

Abstract
Sleep is required for, and sleep loss impairs, normal hippocampal synaptic N-methyl-d-aspartate (NMDA) glutamate receptor function and expression, hippocampal NMDA receptor-dependent synaptic plasticity, and hippocampal-dependent memory function. Although sleep is essential, the signals linking sleep to hippocampal function are not known. One potential signal is growth hormone. Growth hormone is released during sleep, and its release is suppressed during sleep deprivation. If growth hormone links sleep to hippocampal function, then restoration of growth hormone during sleep deprivation should prevent adverse consequences of sleep loss. To test this hypothesis, we examined rat hippocampus for spontaneous excitatory synaptic currents in CA1 pyramidal neurons, long-term potentiation in area CA1, and NMDA receptor subunit proteins in synaptic membranes. Three days of sleep deprivation caused a significant reduction in NMDA receptor-mediated synaptic currents compared with control treatments. When rats were injected with growth hormone once per day during sleep deprivation, the loss of NMDA receptor-mediated synaptic currents was prevented. Growth hormone injections also prevented the impairment of long-term potentiation that normally follows sleep deprivation. In addition, sleep deprivation led to a selective loss of NMDA receptor 2B (NR2B) from hippocampal synaptic membranes, but normal NR2B expression was restored by growth hormone injection. Our results identify growth hormone as a critical mediator linking sleep to normal synaptic function of the hippocampus.
Keywords: hippocampal area CA1, long-term potentiation, NMDA receptor, sleep deprivation
sleep is a critical regulator of biological functions, including brain function, yet the signals linking sleep to brain function are not known. Previous investigations showed that growth hormone, which is normally secreted during sleep and reduced during sleep deprivation (5, 15, 21, 52), regulates synaptic function in the hippocampus (33, 57). Growth hormone could therefore be a link between sleep and brain function, but this possibility has not yet been tested. Growth hormone is released from somatotroph cells in the anterior pituitary and is essential for somatic growth and metabolism (18). Recent studies have shown that growth hormone can cross the blood-brain barrier (9, 19, 42) and that the growth hormone receptor is expressed in several brain regions including the hippocampus (24, 30). Growth hormone effects on brain and cognitive function (41) may involve growth hormone regulation of synaptic plasticity in the hippocampus and hippocampal-dependent learning and memory (25, 27, 43). In the hippocampus, N-methyl-d-aspartate (NMDA) receptors play critical roles in memory formation and induction of long-term potentiation (LTP), a widely used synaptic model of learning and memory (4, 31). Growth hormone supplementation in chronic growth hormone deficiency or aged animals alters expression of specific NMDA receptor (NMDAR) subunit mRNAs in the hippocampus (25, 27), supporting a link between growth hormone and brain function.
Secretion of pituitary growth hormone is strongly related to sleep. A major surge of growth hormone secretion occurs during sleep, and sleep deprivation suppresses growth hormone secretion (5, 15, 21, 52). Sleep deprivation itself has long been associated with impairment of learning and memory (36, 47, 55, 56). Also, like growth hormone, sleep deprivation affects hippocampal synaptic plasticity by altering NMDAR expression and function (7, 23, 37). These sleep deprivation-induced changes in NMDAR function may underlie the LTP deficit and learning and memory impairment following sleep deprivation (6, 12, 20, 37). It seemed likely, therefore, that loss of normal growth hormone might cause the altered hippocampal NMDAR expression and function and impaired NMDAR-dependent synaptic plasticity that occur as a consequence of sleep deprivation. To test this hypothesis, we treated animals with growth hormone during sleep deprivation, and examined hippocampal NMDAR-mediated synaptic currents, LTP, and NMDAR subunit expression in synaptosomal membranes. We found that growth hormone restoration to sleep-deprived animals restored normal NMDAR function and subunit expression, indicating that growth hormone is an essential mediator of sleep effects on hippocampal memory-related functions.
MATERIALS AND METHODS
Animals and treatment.
All procedures were approved by the Institutional Animal Care and Use Committee at Marshall University.
