

Abstract
Nitric oxide (NO), synthesized by endothelial nitric oxide synthase (eNOS), exerts control over vascular function via two distinct mechanisms, the activation of soluble guanylate cyclase (sGC)/cGMP-dependent signaling or through S-nitrosylation of proteins with reactive thiols (S-nitrosylation). Previous studies in cultured endothelial cells revealed that eNOS targeted to the plasma membrane (PM) releases greater amounts of NO compared with Golgi tethered eNOS. However, the significance of eNOS localization to sGC-dependent or -independent signaling is not known. Here we show that PM-targeted eNOS, when expressed in human aortic endothelial cells (HAEC) and isolated blood vessels, increases sGC/cGMP signaling to a greater extent than Golgi-localized eNOS. The ability of local NO production to influence sGC-independent mechanisms was also tested by monitoring the secretion of Von Willebrand factor (vWF), which is tonically inhibited by the S-nitrosylation of N-ethylmaleimide sensitive factor (NSF). In eNOS “knockdown” HAECs, vWF secretion was attenuated to a greater degree by PM eNOS compared with a Golgi-restricted eNOS. Moreover, the PM-targeted eNOS induced greater S-nitrosylation of NSF vs. Golgi eNOS. To distinguish between the amount of NO generated and the intracellular location of synthesis, we expressed Golgi and PM-targeted calcium-insensitive forms of eNOS in HAEC. These constructs, which generate equal amounts of NO regardless of location, produced equivalent increases in cGMP in bioassays and equal inhibition of vWF secretion. We conclude that the greater functional effects of PM eNOS are due to the increased amount of NO produced rather than effects derived from the local synthesis of NO.
Keywords: nitric oxide, endothelial nitric oxide synthase, endothelium, nitrosylation, intracellular location, Von Willebrand factor
cardiovascular disease (CVD) remains the principal cause of death in both developed and developing countries, and almost 800,000 die annually in the United States from CVDs that include atherosclerosis, hypertension, congestive heart failure, and stroke (18). Endothelium-derived nitric oxide (NO) as synthesized by endothelial nitric oxide synthase (eNOS) plays a vital role in maintaining cardiovascular homeostasis and has potent effects on vascular tone, smooth muscle cell proliferation and migration, leukocyte adhesion, and platelet aggregation (9). Numerous studies have shown that eNOS is protective against pathological vascular remodeling, hypertension, and atherosclerosis (25, 34, 40). Moreover, reduced expression and dysregulation of eNOS, which results in the decreased synthesis of NO and the increased production of superoxide instead of NO, increases the severity of CVD (30, 31). However, the mechanisms by which eNOS affords vascular protection are incompletely understood.
Endothelial-derived NO controls vascular function via at least two distinct mechanisms. The first is the well-characterized activation of the soluble guanylate cyclase (sGC)-cyclic guanosine monophosphate (cGMP) signaling pathway (5, 10). This pathway has been established to mediate the NO-dependent relaxation of vascular smooth muscle and the ability of NO to suppress platelet aggregation (11). However, not all of the actions of NO are dependent on sGC/cGMP signaling. A second mechanism involves the ability of NO or its metabolites to react with free thiol groups on cysteine residues of target proteins in a process called S-nitrosylation (42). Protein S-nitrosylation is a posttranslational modification that has been compared with phosphorylation, where the NO-modified cysteine residue can induce allosteric changes in protein function. S-nitrosylation has been proposed to regulate a number of cellular processes such as apoptosis (6, 24), proliferation (20), nitric oxide synthase (NOS) activity (7), ion channel activity (45), transcription factor activity (32), proliferation (20), and protein secretion (28). An established target of S-nitrosylation is the N-ethylmaleimide sensitive factor (NSF), a plasma membrane (PM) protein that regulates the exocytosis of Weibel Palade bodies and ultimately the release of Von Willebrand factor (vWF) (28). The relative ability of PM- or Golgi-targeted eNOS to influence vWF secretion is not known.
It is not yet clear what factors distinguish signaling through cGMP-dependent vs. -independent pathways. Two potential contributing variables are the concentration and location of NO. The affinity of sGC for NO is very high; thus, low amounts of NO are sufficient to activate this pathway. This property of sGC enables endothelium-derived NO to function as an autacoid and activate sGC within adjacent vascular smooth muscle cells. In contrast, higher amounts of NO are required for S-nitrosylation, and, because of the rapid diffusibility of NO (27) and high cellular levels of glutathione, this may allow for the selective nitrosylation of proteins within close proximity to the source of NO synthesis (21). Indeed, eNOS is itself nitrosylated (8), and a number of other nitrosylated proteins can be found in a perinuclear distribution (17), but the respective contribution of eNOS localization and local NO production to sGC-dependent or -independent pathways remains poorly understood.
