

Abstract
Many peptides and other compounds that influence metabolism also influence food intake, and numerous hypotheses explaining the observed effects in terms of energy homeostasis have been suggested over the years. For example, cholecystokinin (CCK), a duodenal peptide secreted during meals that aids in digestion, also reduces ongoing food intake, thereby contributing to satiation; and insulin and leptin, hormones secreted in direct proportion to body fat, act in the brain to help control adiposity by reducing energy intake. These behavioral actions are often considered to be hard-wired, such that negative experiments, in which an administered compound fails to have its purported effect, are generally disregarded. In point of fact, failures to replicate the effects of compounds on food intake are commonplace, and this occurs both between and within laboratories. Failures to replicate have historically fueled heated debate about the efficacy and/or normal function of one or another compound, leading to confusion and ambiguity in the literature. We review these phenomena and their implications and argue that, rather than eliciting hard-wired behavioral responses in the maintenance of homeostasis, compounds that alter food intake are subjected to numerous influences that can render them completely ineffective at times and that a major reason for this variance is that food intake is not under stringent homeostatic control.
Keywords: cholecystokinin, homeostasis, insulin, leptin, satiation
it has been recognized for more than a century that as an integral part of the digestive process the gastrointestinal (GI) tract secretes hormones and other signals in response to the food that is ingested. Each of these signals in turn helps mediate one or more components of the digestive-absorptive process, such as causing enzymes to be secreted into the intestinal lumen or varying the contraction and, hence, tension of GI musculature and, consequently, the rate at which consumed food passes through the system. A rational view, and one that has dominated thinking about food intake, signals, and digestion since the time of Pavlov, is that the secretion of these GI signals is a reflexive response triggered by specific consumed nutrients interacting with specific receptors in the oral cavity, stomach, and intestines. In this way, the specific blend of GI enzymes secreted into the intestinal lumen, and the GI hormones entering the blood, reflects the quality and quantity of the nutrients being ingested; i.e., the cocktail of hormones secreted is customized to optimally facilitate the digestion of the particular nutrients that are being consumed. A more recent revelation has been that, in addition to acting locally on individual components of the digestive system, GI hormones and other compounds secreted during meals have other key functions. Many of them provide a signal to the brain regarding the quantity and quality of nutrients being eaten and thereby engage neural reflexes coordinating activity across the entire GI system. Some also act as incretins, facilitating the secretion of insulin during meals and consequently helping maintain glucose as well as energy homeostasis during and after meals.
GI activity is tightly linked to food taking, and numerous hypotheses have been proposed that presume causality. With regard to factors that influence food intake (i.e., both the initiation and the termination of meals), the prevailing view for much of the 20th Century was that neural signals related to the processing of nutrients in the GI tract are a major determinant of behavior. For example, a popular view, and one that continues to be held by many people, is that stomach distension and contractions initiate the perceptions of both hunger and satiation. Beginning with the work of Cannon (15), neural signals generated by mechanical activity of the stomach have been implicated in the control of eating, and there is considerable supporting evidence. Results from experiments with gastric cannulas, from which ingested fluids can be infused or drained (22, 36, 42, 56), and pyloric cuffs, which can be inflated to prevent food from entering the small intestine (22, 36, 42, 56), indicate that gastric volume and, hence, stretch on the gastric musculature is an adequate stimulus for mechanoreceptors that contributes to the control of eating. Nevertheless, these signals alone have not been found to be sufficient for the normal control of meal size, because supraphysiological volumes of gastric fill are required to reduce food intake in rats. One limitation of the pyloric cuff model in the assessment of the role of gastric mechanosignals in the control of eating is, of course, that it also prevents potential interactions of gastric with postgastric intestinal and even postabsorptive signals. While several findings indicate that such interactions can be influential (63), normal gastric mechanosignals are not thought to be of sufficient magnitude to limit meal size.
The popular reliance on gastric distension cues to explain eating behavior was later overshadowed by the glucostatic theory, which opined that energy derived from glucose utilization by key cells in the hypothalamus determined both the onset and termination of meals (48, 49). The concept was simple and appealing. On the basis of the premise that a major goal of meals is to replenish dwindling energy supplies, the key factors necessary and sufficient to induce eating and satiation included neurons whose output activity to neural circuits guiding food taking is proportional to their rate of metabolizing certain nutrients (reviewed by Refs. 41 and 42). The past few decades have spawned numerous hypotheses linking the metabolism of specific nutrients or a common denominator of their energy content or metabolism in one or another organ to eating. Parenteral administration of metabolic fuels often reduces, whereas pharmacological inhibition of fuel utilization mostly increases, food intake. In some reports, metabolic inhibitors also attenuated the decrease in food intake in response to intravenous nutrient infusions. These experiments are important because they collectively suggest that fluctuations in the availability or utilization of energy-yielding substrates, mainly glucose and fatty acids, or else a common denominator of their utilization, help regulate energy intake as depicted in Fig. 1. The threshold of a decrease in whole body glucose utilization or fatty acid oxidation to generate a signal sufficient to stimulate eating is probably greater than what occurs before spontaneous meals (25), implying that these effects of metabolic inhibitors elicit primarily emergency responses. Nevertheless, local changes in fuel utilization that do not reflect homeostatically relevant alterations in whole body metabolism may well occur in association with the presence of food in the gut and may contribute to the control of food intake (41, 43, 62, 70).
Fig. 1.
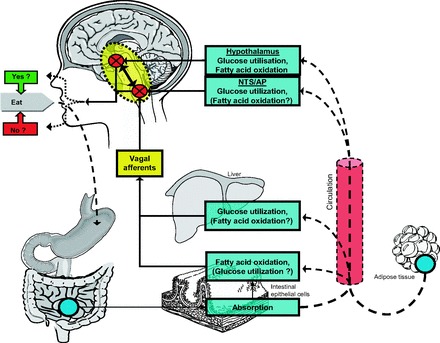
Diagram depicting peripheral and CNS sensors that generate signals in response to the availability and/or utilization of metabolic fuels that affect eating behavior. Circulating metabolic fuels derived from absorption of ingested nutrients or from mobilization of endogenous stores (i.e., glucose from liver and free fatty acids from adipose tissue) may reach the brain directly and/or may trigger signals in vagal afferent or other sensory nerves to the CNS. Metabolic fuels reaching the brain directly may trigger responses in the caudal brain stem, especially in the nucleus of the solitary tract (NTS)/area postrema (AP) regions or else more anteriorly, including regions of the hypothalamus such as the arcuate nucleus. Bidirectional arrow between caudal brain stem and hypothalamus reflects important interconnections in integrating signals from metabolic fuels, from other meal-generated satiation signals, and from adipose stores. [from Langhans and Geary (42)].
Peptides and Food Intake
Toward the end of the 20th Century, a paradigm shift occurred with the recognition that GI hormones, in addition to orchestrating digestion, directly influence eating behavior. It should be noted that the possibility that peptides from the GI tract might directly influence behavior was counter to a long-standing dogma that the nervous and endocrine systems were distinct entities, and also seemed contrary to the notion that hormonal peptides in the blood cannot penetrate into the brain. The roots of the new concept can be traced to several technological advancements. For one, after the pancreatic hormone insulin was discovered and found to lower blood glucose, the observations that its administration also elicited eating in animals as well as a feeling of hunger in people soon followed (46). While these observations obviously helped solidify the tenets of the glucostatic theory, they nonetheless also demonstrated that an increase of a meal-associated hormone could have behavioral consequences, albeit indirectly through insulin's hypoglycemic action. Another development that occurred in the latter half of the 20th Century was the advent of quick and reliable methods to measure the levels of specific peptides and proteins, the first being the radioimmunoassay method for insulin [RIAs (86)]. At the same time, improved extraction and purification techniques for obtaining measurable amounts of peptide hormones, and later their synthesis as well, were forthcoming, enabling the exogenous administration of relatively pure formulations of peptide hormones and allowing scientists to ask more pointed questions about a possible causality vis-à-vis food intake.
