990) Clinical and genetic heterogeneity in retinitis pigmentosa. Hum. Genet. 85, 635–642 [DOI] [PubMed] [Google Scholar]
- 3. Sohocki M. M., Bowne S. J., Sullivan L. S., Blackshaw S., Cepko C. L., Payne A. M., Bhattacharya S. S., Khaliq S., Qasim Mehdi S., Birch D. G., Harrison W. R., Elder F. F., Heckenlively J. R., Daiger S. P. (2000) Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat. Genet. 24, 79–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. den Hollander A. I., Roepman R., Koenekoop R. K., Cremers F. P. (2008) Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 27, 391–419 [DOI] [PubMed] [Google Scholar]
- 5. van der Spuy J., Chapple J. P., Clark B. J., Luthert P. J., Sethi C. S., Cheetham M. E. (2002) The Leber congenital amaurosis gene product AIPL1 is localized exclusively in rod photoreceptors of the adult human retina. Hum. Mol. Genet. 11, 823–831 [DOI] [PubMed] [Google Scholar]
- 6. Das A. K., Cohen P. W., Barford D. (1998) The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein-protein interactions. EMBO J. 17, 1192–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van der Spuy J., Cheetham M. E. (2006) The chaperone function of the LCA protein AIPL1: AIPL1 chaperone function. Adv. Exp. Med. Biol. 572, 471–476 [DOI] [PubMed] [Google Scholar]
- 8. Sohocki M. M., Perrault I., Leroy B. P., Payne A. M., Dharmaraj S., Bhattacharya S. S., Kaplan J., Maumenee I. H., Koenekoop R., Meire F. M., Birch D. G., Heckenlively J. R., Daiger S. P. (2000) Prevalence of AIPL1 mutations in inherited retinal degenerative disease. Mol. Genet. Metab. 70, 142–150 [DOI] [PubMed] [Google Scholar]
- 9. Stone E. M. (2007) Leber congenital amaurosis, a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 144, 791–811 [DOI] [PubMed] [Google Scholar]
- 10. Dharmaraj S., Leroy B. P., Sohocki M. M., Koenekoop R. K., Perrault I., Anwar K., Khaliq S., Devi R. S., Birch D. G., De Pool E., Izquierdo N., Van Maldergem L., Ismail M., Payne A. M., Holder G. E., Bhattacharya S. S., Bird A. C., Kaplan J., Maumenee I. H. (2004) The phenotype of Leber congenital amaurosis in patients with AIPL1 mutations. Arch. Ophthalmol. 122, 1029–1037 [DOI] [PubMed] [Google Scholar]
- 11. Trivellin G., Korbonits M. (2011) AIP and its interacting partners. J. Endocrinol. 210, 137–155 [DOI] [PubMed] [Google Scholar]
- 12. Storer C. L., Dickey C. A., Galigniana M. D., Rein T., Cox M. B. (2011) FKBP51 and FKBP52 in signaling and disease. Trends Endocrinol. Metab. 22, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hidalgo-de-Quintana J., Evans R. J., Cheetham M. E., van der Spuy J. (2008) The Leber congenital amaurosis protein AIPL1 functions as part of a chaperone heterocomplex. Invest. Ophthalmol. Vis. Sci. 49, 2878–2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schwartz M. L., Hurley J. B., Ramamurthy V. (2006) Biochemical function of the LCA-linked protein, aryl hydrocarbon receptor interacting protein like-1 (AIPL1): role of AIPL1 in retina. Adv. Exp. Med. Biol. 572, 89–94 [DOI] [PubMed] [Google Scholar]
- 15. Kosmaoglou M., Schwarz N., Bett J. S., Cheetham M. E. (2008) Molecular chaperones and photoreceptor function. Prog. Retin. Eye Res. 27, 434–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Akey D. T., Zhu X., Dyer M., Li A., Sorensen A., Blackshaw S., Fukuda-Kamitani T., Daiger S. P., Craft C. M., Kamitani T., Sohocki M. M. (2002) The inherited blindness-associated protein AIPL1 interacts with the cell cycle regulator protein NUB1. Hum. Mol. Genet. 