Intracellular localization is a key posttranslational mechanism by which the activity of eNOS is regulated. Within endothelial cells, eNOS is found primarily within cholesterol-rich microdomains of the PM such as caveolae and lipid rafts (39, 41) as well as intracellular membranes of organelles such as the perinuclear/Golgi complex (13, 29, 37). The activity of eNOS is strongly influenced by its subcellular localization, and, as such, restricting expression of eNOS to the cytosol, PM, or Golgi differentially regulates eNOS activity in a phosphorylation-dependent manner (12, 47). eNOS restricted to the PM was constitutively phosphorylated at S1179 and produces significantly more NO. In contrast, the Golgi pool of eNOS, while functional, produces less NO. In blood vessels in vivo, eNOS is also found at both the perinuclear/Golgi complex and the PM (1, 2), and the relative proportion of eNOS in each location can be influenced by the confluency of endothelial cells (15), blood vessel location (1), and shear stress (2). However, the functional significance of these two locations with regard to cGMP signaling or S-nitrosylation is poorly understood.
In the current study, we sought to determine whether localized production of NO exerts preferential control over endothelial function through cGMP pathways or through S-nitrosylation-dependent mechanisms, which was achieved by selective expression of Golgi vs. PM-restricted eNOS in eNOS null blood vessels and eNOS-depleted human endothelial cells.
MATERIALS AND METHODS
Cell culture and transfection.
Human aortic endothelial cells (HAECs or HAVSMC) were purchased from Cascade Biologics and grown in epidermal growth medium-2 or medium 231. COS-7 cells were grown in Dulbecco's modified Eagle's medium containing 100 U/ml penicillin, 100 mg/ml streptomycin, and 10% FCS. For transfection, COS-7 cells were seeded at a density of 2.5 × 105 cells/C12 well dish and transfected the next day using Lipofectamine 2000 (Invitrogen).
Generation of subcellular eNOS targeting fusion proteins.
eNOS fusion proteins that are targeted specifically to either the Golgi or the PM were generated previously (12). To generate eNOS constructs impervious to the RNAi no. 3122, several silent mutations, which do not change the amino acid sequence, were generated as described (47).
Adenoviral generation and transduction.
Replication-deficient adenoviruses encoding RNAi no. 3122 were generated using the Block-iT U6 RNAi Entry Vector system (Invitrogen). Adenoviruses encoding the control viruses green fluorescent protein or β-gal and Golgi and PM targeted eNOS were generated using the pAdDEST adenoviral expression system (Invitrogen). HAECs were seeded at a density of 2.5 × 105 cells/C12 well dish and transduced the next day at a multiplicity of infection (MOI) ranging from 12.5 to 125.
NO release.
Posttransfection or viral transduction (36 h), medium (100 μl) containing NO (primarily NO2−) was ethanol-precipitated to remove proteins and refluxed in sodium iodide/glacial acetic acid to convert NO2− to NO for measurement of the basal NO. Net NO release was calculated by NO-specific chemiluminescence after subtracting unstimulated basal release as described previously (14).
cGMP reporter assay.
HAECs were grown on collagen-treated glass cover slips, and, on the day of the experiment, cover slips were removed and placed over the top of confluent HAVSMCs, a source of sGC. HAECs were stimulated with 1 μM ionomycin in the presence of 100 U/ml superoxide dismutase and 300 μM isobutyl methylxanthine for 10 min. cGMP content was measured in the smooth muscle cells using a cGMP-specific enzyme immunoassay according to the manufacturer's instructions (Cayman).
Measurement of vWF secretion.
eNOS “knockdown” HAECs were transduced with adenoviruses encoding eNOS fusion proteins targeted to PM or Golgi, and, 36 h posttransduction, vWF accumulation in the medium was detected via a human specific vWF enzyme-linked immunosorbent assay (ELISA; American Diagnostica).
Detection of S-nitrosylation.
eNOS knockdown HAECs were transduced with adenoviruses encoding eNOS fusion proteins targeted to PM or Golgi, and, 36 h posttransduction, cells were stimulated with endothelial agonist vascular endothelial growth factor. Cells were then lysed and biotin-labeled using a commercially available assay (Cayman) modified from the procedure of Jaffrey and Snyder (22). Biotin-labeled proteins were pulled down by streptavidin-coated agarose beads (Pierce), size-fractionation via SDS-PAGE, and detected by chemiluminescence.
Adenoviral gene delivery in mouse aortic endothelium.