Sleep deprivation was produced as described previously (14). Male Sprague-Dawley rats (270–350 g) were sleep deprived by confinement on small (10.5 diameter) platforms over water for 3 days. This technique, which very effectively deprives animals of almost all rapid eye movement (REM) sleep and also fragments and reduces non-REM sleep states (17, 32, 39, 53), suppress the normal sleep-related surge in growth hormone release (52). In the first set of experiments, two controls were used: large platform control rats (control) were placed on 28-cm diameter platforms (20), and naive rats were housed individually in standard animal cages. A total of 14 animals were used in this experiment. In the second set of experiments, four groups of animals were examined: animals received either growth hormone (1 mg·kg−1·day−1 sc) or vehicle (sterile saline, 1 ml·kg−1·day−1 sc) injections and were subjected to either sleep deprivation (small platform) or control (large platform) treatment. Our growth hormone injection protocol was chosen based on a review of the literature and should have produced a peak circulating growth hormone concentration comparable to the large-amplitude growth hormone surges that occur during normal sleep (29, 50). Growth hormone-injected, sleep-deprived animals were treated with recombinant human growth hormone (Santa Cruz Biotechnology, Santa Cruz, CA or Cell Sciences, Canton, MA) in sterile saline, and were sleep deprived for 3 days. Growth hormone-injected control animals also received daily growth hormone injections, but were subjected to the large platform control treatment for 3 days. Vehicle-injected, sleep-deprived animals received equivalent volume sterile saline injections during 3 days of sleep deprivation. Vehicle-injected control animals received sterile saline injections and large platform control treatment. A total of 38 animals were used in this experiment. Naive controls were not used in the second set of experiments because the first set of experiments revealed no differences between control and naive treatment (see results). Growth hormone or saline injections were given between 9 and 11 AM on each of the 3 days of sleep deprivation or control treatment. Food and clean water were available ad libitum for all animals throughout all phases of treatment, and all animals were maintained under 12:12-h light-dark conditions with lights on at 8 AM. In preliminary experiments, we verified REM sleep deprivation by the presence of a REM sleep rebound during recovery sleep following confinement on small platforms (as described in Ref. 14) and the absence of REM sleep rebound following confinement on large platforms. To the degree possible, we minimized use of animals by collecting hippocampal and blood samples from the same animals that were used to prepare slices for electrophysiology (see below).
Slice preparation.
Rats were sedated by CO2/air inhalation and decapitated. The brain was removed and placed into chilled artificial cerebrospinal fluid (ACSF) composed of (in mM) 124 NaCl, 26 NaHCO3, 3.4 KCl, 1.2 NaH2PO4, 2.0 CaCl2, 2.0 MgSO4, 10 glucose, pH 7.35, equilibrated with 95% O2-5% CO2. The brain was cut in half along the longitudinal fissure. The hippocampus was removed from one of the hemispheres and immediately frozen on dry ice. The other hemisphere was trimmed and glued to the stage of a vibrating microtome (Campden Instruments), immersed in chilled ACSF, and sectioned into 400-μm thick coronal slices. Transverse hippocampal slices were dissected free from surrounding structures and stored at room temperature (20–22°C) in an interface holding chamber. Individual slices were transferred as needed to a small volume (∼200 μl) interface recording chamber with oxygenated ACSF (35°C, perfusion rate: 1–1.5 ml/min).
Electrophysiology.
Somatic whole cell patch-clamp recordings were obtained from a total of 81 CA1 pyramidal neurons from 81 hippocampal slices, as described previously (33). Patch electrodes (3–4 MΩ) were filled with 140 mM cesium gluconate, 10 mM sodium HEPES, and 3 mM MgCl2 adjusted to 285–290 mOsm, pH 7.2. Recordings were done in the continuous voltage clamp mode of an Axoclamp 2B (Axon Instruments). Signals were amplified (gain 10), low-pass filtered (3 kHz), and then digitized 10–100 kHz (National Instruments) and stored on a personal computer using WinWCP or WinEDR programs (Strathclyde Electrophysiology Software, John Dempster, University of Strathclyde, http://spider.science.strath.ac.uk/sipbs/software_ses.htm). Holding potentials were set to between −70 and −85 mV, corrected for a calculated liquid junction potential of 10 mV, at the beginning of each recording and kept constant for the remainder of the recording. Excitatory postsynaptic currents (EPSCs) were recorded in low-Mg2+ (50 μM) ACSF containing 10 μM bicuculline to block GABAA receptors. EPSCs were recorded for a 5 to 10 min baseline period, and then d(−)-2-amino-5-phosphonopentanoic acid (d-AP5; 50 μM, 10 min) was added to block NMDAR-mediated currents. Finally, washout of d-AP5 was attempted, but was not obtained in all recordings due to the long time (20 min) required for complete washout. Spontaneous EPSCs were detected and analyzed by using the Mini Analysis program (Synaptosoft) with detection threshold set to four times the root mean square baseline noise level. The detection threshold was determined separately for each cell at the beginning of the recording, and the same threshold was used for analyzing all portions of that recording. EPSCs from the final 5-min periods of the baseline recording, d-AP5 application, and washout (when obtained) were aligned and averaged separately. NMDAR-mediated synaptic current was quantified by comparing half-widths of the averaged EPSC in d-AP5 with the averaged baseline EPSC.