All of these developments in the latter half of the 20th Century conspired to set the stage for the breakthrough report in 1973 by Gibbs, Smith, and their colleagues that administering the duodenal peptide cholecystokinin (CCK), or its shorter but active analog CCK-8, at the start of a meal results in animals eating smaller meals (28). Over the ensuing decades, the basic observation has been replicated dozens of times in a wide range of species including humans (see reviews in Refs. 27, 36, 52, 71). It is now recognized that endogenous CCK can have this same satiating action, since administering specific antagonists to the appropriate CCK receptor (CCK-1R) results in larger meals being eaten.
The chain of information passage leading from ingested food in the intestine to CCK secretion to neural circuits influencing food intake in the brain has been well worked out. In brief, as partially digested and heavily acidified nutrients exit the stomach and enter the duodenum, they interact with enteroendocrine I cells interspersed among the enterocytes lining the duodenal lumen. Specific receptors on the I cells detect specific nutrients (especially lipids), and the cells respond by secreting CCK. Some of the CCK enters the hepatic portal blood and circulates to the liver, where it stimulates bile flow, as well as to the exocrine pancreas, where it stimulates the secretion of pancreatic juice. Bile and pancreatic juice pass through ducts to the duodenum, where they neutralize the stomach acid and facilitate the further digestion of the meal. Some of the secreted CCK also acts locally on CCK-1R located on smooth muscle cells of the pyloric sphincter, causing it to constrict the aperture and thereby slow gastric emptying. Locally released CCK also acts on CCK-1R on sensory nerve endings located in close proximity to I cells, generating action potentials that travel via the vagus nerve to the nucleus of the solitary tract (NTS) of the hindbrain.
The importance of the initial findings on CCK cannot be overstated. For one thing, they heralded in the “Age of Peptides” with regard to factors that influence food intake, a movement that continues actively to this day. In fact, the rate at which GI secretions that influence food intake are newly discovered continues to increase as molecular genetic techniques identify novel molecules and receptors on a regular basis. Because many of these newly identified molecules are routinely being screened/evaluated with regard to having a possible therapeutic influence on caloric intake and body weight, it is important to understand the limitations of the underlying assumptions of this endeavor, especially with regard to identifying targets to treat obesity.
Interpreting the ever-increasing tide of experiments in which one or another peptide or other compound hypothesized to mimic or antagonize the action of an endogenous signal is given and consequently found to influence food intake has necessitated the establishment and adoption of a strict set of criteria. As originally proposed by Gibbs and Smith (28) and later modified by Smith (69) and reviewed by Moran (see Ref. 52), criteria for an endogenous meal-generated satiation signal are that 1) the proposed candidate signal should be activated by eating and therefore released during meals; 2) exogenous administration of the candidate signal should elicit a significant and dose-dependent reduction of meal size; 3) the proposed signal should have a rapid onset of action if it is involved in meal termination, as well as a brief duration of action to allow for the frequency of meal-taking behavior; 4) the reduction of food intake caused by administration of the candidate compound should not be secondary to illness, malaise, or incapacitation; 5) the reduction of meal size should be obtained with doses that achieve physiological concentrations at the receptor site; and 6) reducing the activity of the candidate compound at its endogenous receptor should attenuate action of the exogenously administered compound and should also cause an increase of meal size during normal meals. An additional contemporary strategy often applied is to use molecular genetic models (e.g., gene deletions, knock-ins, etc.) to modify endogenous levels of the candidate compound and/or its receptor and determine whether the genetic findings complement pharmacological approaches. The point is that, although it is relatively easy to induce an animal to eat less food in a test situation, nonspecific causes must be ruled out before concluding any link to the endogenous controls over normal eating behavior. Importantly, and as discussed in detail below, another criterion seems to have been informally adopted; i.e., it is important to replicate key observations (both between and within laboratories). The obverse of this is how a failure to replicate should be interpreted.
There are several categories of endogenous signals that that are thought to reduce food intake. Satiation factors such as CCK are meal-generated, acutely acting signals that meet the criteria discussed above. Other signals such as the hormones leptin and insulin are secreted in proportion to body fat. They are considered to be adiposity signals that provide a tone or background that influences the efficacy of satiation signals. For example, an individual who has gained body fat has levels of circulating leptin and insulin that have also been increased. The consequent elevation of adiposity signals reaching the brain is thought to render the individual more sensitive to CCK and other satiation signals, such that less food is eaten in each single meal, ultimately promoting a reduction of the increased body fat. The increased sensitivity to CCK's satiating action has been documented for both insulin and leptin (23, 51, 53, 61). Thus, when considered in isolation, insulin or leptin may not strictly satisfy each of the criteria above (e.g., leptin is not increased during meals, and it is certainly not the case that the administration of either leptin or insulin into the brain results in physiological levels). Nonetheless, their robust interaction with satiation factors enables them to meet the spirit of those criteria.
Problems of Interpretation and Failures to Replicate
It is important to note that the basic tenet of the idea that exogenous peptides could have a meaningful, physiological action with regard to influencing food intake was challenged from the start. As an example, Deutsch and colleagues (19, 20) argued that reduced meal size resulting from exogenous formulations of CCK is an artifact based on the administered peptide stimulating the GI tract in abnormal ways, relying upon recordings of gastric and intestinal contractions to make their point. On another front, Stricker and Verbalis and their colleagues (75, 76) argued that exogenous CCK actually renders people (and by analogy, animals) ill and that malaise is the reason they consequently consume less food. This latter issue was resolved with the development of specific antagonists to the CCK-1R. Administration of these antagonists actually increases meal size, implicating endogenous CCK as suppressing meal size during normal meals in humans who do not report feeling ill (5, 33, 60). Many studies in animals and humans have reported that at doses that reliably reduce food intake CCK does not cause illness or malaise (27, 36, 52, 71). So, whereas there is no doubt that large pharmacological doses of most peptides can elicit some malaise, a general belief that permeates the literature on exogenous CCK and many other “satiation” peptides is that at small, near-physiological doses they reduce meal size, presumably by combining with and mimicking the action of their endogenous counterparts on their critical receptors. Furthermore, it is important to point out that satiation and nausea are not at opposite ends of a continuum reducing food intake but rather represent distinct phenomena related to features of food being consumed on a gradual scale from hunger to overly full.
We are concerned with a different issue that clouds the interpretation of experiments on putative satiation signals. As a point of reference, each of us (S. C. Woods and W. Langhans) has considerable experience investigating the effect of exogenously administered compounds on food intake, and we cite examples from our own laboratories to make important points. Nonetheless, we have reason to believe that our experiences are shared by many other investigators. We refer to the unreliability of replicating findings in which an administered compound alters food intake, and we believe this is a frequent yet generally unreported occurrence both between and within laboratories, one that begs to be brought into the open and discussed.