11, 2723–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramamurthy V., Niemi G. A., Reh T. A., Hurley J. B. (2004) Leber congenital amaurosis linked to AIPL1: a mouse model reveals destabilization of cGMP phosphodiesterase. Proc. Natl. Acad. Sci. U.S.A. 101, 13897–13902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu X., Bulgakov O. V., Wen X. H., Woodruff M. L., Pawlyk B., Yang J., Fain G. L., Sandberg M. A., Makino C. L., Li T. (2004) AIPL1, the protein that is defective in Leber congenital amaurosis, is essential for the biosynthesis of retinal rod cGMP phosphodiesterase. Proc. Natl. Acad. Sci. U.S.A. 101, 13903–13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arshavsky V. Y., Burns M. E. (2012) Photoreceptor signaling: supporting vision across a wide range of light intensities. J. Biol. Chem. 287, 1620–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cote R. H. (2004) Characteristics of photoreceptor PDE (PDE6): similarities and differences to PDE5. Int. J. Impot. Res. 16, S28–33 [DOI] [PubMed] [Google Scholar]
- 21. Farber D. B., Lolley R. N. (1974) Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina. Science 186, 449–451 [DOI] [PubMed] [Google Scholar]
- 22. Bowes C., Li T., Danciger M., Baxter L. C., Applebury M. L., Farber D. B. (1990) Retinal degeneration in the rd mouse is caused by a defect in the β subunit of rod cGMP-phosphodiesterase. Nature 347, 677–680 [DOI] [PubMed] [Google Scholar]
- 23. Pittler S. J., Baehr W. (1991) Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase β-subunit gene of the rd mouse. Proc. Natl. Acad. Sci. U.S.A. 88, 8322–8326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McLaughlin M. E., Ehrhart T. L., Berson E. L., Dryja T. P. (1995) Mutation spectrum of the gene encoding the β subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. U.S.A. 92, 3249–3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dryja T. P., Rucinski D. E., Chen S. H., Berson E. L. (1999) Frequency of mutations in the gene encoding the α subunit of rod cGMP-phosphodiesterase in autosomal recessive retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 40, 1859–1865 [PubMed] [Google Scholar]
- 26. Kolandaivelu S., Huang J., Hurley J. B., Ramamurthy V. (2009) AIPL1, a protein associated with childhood blindness, interacts with α-subunit of rod phosphodiesterase (PDE6) and is essential for its proper assembly. J. Biol. Chem. 284, 30853–30861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Anant J. S., Ong O. C., Xie H. Y., Clarke S., O'Brien P. J., Fung B. K. (1992) In vivo differential prenylation of retinal cyclic GMP phosphodiesterase catalytic subunits. J. Biol. Chem. 267, 687–690 [PubMed] [Google Scholar]
- 28. Ramamurthy V., Roberts M., van den Akker F., Niemi G., Reh T. A., Hurley J. B. (2003) AIPL1, a protein implicated in Leber's congenital amaurosis, interacts with and aids in processing of farnesylated proteins. Proc. Natl. Acad. Sci. U.S.A. 100, 12630–12635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 30. Linnert M., Lin Y. J., Manns A., Haupt K., Paschke A. K., Fischer G., Weiwad M., Lücke C. (2013) The FKBP-type domain of the human aryl hydrocarbon receptor-interacting protein reveals an unusual Hsp90 interaction. Biochemistry, in press [DOI] [PubMed] [Google Scholar]
- 31. Krieger E., Koraimann G., Vriend G. (2002) Increasing the precision of comparative models with YASARA NOVA: a self-parameterizing force field. Proteins 47, 393–402 [DOI] [PubMed] [Google Scholar]
- 32. Wu B., Li P., Liu Y., Lou Z., Ding Y., Shu C., Ye S., Bartlam M., Shen B., Rao Z. (2004) 3D structure of human FK506-binding protein 52: implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. Proc. Natl. Acad. Sci. U.S.A. 101, 8348–8353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 34. Svergun D. I., Petoukhov M. V., Koch M. H. (2001) Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Konarev P. V., Petoukhov M. V., Volkov V. V., Svergun D. I. (2006) ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Petoukhov M. V., Konarev P. V., Kikhney A. G., Svergun D. I. (2007) ATSAS 2.1: towards automated and Web-supported small-angle scattering data analysis. J. Appl. Crystallogr. 40, S223–228 [Google Scholar]