The adenoviral gene delivery technique was based on that described previously (47). Briefly, male C57bl6 mice (15–18 wk old) were anesthetized with ketamine/xylazine and exsanguinated followed by perfusion of physiological saline through the left ventricle. The aorta was cannulated, ∼20 μl of PBS containing different concentrations of adenoviruses (β-gal, Golgi, PM eNOS) were injected in the lumen of the aorta, and the vessel was tied off. The virus-filled vessel was incubated overnight in serum-free DMEM at 37°C, 5% CO2. All protocols using animals have been approved by the Institutional Animal Care and Use Committee, Medical College of Georgia.
Isometric studies.
Aortic rings (1 mm) were mounted on two wires in a 6-ml chamber vessel myograph (Danish Myo Technology), and basal tension was set at 1.0 gram. After a 45-min equilibration, rings were contracted with 3.2 M KCl two times until the contraction was reproducible. Concentration-response curves were then constructed to phenylephrine (PE, 10−9 to 10−5 M). To assess endothelial-dependent vasodilation, vessels were precontracted with a submaximal concentration of PE (10−6 M) before the serial application of the endothelium-dependent agonist, ACh (10−9 to 10−5 M) (43).
Statistical analysis.
Data are expressed as means ± SE. Comparisons were made using two-tailed Student's t-test or analysis of variance with a post hoc test where appropriate. Differences were considered as significant at P < 0.05.
RESULTS
To determine the importance of eNOS subcellular localization to endothelium-dependent relaxation in isolated blood vessels, the aortic endothelium of eNOS knockout mice was transduced via intraluminal delivery of Golgi (S17) or PM (CAAX) adenovirus, and vessels were incubated at 37°C overnight before analysis of vasomotor function. Aortic rings were precontracted with a submaximal concentration of PE (10−6 M), and endothelium-dependent relaxation was initiated with Ach (10−9 to10−5 M). As shown in Fig. 1A, PM eNOS generated greater ACh-induced relaxations compared with Golgi eNOS (EC50 2.63 × 10−8 vs. 3.1 × 10−7) and a greater maximum relaxation (52 ± 4% vs. 33 ± 8%). The ability to generate greater endothelium-dependent relaxations occurred with PM eNOS despite comparable to slightly reduced levels of expression (Fig. 1A, inset). To confirm that PM eNOS generates more biologically active NO, we expressed Golgi and PM-eNOS in HAEC lacking eNOS and measured cGMP accumulation in adjacent human aortic smooth muscle cells in a coculture bioassay. Consistent with the results shown in Fig. 1A, PM eNOS generated greater cGMP accumulation in adjacent smooth muscle cells. This increase in cGMP was completely inhibited by the NOS inhibitor, NG-nitro-l-arginine methyl ester indicating that it was derived from NO (Fig. 1B).
Fig. 1.
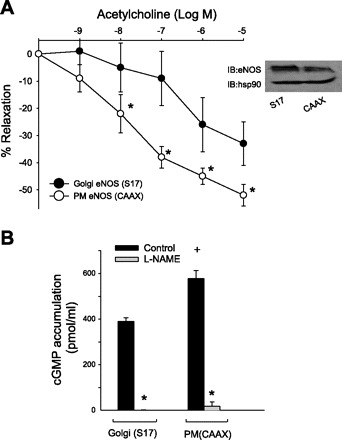
Plasma membrane (PM)-endothelial nitric oxide synthase (eNOS) generates more biologically active nitric oxide (NO) than Golgi eNOS. A: endothelium of eNOS knockout mice was transduced via luminal delivery of adenovirus encoding Golgi (S17) or PM (CAAX) eNOS. Aortic rings were precontracted with phenylephrine (PE, 10−6 M) followed by a dose-response curve to Ach (10−9 to 10−5 M). Data are presented as the percentage of PE-induced tone (%relaxation) (n = 5 experiments). Inset, the relative expression of eNOS protein was determined via Western blot. Heat shock protein (hsp) 90 was used as a loading control. *P < 0.05 for S17 vs. CAAX. B: eNOS “knockdown” human aortic endothelial cells (HAECs) were transduced with PM (CAAX) or Golgi (S17) eNOS adenovirus, and 36 h posttransduction were combined with HAVSMCs in a coculture bioassay in the presence and absence of NG-nitro-l-arginine methyl ester (l-NAME, 1 mM). cGMP content was measured using a cGMP specific enzyme immunoassay (EIA). Data were presented as means ± SE (n = 4). P < 0.05 vs. the control (*) and vs. Golgi (+).