One of us (S. C. Woods) has published multiple reports documenting the ability of insulin, when administered directly into the brain (usually into the 3rd cerebral ventricle, i3vt), to reduce food intake and body weight of experimental animals (64, 81–84). The basic phenomenon has been replicated in multiple laboratories, dozens of reports have appeared over the years (1, 14, 26, 34, 58, 73, 81), and many of the parameters, targets within the brain, and cellular mechanisms of action have been identified. Administering insulin so that it readily accesses the brain also reduces food intake and body weight of humans (6, 32). That said, whenever we embark upon an experiment in which insulin is to be administered into the brain of animals and food intake assessed, there is a probability that the insulin will have no effect whatsoever; i.e., that the animals will respond to insulin as if nothing or vehicle only had been administered.
In Fig. 2, data are presented from two experiments conducted by the same individual in the same lab and with the same strain and age of rats and other parameters, but conducted several months apart. As can be seen, administering i3vt elicited a reliable reduction of food intake in cohort A, yet had no effect whatsoever in cohort B. Importantly, when trying to understand a failure to replicate such as this, the variance of the data should be considered. In point of fact, in most such instances and as indicated in Fig. 2, it is not the case that some animals respond to insulin and others not, implying that using a larger number of subjects would provide sufficient statistical power to reveal the effect. Rather, it is as if all or none of the subjects respond. Furthermore, we included a positive control in both cohorts A and B by administering the same insulin intraperitoneally to other rats and documenting that blood glucose decreased; i.e., the insulin was comparably biologically active in both experiments. This basic phenomenon of a failure of i3vt insulin to have an effect on food intake has occurred (too) many times in our laboratory, and we have ruled out trivial explanations such as that the insulin was no good, was diluted wrongly, was administered into the wrong site, or the like.
Fig. 2.
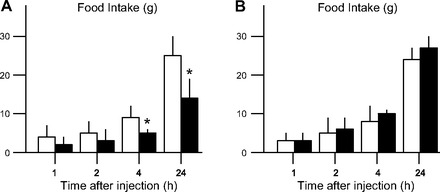
Mean (+SE) food intake (g) following i3vt saline (open bars) or insulin (2 mU; solid bars) in 2 experiments conducted under the same conditions by the same experimenter and using the same strain, age, weight, and source of male rats but several months apart. It is important to note that comparable results (i.e., a failure to replicate when all experimental parameters appear the same) have also been observed with other doses of i3vt insulin. *P < 0.05 relative to saline.
This failure to reliably observe a phenomenon within the same laboratory generalizes to the between-laboratories situation. Many careful investigators have informed us over the years that, try as they might, they simply cannot get intraventricularly administered insulin to reduce food intake in their laboratories, or else that, although insulin sometimes has the anticipated effect, more often than not it doesn't. It is not that they do not believe the phenomenon but that rather they recognize either that the phenomenon is extremely sensitive and that conditions must be exactly correct and/or that it is simply fickle. Others have stated that, as occurs for us, insulin sometimes works and sometimes doesn't and that it seems to be an iffy phenomenon for unknown reasons.
W. Langhans' laboratory has also encountered the same “variable insulin efficacy experience.” In several experiments in his laboratory, i3vt insulin administrations dose-dependently reduced food intake in mice (on about one-third of the trials) but failed to do so in the other cases, confirming the between-laboratory replicability of the failure to replicate a decrease of food intake following i3vt insulin (as well as extending the phenomenon to mice). It is important to reiterate that in each laboratory all trials were performed by the same person, with animals from the same colony, fed the same food, and so on. All attempts to reasonably explain the discordant results by the conduct of the experiments (between- vs. within-subjects designs), the age or body weight of the animals (young vs. old, light vs. heavy) or other more or less arbitrary conditions have so far failed.
Leptin
Another example is important for speaking to the generality of the phenomenon. When located at the University of Washington in the middle 1990s, S. C. Woods and his colleague Randy Seeley, performed several experiments in which leptin, administered i3vt to rats, lowered food intake (67, 68). Some of these findings formed the basis for the seminal observation that leptin's catabolic action in the brain is mediated by the central melanocortin system (68). Woods and Seeley subsequently moved their laboratory to the University of Cincinnati. Importantly, their technician, who had personally conducted the leptin experiments in Seattle, also made the move to Cincinnati along with Woods and Seeley and soon began to try and replicate the leptin experiments in the new surroundings. However, despite using the same source of leptin, the same supplier of and strain of rats, and with the same person conducting the experiment as had occurred in Seattle, leptin had no apparent effect on food intake over numerous attempts (>25 individual experiments and cohorts of rats) and a wide range of doses in Cincinnati. Depicted in Fig. 3 are the data from three of those experiments, and they are typical of all of them. As with the insulin example above, all experimental parameters were the same other than the geographical locale. And also, as occurred many times with i3vt insulin, at no time point was there a difference of intake between rats receiving leptin and those receiving vehicle. For comparison, also depicted are the average data from several published experiments using the same dose of leptin in Seattle. At some inexplicable point and after many negative experiments, leptin was once again found to reduce food intake in our laboratory, and it has been a relatively reliable anorectic agent ever since (e.g., Refs. 2, 16).
Fig. 3.
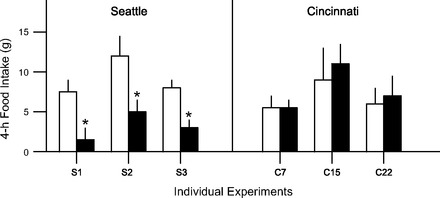
Mean (+SE) 4-h food intake (g) following i3vt saline (open bars) or leptin (3.5 μg; solid bars) in 3 published experiments conducted in Seattle (S1–S3) and 3 unpublished experiments conducted in Cincinnati (C7, C15, C22) by the same experimenter. Although only one dose and one time point are depicted, outcomes were comparable across all doses and times; i.e., i3vt leptin did not reduce food intake in any of the experiments conducted in Cincinnati (C1 through C28). *P < 0.05 from saline. S1 is from Ref. 66; S2 is from Ref. 71; S3 is from Ref. 67.
Adiposity Signals vs. Satiation Signals
Insulin and leptin are functionally related in the sense that each is often considered to be an adiposity signal to the brain; i.e., the levels of each in the circulation are directly proportional to body fat. The widely accepted homeostatic view of the control of food intake and body weight regulation states that adiposity signals enter the brain from the circulation and work in part by influencing the brain's sensitivity to meal-generated satiation signals such as CCK (65, 85). If an animal or person loses weight (e.g., by dieting), reduced levels of insulin and leptin reaching targets in the brain cause reduced sensitivity to CCK and other satiation signals, and larger meals are consequently eaten until weight returns to normal. Conversely, if an individual overeats for several days and extra weight is put on, the consequent elevated levels of adiposity signals increase sensitivity to satiation signals, causing smaller meals to be consumed (65, 85). This homeostatic negative feedback model accounts for the tendency of many individuals to maintain relatively stable body weights over long intervals. Because of the robust nature of the homeostatic regulatory process, nonhomeostatic factors are often thought to be responsible for the upward slippage of body weight referred to as the obesity epidemic.
It is worth considering whether the oft-times failure to replicate reductions of food intake by insulin and leptin is somehow related to their being adiposity signals; i.e., is assessing food intake after administering satiation signals or other compounds that alter individual meal size subject to the same phenomenon? In point of fact, at around the same time that CCK was first reported to meet the criteria for being an endogenous meal-generated satiation factor by some laboratories, other laboratories reported that it had no effect on food intake (29, 38). Still others found exogenous CCK-induced hypophagia to be an example of habituation and that its ability to reduce food intake was not consistent over days in the same animals (50). The point is that over a several-year period before CCK became widely entrenched as a reliable satiation factor in the literature, there were contrary reports. These days, no one would question whether exogenous CCK can in fact reduce food intake.