LTP was assessed by field potential recordings from a total of 35 slices. Extracellular potentials were recorded through low-impedance (3–4 MΩ) glass micropipettes filled with ACSF and placed into the stratum radiatum of area CA1. Signals were amplified (gain 100) and filtered (0.05–3,000 Hz or 0.1–10,000 Hz) and then digitized (10–100 kHz; National Instruments) and stored on a personal computer using WinWCP. Baseline responses were recorded for 15 min, and then theta burst stimulation (20 bursts, each consisting of 4 stimuli at 100 Hz, at 200 ms interburst intervals) was given to induce LTP. Recordings were continued for 60 min following LTP induction. For each slice, EPSP slopes were measured and normalized relative to the mean slope during the pretetanus baseline and then expressed as percentage change from the baseline. For statistical analysis, the percent change in EPSP slope was averaged over the 50–60 min posttetanus periods, and comparisons were made between slices from control animals and slices from sleep-deprived animals.
Synaptic membrane preparation and Western blot analysis.
A total of 20 animals were used. Synaptic membranes were obtained as described earlier (16). Dounce homogenates were prepared from hippocampal tissues in sucrose buffer containing 10 mM Tris·HCl (pH 7.4), 320 mM sucrose, and phosphatase and protease inhibitor cocktails (Sigma, Roche). Homogenates were centrifuged at 1,000 g for 10 min to remove nuclei and large debris (P1). The supernatant (S1) was centrifuged at 10,000 g for 15 min to obtain a crude synaptosomal membrane fraction (P2). The P2 fraction was lysed hypoosmotically in water containing phosphatase and protease inhibitors for 30 min and then centrifuged at 25,000 g for 20 min to yield the synaptosomal membrane fraction (LP1). Pellets were rinsed with cold sucrose buffer after each centrifugation. Synaptosomal membranes were resuspended in RIPA buffer (150 mM NaCl, 50 mM Tris, 10 mM EDTA, 0.5% sodium deoxycholate, 1% Igepal CA-630, 0.1% SDS) and frozen at −80°C for later analysis.
For Western blot analysis, 30 μg of synaptosomal membrane protein were loaded onto 8% SDS-PAGE gels. Proteins were separated by electrophoresis and transferred to nitrocellulose membranes. Membranes were blocked at room temperature with 2% Advance blocking reagent (Amersham/GE Healthcare) in TTS (0.5% Tween 20, 10 mM Tris·HCl, pH 8.0, 150 mM NaCl, 0.2 mM EDTA). Membranes were then incubated with primary antibody to NMDA receptor subunit 1 (NR1; BD Biosciences), NMDA receptor subunit 2A (NR2A; Chemicon/Millipore), or NR2B (BD Biosciences) diluted in TTS either at room temperature for 1 h or overnight at 4°C. After being washed in TTS, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies in TTS for 1 h at room temperature or overnight at 4°C. The blot was washed, and proteins were detected on X-ray film using the ECL Advance system (Amersham/GE Healthcare). Films were scanned and analyzed using Image J software (Wayne Rasband, National Institute of Mental Health, Washington, DC). Blots were stripped (Pierce Restore stripping buffer) and reprobed for other NMDAR subunits and for postsynaptic density protein-95 (PSD-95; Upstate/Millipore, Billerica, MA).
Measurement of serum corticosterone and IGF-I.
Trunk blood was collected at the time of death of animals, kept on ice for 30 min, and then centrifuged at 1,000 g for 15 min to separate serum. Serum corticosterone (AC-14F1; Immunodiagnostic Systems, Scottsdale, AZ) and IGF-1 (AC-18F1; Immunodiagnostic Systems) were measured by ELISA, following the manufacturer's instructions.
Statistical analysis.