We next investigated the role of PM and Golgi eNOS in the NO-dependent, but cGMP-independent, secretion of vWF. Knockdown HAECs were transduced with adenoviruses encoding Golgi (S17) and PM (CAAX) eNOS or β-gal control (MOI of 100), 36 h posttransduction, vWF was measured in the medium using ELISA, and the relative expression of eNOS and heat shock protein (hsp) 90 were determined via Western blot. eNOS expression was absent from eNOS knockdown HAECs transduced with the control (Bgal) adenovirus, whereas the level of eNOS (with overall protein loading controlled for by hsp90) was consistent between Golgi and PM eNOS (Fig. 2A, bottom). vWF production was greatly attenuated in HAECs expressing PM eNOS compared with cells expressing Golgi-restricted eNOS (Fig. 2A), which was coincident with enhanced NO production caused by expression of PM eNOS relative to Golgi eNOS (Fig. 2B). These data suggest that eNOS at the PM more effectively inhibits vWF production than when localized at the Golgi, which may be a consequence of increased NO production at the PM.
Fig. 2.
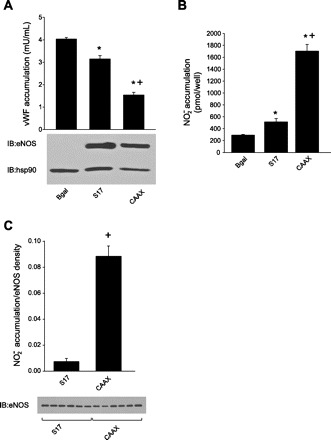
PM eNOS is more effective at inhibiting Von Willebrand factor (vWF) secretion vs. Golgi eNOS. Knockdown HAECs were transduced with adenoviruses encoding Golgi (S17) and PM (CAAX) eNOS or β-gal control [multiplicity of infection (MOI) of 100]. A: 36 h posttransduction, vWF levels were measured in culture medium via enzyme-linked immunosorbent assay (ELISA), and the relative expression of eNOS and hsp90 was determined via Western blot (bottom). B: basal (unstimulated) NO release was measured by NO-specific chemiluminescence. Data are presented as means ± SE (n = 4). P < 0.05 vs. β-gal control (*) and vs. Golgi (+). C: densitometric analysis of NO released from eNOS knockdown HAECs expressing Golgi (S17) or PM (CAAX). Data are presented as means ± SE (n = 6). +P < 0.05 vs. Golgi.
To determine if the effects of NO on vWF secretion were dependent on the location of synthesis or the amount, we next expressed calcium-insensitive forms of eNOS that produce equal amounts of NO regardless of their intracellular location in eNOS knockdown HAEC (3, 23). These constructs are derived from the deletion of two key autoinhibitory domains of 45 and 14 amino acids in length and thus are labeled Δ45/Δ14 eNOS. As shown in Fig. 3A, 36 h posttransduction there was no difference between Golgi and PM-targeted eNOS in their ability to inhibit vWF secretion. The expression levels of Golgi and PM Δ45/Δ14 eNOS and hsp90 loading control were equivalent (Fig. 3A, bottom). To confirm that these constructs produce equivalent amounts of NO, we measured NO release from reconstituted HAEC using chemiluminescence. As shown in Fig. 3B, there was no difference in the NO released from HAEC-expressing PM eNOS vs. Golgi eNOS. Because this assay measures metabolites of NO and is an index of overall NOS activity (48), we next determined the ability of Golgi- and PM-restricted calcium-insensitive eNOS to deliver biologically active NO to adjacent cells. As shown in Fig. 3C, the ability of Golgi and PM Δ45/Δ14 eNOS to elevate cGMP in HAVSMC was similar. The ability of S17 and CAAX eNOS to repress vWF secretion was not due to effects on the sGC/cGMP signaling pathway, since the sGC inhibitor ODQ did not modify this ability (Fig. 4A) or influence NO release (Fig. 4B).
Fig. 3.
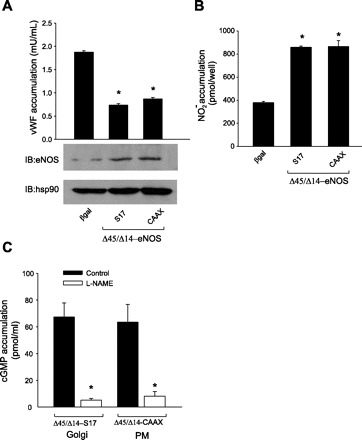
Amount of NO produced is more important for the biological effects of NO than the location of its synthesis. eNOS knockdown HAECs were transduced with calcium/calmodulin (CaM)-insensitive eNOS constructs targeted to the Golgi (Δ45/Δ14 S17) or PM (Δ45/Δ14 CAAX) as well as β-gal control. A: 36 h posttransduction, vWF was measured in the medium via ELISA, and the relative expressions of eNOS and hsp90 were determined via Western blot (bottom). B: 36 h posttransduction, basal NO release was measured by NO-specific chemiluminescence. Data are presented as means ± SE (n = 4). *P < 0.05 vs. the β-gal control. C: eNOS knockdown HAECs were grown on cover slips and transduced with Golgi- and PM-targeted calcium/calmodulin-independent (Δ45/Δ14) eNOS. Posttransfection (36 h), the cover slips were placed over the top of confluent HAVSMCs in the presence or absence of l-NAME (1 mM). cGMP levels in the reporter cells were quantified via EIA. Data were presented as means ± SE (n = 4). *P < 0.05 vs. the control.