In 2002, Bloom's laboratory reported in Nature that peptide YY (PYY), when administered to rats, mice, and even humans, reduced food intake (4). Because of the translational importance of this finding, many laboratories soon tried to replicate the basic observation, and in many of those experiments PYY failed to have an anorectic effect relative to vehicle controls. The failure to replicate was sufficiently widespread that it was discussed among colleagues from different institutions, and one consequence was that a jointly written report that included 42 authors from 15 different laboratories appeared claiming that the original conclusions were in error and likely artifactual (13, 74). Since then, many other groups have been able to replicate the original observation from the Bloom laboratory, and although there was a certain amount of heated rhetoric over the ensuing months, it is now generally accepted that exogenous PYY does in fact reduce food intake. Nonetheless, the entire episode is troubling, because as it unfolded it called into question the methods and even the integrity of respected scholars.
As another example, in 2005 it was reported in Science that the peptide obestatin reduces food intake (88). Obestatin comes from the same parent prohormone as ghrelin, a peptide hormone that increases food intake when exogenously administered. Thus, two fragments of the same parent peptide purportedly elicit opposite effects on food intake, perhaps analogous to β-endorphin and α-MSH, both being fragments of the parent peptide POMC, and that have opposite effects on food intake. Although a second group soon replicated the basic finding of hypophagia in animals administered obestatin (39), several other laboratories could not replicate it. Once again, a joint report from many laboratories and authors was published presenting their negative findings with respect to obestatin on food intake (55). It is almost as if laboratories deliberate and then take a vote on whether a given peptide has an effect on food intake or not. In the case of obestatin, a retraction was subsequently made for one of the earlier reports that it elicited hypophagia, with a stated reason being that the basic findings “could not be reproduced using the same experimental protocol” (40). This seems like an overly strict criterion and one which if universally applied might lead to hundreds of articles on the effects of peptides on food intake being retracted.
The ease of rapid communication among widespread laboratories and investigators that science currently enjoys has many consequences. Important findings are often widely distributed before they are officially published. This allows early attempts at replication and creates a window of opportunity during which opposing findings can be aired, especially when bolstered by the strength of many investigators. At some point, dogma sets in on one side or the other of an issue, and contrary data are no longer newsworthy. We have found over the years that, even though a laboratory may not be able to observe an effect of a compound on food intake that others, or themselves, have previously observed and/or reported, the members of that laboratory nevertheless can be very creative at generating potential explanations for the failure (Table 1).
Table 1.
Explanations often offered to rationalize a failure to replicate one's own findings or the findings of others related to whether or not an administered compound is found to influence food intake
| Explanations/Rationalizations |
| Inactive compound; wrong supplier |
| Miscalculated dose |
| Wrong diluent/solvent |
| Animals not properly prepped/handled |
| Poor lab conditions |
| One lab doesn't know head from hole in ground |
| Wrong time of year/temperature/humidity |
| Sickness or infection in the colony |
| Animals stressed for unknown reasons |
| Disturbance by animal care personnel |
| etc., etc. |
These are not isolated cases. We know of no compound for which there has been universal agreement as to its effect on food intake, including, as discussed above, the prototypical hypophagic factor CCK. In some instances, there are reports that a compound actually elicits the opposite effect from what is reported by others. Again, to use insulin as an example, in addition to dozens of reports that its central administration reduces food intake, there are reports that it has no effect (e.g., Ref. 47) and at least one in which it was actually found to increase food intake (78). Others have reported that administering insulin into the brain reduces food intake in some experiments or conditions but not in others (57, 58). Jessen et al. (35) recently reported the results of several carefully executed experiments in which they did not observe an effect of central insulin to reduce food intake, stating, “Although we varied rat strain, stereotactic coordinates, formulations of insulin and vehicle, dose, volume, and time of injection, the anorectic effect of intracerebroventricular insulin could not be replicated” (p. R43). Sodersten's laboratory investigated the ability of intraventricular leptin and NPY to alter food intake and found, like many others, that, whereas NPY increases and leptin decreases food intake in many situations, these actions are actually reversed in others, with leptin increasing and NPY decreasing intake (3). Divergent results following NPY administration are important because they generalize the phenomenon to compounds that increase food intake (and see Refs. 9, 66).
Metabolic Signals
Adiposity signals and satiation signals are thought to be integrated at several levels of the neuraxis to provide a homeostatic influence over food intake and the amount of fat stored in the body. Other types of homeostatic signals also influence eating. For example, and as discussed above, administering compounds that interfere with either glucose or fatty acid metabolism can increase food intake (Fig. 1). Mercaptoacetate (MA) is a drug that inhibits mitochondrial fatty acid oxidation, and when it is administered to experimental animals, they eat more food, in particular when fed a fat-enriched diet. Following the original report by Scharrer and Langhans (62), the eating-stimulatory effect of MA has been confirmed for most species tested and in a variety of conditions (for review see Ref. 44). Although the effect is robust and well established, several students and post docs in W. Langhans' laboratory have more recently been unable to replicate the hyperphagic effect of MA. After numerous failed experiments with different lots of MA from the same supplier, different suppliers, in-house purification of the compound, and confirmation of MA's metabolic activity in other animals in vivo, no satisfying explanation occurred. After a hiatus of many months, the eating-stimulatory effect of MA somewhat mysteriously “reappeared” in its full magnitude, leaving us to speculate about possible reasons for the experienced difficulties and contributing to our frustration to understand these events.
There is no need to belabor the point further. Administering compounds and assessing food intake can be an uncertain and even perilous endeavor, especially for the graduate student or untenured assistant professor. The question is: what might account for the ubiquitous phenomenon of the lack of reliability with which administered factors alter food intake? We consider several possibilities.
Normal Variation
Is a failure to replicate the effect of a compound on food intake simply a statistical error based on insufficient power? Normal variation should dictate that failures to replicate should occasionally occur, even with otherwise robust phenomena. This seems unlikely, since, as noted above, the failures are not associated with increased variance, and they often persist across numerous experiments. In point of fact, there is no way to easily ascertain the incidence of the phenomenon across laboratories. In this regard, it is important to note that a failure to observe an effect of a compound on food intake would not be likely to be published, especially once a finding is considered dogma, and that many such instances no doubt go unwritten and consequently unreported.
Food Intake vs. Other Ingestive Behaviors
We considered whether the failure to replicate changes of food intake following the administration of compounds is unique or rather an example of a more general phenomenon. Many compounds influence water intake, the majority of them eliciting rather precise adjustments in bodily hydration. Perhaps the most-studied dipsogen is the hormone angiotensin-II (ANG II), which potently and with short latency elicits water intake when administered into the brain. Beginning with the first report in 1969 [Epstein et al. (24)], more than 3,000 references appear on PubMed in response to the input angiotensin and water intake as of this writing. We could not find one article in which ANG II failed to increase water intake or in which the basic premise was challenged. Similarly, when we inserted angiotensin and sodium intake into PubMed, more than 2,000 references appeared, beginning with W. D. Reid (59) (1965), with no apparent dissensions.