- 40. Petoukhov M. V., Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kozin M. B., Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 42. Putnam C. D., Hammel M., Hura G. L., Tainer J. A. (2007) X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 40, 191–285 [DOI] [PubMed] [Google Scholar]
- 43. Chacón P., Díaz J. F., Morán F., Andreu J. M. (2000) Reconstruction of protein form with x-ray solution scattering and a genetic algorithm. J. Mol. Biol. 299, 1289–1302 [DOI] [PubMed] [Google Scholar]
- 44. Sinars C. R., Cheung-Flynn J., Rimerman R. A., Scammell J. G., Smith D. F., Clardy J. (2003) Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. Proc. Natl. Acad. Sci. U.S.A. 100, 868–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Clarke S. (1992) Protein isoprenylation and methylation at carboxyl-terminal cysteine residues. Annu. Rev. Biochem. 61, 355–386 [DOI] [PubMed] [Google Scholar]
- 46. Casey P. J., Seabra M. C. (1996) Protein prenyltransferases. J. Biol. Chem. 271, 5289–5292 [DOI] [PubMed] [Google Scholar]
- 47. Van Duyne G. D., Standaert R. F., Karplus P. A., Schreiber S. L., Clardy J. (1993) Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229, 105–124 [DOI] [PubMed] [Google Scholar]
- 48. Göthel S. F., Marahiel M. A. (1999) Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell. Mol. Life Sci. 55, 423–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li J., Zoldak G., Kriehuber T., Soroka J., Schmid F. X., Richter K., Buchner J. (2013) Unique proline-rich domain regulates the chaperone function of AIPL1. Biochemistry, in press [DOI] [PubMed] [Google Scholar]
- 50. Whitmore L., Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sreerama N., Woody R. W. (1993) A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem. 209, 32–44 [DOI] [PubMed] [Google Scholar]
- 52. van Stokkum I. H., Spoelder H. J., Bloemendal M., van Grondelle R., Groen F. C. (1990) Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 191, 110–118 [DOI] [PubMed] [Google Scholar]
- 53. Gallon V. A., Wilkie S. E., Deery E. C., Newbold R. J., Sohocki M. M., Bhattacharya S. S., Hunt D. M., Warren M. J. (2004) Purification, characterisation and intracellular localisation of aryl hydrocarbon interacting protein-like 1 (AIPL1) and effects of mutations associated with inherited retinal dystrophies. Biochim. Biophys. Acta 1690, 141–149 [DOI] [PubMed] [Google Scholar]
- 54. Galvin J. A., Fishman G. A., Stone E. M., Koenekoop R. K. (2005) Evaluation of genotype-phenotype associations in Leber congenital amaurosis. Retina 25, 919–929 [DOI] [PubMed] [Google Scholar]
- 55. Karan S., Zhang H., Li S., Frederick J. M., Baehr W. (2008) A model for transport of membrane-associated phototransduction polypeptides in rod and cone photoreceptor inner segments. Vision Res. 48, 442–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang H., Liu X. H., Zhang K., Chen C. K., Frederick J. M., Prestwich G. D., Baehr W. (2004) Photoreceptor cGMP phosphodiesterase Δ subunit (PDEΔ) functions as a prenyl-binding protein. J. Biol. Chem. 279, 407–413 [DOI] [PubMed] [Google Scholar]
- 57. Gopalakrishna K. N., Doddapuneni K., Boyd K. K., Masuho I., Martemyanov K. A., Artemyev N. O. (2011) Interaction of transducin with uncoordinated 119 protein (UNC119): implications for the model of transducin trafficking in rod photoreceptors. J. Biol. Chem. 286, 28954–28962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ismail S. A., Chen Y. X., Rusinova A., Chandra A., Bierbaum M., Gremer L., Triola G., Waldmann H., Bastiaens P. I., Wittinghofer A. (2011) Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nat. Chem. Biol. 7, 942–949 [DOI] [PubMed] [Google Scholar]
- 59. Catty P., Deterre P. (1991) Activation and solubilization of the retinal cGMP-specific phosphodiesterase by limited proteolysis: role of the C-terminal domain of the β-subunit. Eur. J. Biochem. 199, 263–269 [DOI] [PubMed] [Google Scholar]