Fig. 4.
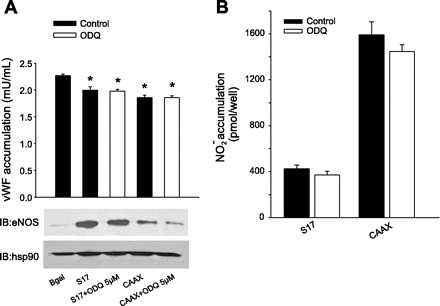
PM and Golgi eNOS-dependent suppression of vWF secretion is independent of sGC. A: eNOS knockdown HAECs were transduced with adenoviruses encoding Golgi (S17) and PM (CAAX) eNOS or β-gal control. Cells were then incubated with or without the sGC inhibitor ODQ (5 μM). Posttransduction (36 h), vWF was measured in culture medium using a vWF specific ELISA, and the relative expressions of eNOS and hsp90 were determined via Western blot (bottom). Data are presented as means ± SE (n = 4). *P < 0.05 vs. the β-gal control. B: basal NO release 36 h posttransduction was measured by NO-specific chemiluminescence. Data are presented as means ± SE (n = 6).
Finally, to address whether the ability of PM eNOS to elicit greater inhibition of vWF secretion was due to its ability to nitrosylate NSF, we performed a biotin-switch assay in a heterologous expression system in COS-7 cells. COS-7 cells were cotransfected with S17 or CAAX eNOS together with NSF. We found that the S-nitrosylation of NSF was greater in cells expressing PM vs. Golgi eNOS (Fig. 5, top) relative to the level of total NSF (Fig. 5, bottom).
Fig. 5.
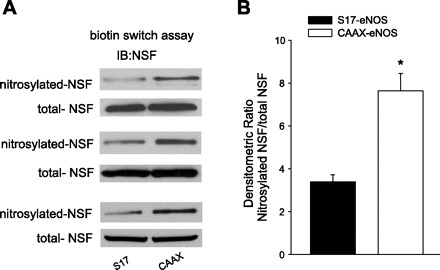
PM eNOS induces greater S-nitrosylation of N-ethylmaleimide sensitive factor (NSF) vs. Golgi eNOS. A: COS-7 cells were cotransfected with cDNAs encoding Golgi (S17) or PM (CAAX) eNOS together with HA-NSF. Posttransfection (36 h), cells were stimulated with ionomycin (1 μM) for 10 min, and S-nitrosylated proteins were detected using the biotin-switch assay (top) followed by immunoblotting for total precipitated NSF (bottom). B: densitometric analysis of NSF nitrosylation vs. total NSF. Data are presented as means ± SE (n = 4). *P < 0.05 vs. the Golgi eNOS.
DISCUSSION
eNOS is a dually acylated protein that is modified by cotranslational NH2-myristoylation and posttranslational palmitoylation. These modifications are responsible for targeting eNOS to the PM and also to intracellular membranes, including the Golgi (26, 33, 36, 39). In endothelial cells in culture and also in isolated blood vessels, the majority of eNOS is present in these two locations (1, 13, 16, 44). The goal of the current study was to identify whether residence at the Golgi or the PM influences the ability of eNOS to elicit NO signaling via cGMP-dependent or -independent mechanisms in endothelial cells.