Thus, there may be something unique about food intake. Elicited water intake or sodium intake are generally regarded as directly addressing and correcting a detected hydromineral/osmotic imbalance. If such deviations from ideal remain uncorrected for prolonged intervals, most bodily functions are compromised and life is threatened. This scenario reflects homeostatic regulation as it is described in textbooks. When a deviation from optimal occurs, it is quickly detected and precisely corrected. In contrast, it is extremely rare that the intake of an individual meal would make sweeping corrections with regard to ongoing energy homeostasis or body weight. Rather, unlike the case for body water, body energy can be stored and made available when needed. Consequently, when body fat has been reduced, it often requires several days for the homeostatic controllers to counter the change (12, 18). On the other hand, there are some circumstances in which food intake does appear to be a rapid, homeostatically driven response, for example when blood glucose or glucose utilization is suddenly reduced. Administering insulin peripherally to lower glucose, or administering drugs like 2-deoxyglucose that compromise glucose utilization (72), elicits a prompt corrective response, one that has apparently never been widely questioned. So perhaps there is something unique about assessing food intake in an individual at a time when a homeostatically controlled variable such as glucose utilization is not under immediate threat and a metabolically active compound is administered.
Environmental and Related Factors
For most individuals, the intake of food, especially the onset and size of individual meals, has many possible influences or causes (79, 80). It is often habitual, occurring at the same time every day, the exact times being subject to environmental circumstances and experience. It can be opportunistic, when a desirable food source is unexpectedly encountered. It can be social, when conspecifics gather and food is available. It can be reflexive, resulting from a sudden reduction in the availability of energy at a critical site in the body or brain. In principle, it can also be reflexive to a severely depleted amount of stored fat or other form of stored energy. The amount eaten as well as the initiation of meals can be modified by emotions, by stressors, by palatability and other hedonic qualities, by food variety, and by other environmental factors. Within the brain, each of these influences is mediated by specific neuronal circuits with their unique sensory inputs and outputs, and the various pathways interact in myriad ways [e.g., (10, 11)].
Learning
Each of these same factors in turn can be modified by experience or learning. To cite a pertinent example, although CCK reliably reduces meal size in most experiments, when animals are consistently administered CCK in a specific stimulus situation, they learn to ignore it as a satiating factor to the point that its exogenous administration no longer reduces food intake (21, 30). Thus, the specific metabolic history of an individual might render it more or less sensitive to one or more satiating signals in some environmental situations. Perhaps analogously, there are experimental conditions where NPY elicits food intake in rats, and others where it has no effect in the same rats (66), and these differences have been found to be due to prior experience with the paradigm (9). The ability of melanocortins to reduce food intake can also be modified by particular prior experiences with the assessment paradigm (8), and both insulin and leptin depend on melanocortin signaling for their anorectic action (7, 68). The key point is that nonhomeostatic factors can intervene, often as the result of experience or learning, but presumably due to other factors as well, to completely negate the impact of presumed homeostatic signals on food intake. It is important to remember that exogenous compounds given to alter food intake are thought to act by mimicking endogenous signals or activity at their receptors or sensor sites such that they consequently and necessarily are influenced by the individual's past experience with activity in those neural circuits.
Interacting Neural Circuits
The ultimate decision to start or to end a meal is thus a complex calculus of multiple interacting neuronal circuits, and numerous factors are positioned to enhance, reduce, or override others. For example, and as discussed above, rather than directly influencing meal size, insulin and leptin are thought to exert their anorectic action by increasing the sensitivity of the brain to satiating signals such as CCK (23, 51, 53, 61). Analogously, the satiating action of apolipoprotein A-IV relies upon CCK (45), whereas mice lacking apolipoprotein A-IV have enhanced sensitivity to CCK's satiating action (87). Thus, satiating factors can interact in complex ways to enhance or perhaps interfere with the actions of others. Some stressors cause reduced food intake and others cause increased food intake (31, 37, 54). The important point is that it is likely that most factors that can influence food ingestion interact in complex and often unknown ways.
Experimental Paradigms
In most experiments designed to investigate the effect of a compound on food intake in rodents, the subjects are either adapted to a particular feeding schedule where they are fasted for a short time each day and then given food (often at a time convenient for the investigator but arbitrary to the subjects) and/or are assessed at the time the lights in the room are turned off and the animals tend to eat the largest meals of the day. It is easy to conceive that some subtle environmental condition may exist that could allow, or disallow, the influence of an exogenously administered compound on food intake, and that it influences all of the animals in a group comparably. As an example, in any given experiment there could be a nonobvious stressor present, less-than-optimal humidity/temperature, bedding with a peculiar odor, a seasonal effect, or whatever, and the consequence is to attenuate or compromise the ability of certain circuits from influencing food intake. Alternatively, the individuals could have had common but specific experiences that render them functionally blind to the satiating action of a peptide.
What to do?
While this is all speculation, there must nonetheless be some common factor that impacts all of the animals in one group comparably to account for the empirical observation that a compound has an effect in one experiment or on one occasion and not in another, since it is less a phenomenon of individuals than of cohorts of individuals. We suspect that the very nature of food ingestion, and the complexity of influences over it, is key, but until or unless experiments designed to dissect the phenomenon are completed and understood, failing to elicit an anticipated change of food intake will remain a possibility in any given situation. The outcome will always be probabilistic rather than certain.
So what can be done? Replication is obviously important, and when a negative finding occurs that fails to replicate a prior positive finding, it is imperative to include a positive control in order to rule out certain trivial explanations. That is, it is important to make certain that the administered compound exerts some other, hopefully less probabilistic, effect. Pay attention to, and control as much as possible, environmental and experiential factors. It would probably also be helpful to include negative experiments in published reports to begin to establish a database, since it is likely that efficacy of some administered compounds on food intake is more prone to influences that block or otherwise interfere with their effect than others. Based on our laboratories' experiences, the probability that insulin or leptin will fail to reduce food intake is greater than the probability that CCK will. Since adiposity signals such as leptin and insulin influence intake upstream of satiation signals such as CCK, this might imply that the nearer a circuit is to the final common pathway to influence behavior by stopping a meal, the greater its probability of actually having an effect (Fig. 4). In principle, when neural activity starts down the final satiating circuit, it can no longer be influenced. However, the probability that some particular factor will exert sufficient influence to stimulate the final common pathway depends on the number and importance of possible upstream intervening pathways. For example, it is known that for individual meals insulin and leptin are upstream of CCK and other satiation peptides. The relative positioning of metabolic signals vis-à-vis others is not well known; and inputs from habits, stressors, social factors, palatability and a host of others are also not known. Thus, whether or not a distant input, such as insulin or leptin, is effective can be at the mercy of any of a number of other more proximal factors.
Fig. 4.
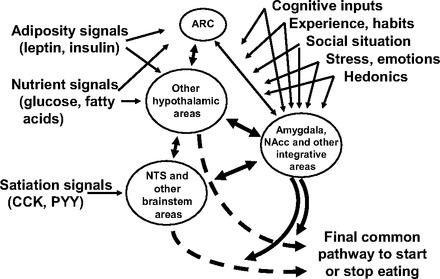
The ultimate decision to initiate or stop meals is influenced by multiple, diverse categories of signals. As a generality, homeostatic signals, including adiposity signals such as leptin and insulin, as well as nutrient signals, stimulate and are integrated in the hypothalamic arcuate nucleus (ARC), whereas signals related to the meal being eaten (satiation signals) are handled more rostrally, in the nucleus of the solitary tract (NTS) and other brain stem areas. Superimposed on these signals are inputs from diverse brain areas, and these can modulate and potentially override the impact of homeostatic signals. It should be noted that only a subset of interacting factors that can influence the decision to eat are depicted. NAcc, nucleus accumbens.
It is not known whether there is a single underlying mechanism (as yet unknown) that accounts for all of the examples we cite or whether diverse mechanisms can come into play depending upon idiosyncratic features of some laboratories or experiments. If understood and remediable, the former would facilitate far better appreciation of the calculus that influences meal taking. It would likely also make this area of research more attractive to some investigators. For example, if the explanation were that even a very mild food deprivation, such as is often used to guarantee a high baseline intake, can be sufficient to negate the impact of a satiation signal and that this is exacerbated in not-yet-fully-mature animals, these factors could be controlled and eliminated. We have, however, systematically considered several such possibilities without arriving at a definitive answer, but the possibility nonetheless exists.