The intracellular location of eNOS is a dynamic regulator of its activity. The loss of fatty acylation prevents proper membrane targeting and reduces eNOS activity (36). Furthermore, the targeting of eNOS to distinct regions of the cell can greatly increase or decrease its activity. The PM is the site of highest eNOS activity followed by the Golgi, and activity is considerably blunted in areas where eNOS is normally absent, including the cytosol, mitochondria, and nucleus (12, 23, 47). Thus location is clearly an important factor for regulating eNOS activity, but much less is known about the contribution of the eNOS intracellular location to downstream NO signal transduction. In the current study, we found that, compared with the Golgi, the PM location of eNOS results in a greater ability to elicit cGMP-dependent signaling and endothelium-dependent relaxation of isolated blood vessels. The ability of PM eNOS to elicit more effective endothelium-dependent relaxations and greater increases in cGMP accumulation is perhaps not surprising given the extraordinary sensitivity of sGC for NO (35) and most likey reflects the increased NO production from this location (12). Although a PM location favors the highest output of NO from eNOS, it is also the most susceptible to extracellular influences such as oxidized low-density lipoprotein (LDL), which selectively reduces PM NO release (38, 47). eNOS restricted to the Golgi is resistant to the actions of oxidized LDL and in this report we show for the first time that a Golgi-restricted eNOS is also capable of supplying biologically active NO to adjacent smooth muscle cells and mediating endothelium-dependent relaxation. Although eNOS is generally regarded as being protective in murine models of atherosclerosis (25), it is not yet known whether a Golgi location of eNOS would offer more protection against lesion formation vs. the PM.
In addition to cGMP signaling, we also found that the PM eNOS was more effective at supressing the secretion of vWF from endothelial cells compared with Golgi-targeted eNOS. Previous studies have shown that eNOS activity in the Golgi results in the compartmentalization of NO and S-nitrosylation of local proteins (21). This has prominent effects on the vessicular traffic of local proteins and suggests that the location of eNOS can influence discrete signaling events. Compartmentalization of protein S-nitrosylation is also evident in neurons where a number of accessory proteins in proximity to neuronal NOS are selectively S-nitrosylated (19). In our study, the PM-targeted eNOS was able to induce greater S-nitrosylation of NSF, which reduces the exocytosis of vWF (28). However, one of the most robust influences on the S-nitrosylation of proteins is also the amount or concentration of NO. Endogenous levels of S-nitrosylation are difficult to detect and are far below that which can be achieved using an extracellular NO donor (22), and, in endothelial cells, PM eNOS releases more NO than the Golgi eNOS.
To distinguish between effects dependent on the amount of NO vs. the location of its synthesis, we expressed Golgi- and PM-targeted calcium-insensitive forms of eNOS in HAECs. These constructs produce equal amounts of NO regardless of intracellular location (3, 23), and, when the amount of NO produced is equalized between the Golgi and PM, the ehanced ability of PM eNOS to elicit cGMP signaling or inhibit vWF secretion is lost. Although these results provide evidence against the importance of location in NO signaling, it is possible that the greater amounts of NO generated by PM eNOS vs. Golgi eNOS in our study result in a larger sphere of influence and less compartmentalization of protein S-nitrosylation. It is also possible that the Golgi- and PM-targeted calcium-insensitive eNOS constructs act on distinct local targets to produce the same inhibitory effect on vWF secretion, although evidence to this effect is lacking. There is also considerable evidence that the protein S-nitrosylation in response to extracellular NO donors exhibits a perference for certain areas of the cell, particularly in the perinuclear Golgi/ER and mitochrondria (17, 46). Others have shown that the S-nitrosylation of eNOS itself varies with location and eNOS activity. Cytosolic eNOS, which produces much less NO, is hyponitrosylated compared with wild-type eNOS, which itself is less nitrosylated compared with a more active, membrane-targeted eNOS (8). Thus, although location is an important factor in the selective S-nitroslyation of proteins, there are clearly other factors at play. One of the reasons that higher concentrations of NO are needed for S-nitrosylation is the high level of free thiols in the cell (4); thus, it is reasonable to speculate that the selective nitrosylation of proteins such as NSF is more likely derived from the combined effects of the amount of NO produced and the location of the target protein rather than the location of NO synthesis.
In conclusion, the functional significance of the increased NO produced by the PM eNOS is reflected in the greater ability to elicit endothelium-dependent relaxation through the sGC-cGMP pathway and the greater suppression of vWF secretion through S-nitrosylation. Mechanistically, these effects of PM eNOS are due to the amount of NO produced rather than the PM location of its synthesis.
DISCLOSURES
No conflicts of interest are declared by the authors.