Note that such explanations may be quite different for water intake, at least when it is homeostatically activated by angiotensin. It may therefore be the case that the most fragile responses are behaviors that are not directly driven by homeostatic negative feedback considerations. An interesting complementary observation was reported by Crabbe et al. several years ago. Three different laboratories obtained mice from the same source and put them through a battery of (ostensibly) identical physical assessments and behavioral challenges (17). For many variables, and especially behavioral responses, there was poor agreement among the laboratories. That same group also found that changing experimental sites or even very small environmental features could have a large impact on behavioral assessments (77). The point is that subtle and for the most part unknown factors come to play when assessing behaviors that are not immediately critical for life.
Conclusions
Many compounds, including many metabolically important peptides, have been demonstrated to influence food intake in experimental situations. However, the actual effect appears to be probabilistic rather than certain. An important implication is that, except in rare, usually experimentally introduced circumstances, food intake should not be considered a homeostatic response. Rather, both the onset and termination of meals can be quite flexible, allowing for myriad influences to come into play. Over long intervals, of perhaps several days or more in humans, homeostatic considerations can be seen to have an effect, but in the short term, the impact of the same homeostatic signals can be variable.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-017844 and DK-078201 and Swiss National Science Foundation Grant 31_130665.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.C.W. and W.L. conception and design of research; S.C.W. and W.L. interpreted results of experiments; S.C.W. and W.L. prepared figures; S.C.W. and W.L. drafted manuscript; S.C.W. and W.L. edited and revised manuscript; S.C.W. and W.L. approved final version of manuscript.
REFERENCES
- 1.Air EL, Benoit SC, Blake Smith KA, Clegg DJ, Woods SC. Acute third ventricular administration of insulin decreases food intake in two paradigms. Pharmacol Biochem Behav 72: 423–429, 2002 [DOI] [PubMed] [Google Scholar]
- 2.Air EL, Benoit SC, Clegg DJ, Seeley RJ, Woods SC. Insulin and leptin combine additively to reduce food intake and body weight in rats. Endocrinology 143: 2449–2452, 2002 [DOI] [PubMed] [Google Scholar]
- 3.Ammar AA, Sederholm F, Saito TR, Scheurink AJ, Johnson AE, Sodersten P. NPY-leptin: opposing effects on appetitive and consummatory ingestive behavior and sexual behavior. Am J Physiol Regul Integr Comp Physiol 278: R1627–R1633, 2000 [DOI] [PubMed] [Google Scholar]
- 4.Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, Cone RD, Bloom SR. Gut hormone PYY(3–36) physiologically inhibits food intake. Nature 418: 650–654, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Beglinger C, Degen L, Matzinger D, D'Amato M, Drewe J. Loxiglumide, a CCK-A receptor antagonist, stimulates calorie intake and hunger feelings in humans. Am J Physiol Regul Integr Comp Physiol 280: R1149–R1154, 2001 [DOI] [PubMed] [Google Scholar]
- 6.Benedict C, Kern W, Schultes B, Born J, Hallschmid M. Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J Clin Endocrinol Metab 93: 1339–1344, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Benoit SC, Air EL, Coolen LM, Strauss R, Jackman A, Clegg DJ, Seeley RJ, Woods SC. The catabolic action of insulin in the brain is mediated by melanocortins. J Neurosci 22: 9048–9052, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benoit SC, Clegg DJ, Barrera JG, Seeley RJ, Woods SC. Learned meal initiation attenuates the anorexic effects of the melanocortin agonist MTII. Diabetes 52: 2684–2688, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Benoit SC, Clegg DJ, Woods SC, Seeley RJ. The role of previous exposure in the appetitive and consummatory effects of orexigenic neuropeptides. Peptides 26: 751–757, 2005 [DOI] [PubMed] [Google Scholar]
- 10.Berthoud HR. Metabolic and hedonic drives in the neural control of appetite: who is the boss? Curr Opin Neurobiol, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berthoud HR. Neural control of appetite: cross-talk between homeostatic and non-homeostatic systems. Appetite 43: 315–317, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Birch LL, Johnson SL, Andresen G, Peters JC, Schulte MC. The variability of young childrens energy-intake—reply. New Engl J Med 324: 1817–1817, 1991 [DOI] [PubMed] [Google Scholar]
- 13.Boggiano MM, Chandler PC, Oswald KD, Rodgers RJ, Blundell JE, Ishii Y, Beattie AH, Holch P, Allison DB, Schindler M, Arndt K, Rudolf K, Mark M, Schoelch C, Joost HG, Klaus S, Thone-Reineke C, Benoit SC, Seeley RJ, Beck-Sickinger AG, Koglin N, Raun K, Madsen K, Wulff BS, Stidsen CE, Birringer M, Kreuzer OJ, Deng XY, Whitcomb DC, Halem H, Taylor J, Dong J, Datta R, Culler M, Ortmann S, Castaneda TR, Tschop M. PYY3–36 as an anti-obesity drug target. Obes Rev 6: 307–322, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Brown LM, Clegg DJ, Benoit SC, Woods SC. Intraventricular insulin and leptin reduce food intake and body weight in C57BL/6J mice. Physiol Behav 89: 687–691, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Cannon WB, Washburn AL. An explanation of hunger. Am J Physiol 29: 441–445, 1912 [Google Scholar]
- 16.Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Hypothalamic mTOR signaling regulates food intake. Science 312: 927–930, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Crabbe JC, Wahlsten D, Dudek BC. Genetics of mouse behavior: Interactions with laboratory environment. Science 284: 1670–1672, 1999 [DOI] [PubMed] [Google Scholar]
- 18.DeCastro JM. How can eating behavior be regulated in the complex environments of free-living humans? Neurosci Biobehav Rev 20: 119–131, 1996 [DOI] [PubMed] [Google Scholar]
- 19.Deutsch JA, Hardy WT. Cholecystokinin produces bait shyness in rats. Nature 266: 196, 1977 [DOI] [PubMed] [Google Scholar]
- 20.Deutsch JA, Thiel TR, Greenberg LH. Duodenal motility after cholecystokinin injection or satiety. Behav Biol 24: 393–399, 1978 [DOI] [PubMed] [Google Scholar]
- 21.Duncan EA, Davita G, Woods SC. Changes in the satiating effect of cholecystokinin over repeated trials. Physiol Behav 85: 387–393, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Eisen S, Davis JD, Rauhofer E, Smith GP. Gastric negative feedback produced by volume and nutrient during a meal in rats. Am J Physiol Regul Integr Comp Physiol 281: R1201–R1214, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Emond M, Schwartz GJ, Ladenheim EE, Moran TH. Central leptin modulates behavioral and neural responsivity to CCK. Am J Physiol Regul Integr Comp Physiol 276: R1545–R1549, 1999 [DOI] [PubMed] [Google Scholar]
- 24.Epstein AN, Fitzsimons JT, Simons BJ. Drinking caused by the intracranial injection of angiotensin into the rat. J Physiol 200: 98P–100P, 1969 [PubMed] [Google Scholar]
- 25.Even P, Nicolaidis S. Spontaneous and 2DG induced metabolic changes and feeding: the ischymetric hypothesis. Brain Res Bull 15: 429–435, 1985 [DOI] [PubMed] [Google Scholar]
- 26.Florant GL, Singer L, Scheurink AJ, Park CR, Richardson RD, Woods SC. Intraventricular insulin reduces food intake and body weight of marmots during the summer feeding period. Physiol Behav 49: 335–338, 1991 [DOI] [PubMed] [Google Scholar]
- 27.Geary N. Endocrine controls of eating: CCK, leptin, and ghrelin. Physiol Behav 81: 719–733, 2004 [DOI] [PubMed] [Google Scholar]
- 28.Gibbs J, Young RC, Smith GP. Cholecystokinin decreases food intake in rats. J Comp Physiol Psychol 84: 488–495, 1973 [DOI] [PubMed] [Google Scholar]
- 29.Glick Z, Thomas DW, Mayer J. Absence of effect of injections of the intestinal hormones secretin and choecystokinin-pancreozymin upon feeding behavior. Physiol Behav 6: 5–8, 1971 [DOI] [PubMed] [Google Scholar]
- 30.Goodison T, Siegel S. Learning and tolerance to the intake suppressive effect of cholecystokinin in rats. Behav Neurosci 109: 62–70, 1995 [PubMed] [Google Scholar]
- 31.Greeno CG, Wing RR. Stress-induced eating. Psychol Bull 115: 444–464, 1994 [DOI] [PubMed] [Google Scholar]
- 32.Hallschmid M, Benedict C, Born J, Kern W. Targeting metabolic and cognitive pathways of the CNS by intranasal insulin administration. Expert Opin Drug Deliv 4: 319–322, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Hewson G, Leighton GE, Hill RG, Hughes J. The cholecystokinin receptor antagonist L364,718 increases food-intake in the rat by attenuation of the action of endogenous cholecystokinin. Brit J Pharmacol 93: 79–84, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Honda K, Kamisoyama H, Saneyasu T, Sugahara K, Hasegawa S. Central administration of insulin suppresses food intake in chicks. Neurosci Lett 423: 153–157, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Jessen L, Clegg DJ, Bouman SD. Evaluation of the lack of anorectic effect of intracerebroventricular insulin in rats. Am J Physiol Regul Integr Comp Physiol 298: R43–R50, 2010 [DOI] [PubMed] [Google Scholar]
- 36.Kaplan JM, Moran TH. Gastrointestinal signaling controlling food intake. In: Neurobiology of Food and Fluid Intake, edited by Stricker EM, Woods SC. New York: Kluwer Academic/Plenum, 2004, p. 275–305. [Google Scholar]
- 37.Koob GF, Heinrichs SC. A role for corticotropin releasing factor and urocortin in behavioral responses to stressors. Brain Res 848: 141–152, 1999 [DOI] [PubMed] [Google Scholar]
- 38.Koopmans HS, Deutsch JA, Branson PJ. The effect of cholecystokinin pancreozymin on hunger and thirst in mice. Behav Biol 7: 441–444, 1972 [DOI] [PubMed] [Google Scholar]
- 39.Lagaud GJ, Young A, Acena A, Morton MF, Barrett TD, Shankley NP. Obestatin reduces food intake and suppresses body weight gain in rodents. Biochem Biophys Res Commun 357: 264–269, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Lagaud GJ, Young A, Acena A, Morton MF, Barrett TD, Shankley NP. Obestatin reduces food intake and suppresses body weight gain in rodents (Retraction of vol 357, p. 264, 2007). Biochem Biophys Res Commun 388: 619–619, 2009 [DOI] [PubMed] [Google Scholar]
- 41.Langhans W. The enterocyte as an energy flow sensor in the control of eating. Forum Nutr 63: 75–84, 2010 [DOI] [PubMed] [Google Scholar]
- 42.Langhans W, Geary N. Overview of the physiological control of eating. Forum Nutr 63: 9–53, 2010 [DOI] [PubMed] [Google Scholar]
- 43.Langhans W, Leitner C, Arnold M. Dietary fat sensing via fatty acid oxidation in enterocytes: possible role in the control of eating. Am J Physiol Regul Integr Comp Physiol 300: R554–R565, 2011 [DOI] [PubMed] [Google Scholar]
- 44.Leonhardt M, Langhans W. Fatty acid oxidation and control of food intake. Physiol Behav 83: 645–651, 2004 [DOI] [PubMed] [Google Scholar]
- 45.Lo CM, Zhang DM, Pearson K, Ma L, Sun W, Sakai RR, Davidson WS, Liu M, Raybould HE, Woods SC, Tso P. Interaction of apolipoprotein AIV with cholecystokinin on the control of food intake. Am J Physiol Regul Integr Comp Physiol 293: R1490–R1494, 2007 [DOI] [PubMed] [Google Scholar]
- 46.MacKay EM, Callaway JW, Barnes RH. Hyperalimentation in normal animals produced by protamine insulin. J Nutr 20: 59–66, 1940 [Google Scholar]
- 47.Manin M, Balage M, Larue-Achagiotis C, Grizard J. Chronic intracerebroventricular infusion of insulin failed to alter brain insulin-binding sites, food intake, and body weight. J Neurochem 51: 1689–1695, 1988 [DOI] [PubMed] [Google Scholar]
- 48.Mayer J. Glucostatic mechanism of regulation of food intake. New Engl J Med 249: 13–16, 1953 [DOI] [PubMed] [Google Scholar]
- 49.Mayer J, Thomas DW. Regulation of food intake and obesity. Science 156: 328–337, 1967 [DOI] [PubMed] [Google Scholar]
- 50.Mineka S, Snowdon CT. Inconsistency and possible habituation of CCK-induced satiety. Physiol Behav 21: 65–72, 1978 [DOI] [PubMed] [Google Scholar]
- 51.Moran TH, Aja S, Ladenheim EE. Leptin modulation of peripheral controls of meal size. Physiol Behav 89: 511–516, 2006 [DOI] [PubMed] [Google Scholar]
- 52.Moran TH, Kinzig KP. Gastrointestinal satiety signals. II. Cholecystokinin. Am J Physiol Gastrointest Liver Physiol 286: G183–G188, 2004 [DOI] [PubMed] [Google Scholar]
- 53.Morton GJ, Blevins JE, Williams DL, Niswender KD, Gelling RW, Rhodes CJ, Baskin DG, Schwartz MW. Leptin action in the forebrain regulates the hindbrain response to satiety signals. J Clin Invest 115: 703–710, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nieuwenhuizen AG, Rutters F. The hypothalamic-pituitary-adrenal-axis in the regulation of energy balance. Physiol Behav 94: 169–177, 2008 [DOI] [PubMed] [Google Scholar]
- 55.Nogueiras R, Pfluger P, Tovar S, Arnold M, Mitchell S, Morris A, Perez-Tilve D, Vazquez MJ, Wiedmer P, Castaneda TR, DiMarchi R, Tschop M, Schurmann A, Joost HG, Williams LM, Langhans W, Dieguez C. Effects of obestatin on energy balance and growth hormone secretion in rodents. Endocrinology 148: 21–26, 2007 [DOI] [PubMed] [Google Scholar]