Footnotes
J. Qian and Q. Zhang contributed equally to this work.
REFERENCES
- 1.Andries LJ, Brutsaert DL, Sys SU Nonuniformity of endothelial constitutive nitric oxide synthase distribution in cardiac endothelium. Circ Res 82: 195–203, 1998 [DOI] [PubMed] [Google Scholar]
- 2.Cheng C, van Haperen R, de Waard M, van Damme LC, Tempel D, Hanemaaijer L, van Cappellen GW, Bos J, Slager CJ, Duncker DJ, van der Steen AF, de Crom R, Krams R Shear stress affects the intracellular distribution of eNOS: direct demonstration by a novel in vivo technique. Blood 106: 3691–3698, 2005 [DOI] [PubMed] [Google Scholar]
- 3.Church JE, Fulton D Differences in eNOS activity because of subcellular localization are dictated by phosphorylation state rather than the local calcium environment. J Biol Chem 281: 1477–1488, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Clementi E, Brown GC, Feelisch M, Moncada S Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA 95: 7631–7636, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Denninger JW, Marletta MA Guanylate cyclase and the NO/cGMP signaling pathway. Biochim Biophys Acta 1411: 334–350, 1999 [DOI] [PubMed] [Google Scholar]
- 6.Dimmeler S, Haendeler J, Nehls M, Zeiher AM Suppression of apoptosis by nitric oxide via inhibition of interleukin-1beta-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J Exp Med 185: 601–607, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erwin PA, Lin AJ, Golan DE, Michel T Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem 280: 19888–19894, 2005 [DOI] [PubMed] [Google Scholar]
- 8.Erwin PA, Mitchell DA, Sartoretto J, Marletta MA, Michel T Subcellular targeting and differential S-nitrosylation of endothelial nitric-oxide synthase. J Biol Chem 281: 151–157, 2006 [DOI] [PubMed] [Google Scholar]
- 9.Forstermann U, Munzel T Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113: 1708–1714, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Friebe A, Koesling D Regulation of nitric oxide-sensitive guanylyl cyclase. Circ Res 93: 96–105, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Friebe A, Mergia E, Dangel O, Lange A, Koesling D Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. Proc Natl Acad Sci USA 104: 7699–7704, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fulton D, Babbitt R, Zoellner S, Fontana J, Acevedo L, McCabe TJ, Iwakiri Y, Sessa WC Targeting of endothelial nitric-oxide synthase to the cytoplasmic face of the Golgi complex or plasma membrane regulates Akt- vs. calcium-dependent mechanisms for nitric oxide release. J Biol Chem 279: 30349–30357, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Fulton D, Fontana J, Sowa G, Gratton JP, Lin M, Li KX, Michell B, Kemp BE, Rodman D, Sessa WC Localization of endothelial nitric-oxide synthase phosphorylated on serine 1179 and nitric oxide in Golgi and plasma membrane defines the existence of two pools of active enzyme. J Biol Chem 277: 4277–4284, 2002 [DOI] [PubMed] [Google Scholar]
- 14.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399: 597–601, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Govers R, Bevers L, de Bree P, Rabelink TJ Endothelial nitric oxide synthase activity is linked to its presence at cell-cell contacts. Biochem J 361: 193–201, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Govers R, van der Sluijs P, van Donselaar E, Slot JW, Rabelink TJ Endothelial nitric oxide synthase and its negative regulator caveolin-1 localize to distinct perinuclear organelles. J Histochem Cytochem 50: 779–788, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Greco TM, Hodara R, Parastatidis I, Heijnen HF, Dennehy MK, Liebler DC, Ischiropoulos H Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc Natl Acad Sci USA 103: 7420–7425, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heron M, Hoyert DL, Murphy SL, Xu J, Kochanek KD, Tejada-Vera B Deaths: Final Data for 2006 National Vital Statistics. Reports 57: 1–80, 2009 [PubMed] [Google Scholar]
- 19.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 6: 150–166, 2005 [DOI] [PubMed] [Google Scholar]
- 20.Ignarro LJ, Buga GM, Wei LH, Bauer PM, Wu G, del Soldato P Role of the arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc Natl Acad Sci USA 98: 4202–4208, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, Groszmann RJ, Shah VH, Sessa WC Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci USA 103: 19777–19782, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaffrey SR, Snyder SH The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE 2001: PL1, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Jagnandan D, Sessa WC, Fulton D Intracellular location regulates calcium-calmodulin-dependent activation of organelle-restricted eNOS. Am J Physiol Cell Physiol 289: C1024–C1033, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, Mukhopadhyay D, Shah V Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. J Cell Sci 120: 492–501, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation 104: 448–454, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Hughes TE, Sessa WC The first 35 amino acids and fatty acylation sites determine the molecular targeting of endothelial nitric oxide synthase into the Golgi region of cells: a green fluorescent protein study. J Cell Biol 137: 1525–1535, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Miller MJ, Joshi MS, Sadowska-Krowicka H, Clark DA, Lancaster JR, Jr Diffusion-limited reaction of free nitric oxide with erythrocytes. J Biol Chem 273: 18709–18713, 1998 [DOI] [PubMed] [Google Scholar]