- 56.Phillips RJ, Powley TL. Gastric volume rather than nutrient content inhibits food intake. Am J Physiol Regul Integr Comp Physiol 271: R766–R769, 1996 [DOI] [PubMed] [Google Scholar]
- 57.Plata-Salaman CR, Oomura Y. Effect of intra-third ventricular administration of insulin on food intake after food deprivation. Physiol Behav 37: 735–739, 1986 [DOI] [PubMed] [Google Scholar]
- 58.Plata-Salaman CR, Oomura Y, Shimizu N. Dependence of food intake on acute and chronic ventricular administration of insulin. Physiol Behav 37: 717–734, 1986 [DOI] [PubMed] [Google Scholar]
- 59.Reid WD, Laragh JH. Sodium and potassium intake, blood pressure, and pressor response to angiotensin. Proc Soc Exp Biol Med 120: 26–29, 1965 [DOI] [PubMed] [Google Scholar]
- 60.Reidelberger RD, Orourke MF. Potent cholecystokinin antagonist L-364718 stimulates food-intake in rats. Am J Physiol Regul Integr Comp Physiol 257: R1512–R1518, 1989 [DOI] [PubMed] [Google Scholar]
- 61.Riedy CA, Chavez M, Figlewicz DP, Woods SC. Central insulin enhances sensitivity to cholecystokinin. Physiol Behav 58: 755–760, 1995 [DOI] [PubMed] [Google Scholar]
- 62.Scharrer E, Langhans W. Control of food intake by fatty acid oxidation. Am J Physiol Regul Integr Comp Physiol 250: R1003–R1006, 1986 [DOI] [PubMed] [Google Scholar]
- 63.Schwartz GJ. Brainstem integrative function in the central nervous system control of food intake. Forum Nutr 63: 141–151, 2010 [DOI] [PubMed] [Google Scholar]
- 64.Schwartz MW, Figlewicz DP, Baskin DG, Woods SC, Porte D., Jr Insulin in the brain: a hormonal regulator of energy balance. Endocr Rev 13: 387–414, 1992 [DOI] [PubMed] [Google Scholar]
- 65.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature 404: 661–671, 2000 [DOI] [PubMed] [Google Scholar]
- 66.Seeley RJ, Payne CJ, Woods SC. Neuropeptide Y fails to increase intraoral intake in rats. Am J Physiol Regul Integr Comp Physiol 268: R423–R427, 1995 [DOI] [PubMed] [Google Scholar]
- 67.Seeley RJ, van Dijk G, Campfield LA, Smith FJ, Burn P, Nelligan JA, Bell SM, Baskin DG, Woods SC, Schwartz MW. Intraventricular leptin reduces food intake and body weight of lean rats but not obese Zucker rats. Horm Metab Res 28: 664–668, 1996 [DOI] [PubMed] [Google Scholar]
- 68.Seeley RJ, Yagaloff KA, Fisher SL, Burn P, Thiele TE, van Dijk G, Baskin DG, Schwartz MW. Melanocortin receptors in leptin effects. Nature 390: 349, 1997 [DOI] [PubMed] [Google Scholar]
- 69.Smith GP. Introduction to the reviews on peptides and the control of food intake and body weight. Neuropeptides 33: 323–328, 1999 [DOI] [PubMed] [Google Scholar]
- 70.Smith GP, Epstein AN. Increased feeding in response to decreased glucose utilization in the rat and monkey. Am J Physiol 217: 1083–1087, 1969 [DOI] [PubMed] [Google Scholar]
- 71.Smith GP, Gibbs J. Satiating effect of cholecystokinin. Ann NY Acad Sci 713: 236–241, 1994 [DOI] [PubMed] [Google Scholar]
- 72.Smith GP, Root AW. Effect of feeding on hormonal responses to 2-Deoxy-d-glucose in conscious monkeys. Endocrinology 85: 963–966, 1969 [DOI] [PubMed] [Google Scholar]
- 73.Thombre DP, Bharathi S, Krishnamurthy N. Effects of central administration of insulin in normal and VMH (ventromedial hypothalamus) lesioned rats on food intake. Indian J Physiol Pharmacol 33: 207–210, 1989 [PubMed] [Google Scholar]
- 74.Tschop M, Castaneda TR, Joost HG, Thone-Reineke C, Ortmann S, Klaus S, Hagan MM, Chandler PC, Oswald KD, Benoit SC, Seeley RJ, Kinzig KP, Moran TH, Beck-sickinger AG, Koglin N, Rodgers RJ, Blundell JE, Ishii Y, Beattie AH, Holch P, Allison DB, Raun K, Madsen K, Wulff BS, Stidsen CE, Birringer M, Kreuzer OJ, Schindler M, Arndt K, Rudolf K, Mark M, Deng XY, Whitcomb DC, Halem H, Taylor J, Dong J, Datta R, Culler M, Craney S, Flora D, Smiley D, Heiman ML. Physiology: does gut hormone PYY3–36 decrease food intake in rodents? Nature 430: 1 p following 165; discussion 162; p following 165, 2004 [DOI] [PubMed] [Google Scholar]
- 75.Verbalis JG, McCann MJ, McHale CM, Stricker EM. Oxytocin secretion in response to cholecystokinin and food: differentiation of nausea from satiety. Science 232: 1417–1419, 1986 [DOI] [PubMed] [Google Scholar]
- 76.Verbalis JG, Richardson DW, Stricker EM. Vasopressin release in response to nausea-producing agents and cholecystokinin in monkeys. Am J Physiol Regul Integr Comp Physiol 252: R749–R753, 1987 [DOI] [PubMed] [Google Scholar]
- 77.Wahlsten D, Bachmanov A, Finn DA, Crabbe JC. Stability of inbred mouse strain differences in behavior and brain size between laboratories and across decades. Proc Natl Acad Sci USA 103: 16364–16369, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Walls EK, Wishart TB. Influence of intraventricular insulin on hypothalamic unit activity and deprivation-induced feeding. Fed Proc 42: 273, 1982 [Google Scholar]
- 79.Woods SC. The control of food intake: behavioral versus molecular perspectives. Cell Metab 9: 489–498, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Woods SC. The eating paradox: how we tolerate food. Psychol Rev 98: 488–505, 1991 [DOI] [PubMed] [Google Scholar]
- 81.Woods SC, Lotter EC, McKay LD, Porte D., Jr Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 282: 503–505, 1979 [DOI] [PubMed] [Google Scholar]
- 82.Woods SC, Lutz TA, Geary N, Langhans W. Pancreatic signals controlling food intake; insulin, glucagon and amylin. Philos Trans R Soc Lond B Biol Sci 361: 1219–1235, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Woods SC, Porte D, Jr, Bobbioni E, Ionescu E, Sauter JF, Rohner-Jeanrenaud F, Jeanrenaud B. Insulin: its relationship to the central nervous system and to the control of food intake and body weight. Am J Clin Nutr 42: 1063–1071, 1985 [DOI] [PubMed] [Google Scholar]
- 84.Woods SC, Seeley RJ. Insulin as an adiposity signal. Int J Obes Relat Metab Disord 25, Suppl 5: S35–S38, 2001 [DOI] [PubMed] [Google Scholar]
- 85.Woods SC, Seeley RJ, Porte D, Jr, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science 280: 1378–1383, 1998 [DOI] [PubMed] [Google Scholar]
- 86.Yalow RS, Berson SA. Immunoassay of plasma insulin in man. Diabetes 10: 339–344, 1961 [DOI] [PubMed] [Google Scholar]
- 87.Yoshimichi G, Lo CMC, Tamashiro KLK, Ma LY, Lee DM, Begg DP, Liu M, Sakai RR, Woods SC, Yoshimatsu H, Tso P. Effect of peripheral administration of cholecystokinin on food intake in apolipoprotein AIV knockout mice. Am J Physiol Gastrointest Liver Physiol 302: G1336–G1342, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang JV, Ren PG, Avsian-Kretchmer O, Luo CW, Rauch R, Klein C, Hsueh AJ. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin's effects on food intake. Science 310: 996–999, 2005 [DOI] [PubMed] [Google Scholar]