- 28.Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, Hara MR, Quick RA, Cao W, O'Rourke B, Lowenstein JM, Pevsner J, Wagner DD, Lowenstein CJ Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell 115: 139–150, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Brien AJ, Young HM, Povey JM, Furness JB Nitric oxide synthase is localized predominantly in the Golgi apparatus and cytoplasmic vesicles of vascular endothelial cells. Histochem Cell Biol 103: 221–225, 1995 [DOI] [PubMed] [Google Scholar]
- 30.Oemar BS, Tschudi MR, Godoy N, Brovkovich V, Malinski T, Luscher TF Reduced endothelial nitric oxide synthase expression and production in human atherosclerosis. Circulation 97: 2494–2498, 1998 [DOI] [PubMed] [Google Scholar]
- 31.Ozaki M, Kawashima S, Yamashita T, Hirase T, Namiki M, Inoue N, Hirata K, Yasui H, Sakurai H, Yoshida Y, Masada M, Yokoyama M Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J Clin Invest 110: 331–340, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palmer LA, Gaston B, Johns RA Normoxic stabilization of hypoxia-inducible factor-1 expression and activity: redox-dependent effect of nitrogen oxides. Mol Pharmacol 58: 1197–1203, 2000 [DOI] [PubMed] [Google Scholar]
- 33.Robinson LJ, Michel T Mutagenesis of palmitoylation sites in endothelial nitric oxide synthase identifies a novel motif for dual acylation and subcellular targeting. Proc Natl Acad Sci USA 92: 11776–11780, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS, Sessa WC Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J Clin Invest 101: 731–736, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russwurm M, Behrends S, Harteneck C, Koesling D Functional properties of a naturally occurring isoform of soluble guanylyl cyclase. Biochem J 335: 125–130, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sessa WC, Barber CM, Lynch KR Mutation of N-myristoylation site converts endothelial cell nitric oxide synthase from a membrane to a cytosolic protein. Circ Res 72: 921–924, 1993 [DOI] [PubMed] [Google Scholar]
- 37.Sessa WC, Garcia-Cardena G, Liu J, Keh A, Pollock JS, Bradley J, Thiru S, Braverman IM, Desai KM The Golgi association of endothelial nitric oxide synthase is necessary for the efficient synthesis of nitric oxide. J Biol Chem 270: 17641–17644, 1995 [DOI] [PubMed] [Google Scholar]
- 38.Shaul PW Regulation of endothelial nitric oxide synthase: location, location, location. Annu Rev Physiol 64: 749–774, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Shaul PW, Smart EJ, Robinson LJ, German Z, Yuhanna IS, Ying Y, Anderson RG, Michel T Acylation targets emdothelial nitric-oxide synthase to plasmalemmal caveolae. J Biol Chem 271: 6518–6522, 1996 [DOI] [PubMed] [Google Scholar]
- 40.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA 93: 13176–13181, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sowa G, Pypaert M, Sessa WC Distinction between signaling mechanisms in lipid rafts vs. caveolae Proc Natl Acad Sci USA 98: 14072–14077, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stamler JS, Lamas S, Fang FC Nitrosylation, the prototypic redox-based signaling mechanism. Cell 106: 675–683, 2001 [DOI] [PubMed] [Google Scholar]
- 43.Stepp DW, Tulenko TN Alterations in basal and serotonin-stimulated calcium permeability and vasoconstriction in atherosclerotic aorta. Arterioscler Thromb 14: 1854–1859, 1994 [DOI] [PubMed] [Google Scholar]
- 44.van Haperen R, Cheng C, Mees BM, van Deel E, de Waard M, van Damme LC, van Gent T, van Aken T, Krams R, Duncker DJ, de Crom R Functional expression of endothelial nitric oxide synthase fused to green fluorescent protein in transgenic mice. Am J Pathol 163: 1677–1686, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu L, Eu JP, Meissner G, Stamler JS Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279: 234–237, 1998 [DOI] [PubMed] [Google Scholar]
- 46.Yang Y, Loscalzo J S-nitrosoprotein formation and localization in endothelial cells. Proc Natl Acad Sci USA 102: 117–122, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Q, Church JE, Jagnandan D, Catravas JD, Sessa WC, Fulton D Functional relevance of Golgi- and plasma membrane-localized endothelial NO synthase in reconstituted endothelial cells. Arterioscler Thromb Vasc Biol 26: 1015–1021, 2006 [DOI] [PubMed] [Google Scholar]
- 48.Zhang Q, Malik P, Pandey D, Gupta S, Jagnandan D, Belin de Chantemele E, Banfi B, Marrero MB, Rudic RD, Stepp DW, Fulton DJ Paradoxical activation of endothelial nitric oxide synthase by NADPH oxidase. Arterioscler Thromb Vasc Biol 28: 1627–1633, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]