All data are presented as means ± 1 SE. For tests of statistical significance, we used one- or two-way ANOVA as appropriate. When the ANOVA indicated a significant effect, post hoc pairwise comparisons were made using the Bonferroni method. An alpha level of 0.05 was considered significant. Analysis was performed using Gnumeric (http://www.gnome.org/projects/gnumeric/), and R (http://www.r-project.org/) software.
RESULTS
Synaptic NMDAR function was impaired by sleep deprivation.
In an initial set of experiments, we assessed NMDAR synaptic function in sleep-deprived, control, and naive animals. To assess NMDAR synaptic function, we examined spontaneous EPSCs (sEPSCs) in CA1 pyramidal neurons. sEPSCs were recorded from slices in low-Mg2+ (50 μM) ACSF with GABA receptors blocked by the combination of extracellular bicuculline and intracellular Cs+. sEPSCs recorded under these conditions contained both fast, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR)-mediated, and slow, NMDAR-mediated components (see Fig. 1). Application of the NMDAR antagonist, d-AP5 (50 μM), allowed assessment of NMDAR synaptic function by comparing the difference in sEPSCs duration at half amplitude (half-width) before and after block of NMDARs.
Fig. 1.
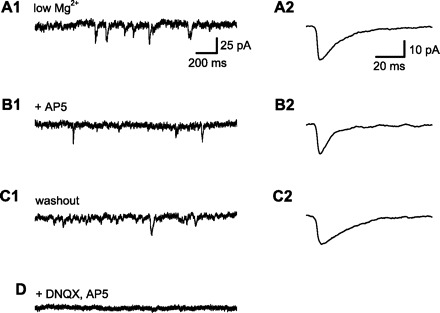
Measurement of N-methyl-d-aspartate receptor (NMDAR)-dependent component of spontaneous synaptic current. All data in this figure are from a single CA1 pyramidal neuron, clamped near the normal resting membrane potential (−70 mV). Spontaneous excitatory postsynaptic currents (sEPSCs) were recorded with low extracellular Mg2+ (50 μM) to allow NMDAR current flow at the negative holding potential and with GABA receptors blocked by a combination of extracellular bicuculline (10 μM) and intracellular Cs+. A1: 2-s sample of membrane current. sEPSCs were identified using an automated method and verified by visual inspection. A2: averaged sEPSCs from a 5-min baseline period. sEPSC amplitude, rise time (10–90%), and duration at half amplitude (half-width) were measured . B1: membrane current after application of d(−)-2-amino-5-phosphonopentanoic acid (d-AP5; 50 μM) to block NMDARs. B2: averaged sEPSCs from a 5-min period after wash-in of d-AP5. Note the decrease in EPSC half-width (from 14.7 to 6.9 ms) with little change in sEPSC amplitude, consistent with the selective block of slower NDMAR-mediated synaptic currents by d-AP5. C1: washout of d-AP5 restored sEPSCs to baseline. C2: averaged sEPSC after d-AP5 washout. Half-width (19.7 ms) was similar to baseline, indicating recovery of NMDAR-mediated synaptic current. D: application of both 6,7-dinitroquinoxaline-2,3(1H, 4H)-dione (DNQX) and d-AP5 abolished all synaptic currents.
We applied this technique to examine effects of sleep deprivation (see Fig. 2). In cells from both naive and control animals, d-AP5 caused substantial decreases in sEPSC half-width. For cells from naive animals, sEPSC half-width was 10.9 ± 1.0 ms before and 7.6 ± 0.7 after d-AP5 application (mean change of −27.6 ± 5.7%; n = 11). For cells from control animals, sEPSC half-width was 12.1 ± 1.0 ms before and 8.1 ± 0.5 ms after d-AP5 application (−31.1 ± 3.4%; n = 12). In contrast, sEPSCs in cells from sleep-deprived animals were much less sensitive to the NMDA receptor antagonist, with sEPSC half-width averaging 8.3 ± 0.5 ms before d-AP5 and 7.1 ± 0.6 ms after d-AP5 (−12.6 ± 6.1%; n = 9). ANOVA indicated a significant overall difference among the three groups (P < 0.05), with pairwise comparisons showing a significant difference between control and sleep-deprived (P < 0.02), but no difference between control and naive. These findings are in agreement with a previous report (37) that examined evoked NMDAR-mediated synaptic currents and NMDA-induced currents in outside-out patches from CA1 dendrite membranes. sEPSC rise times (10–90%) appeared slightly reduced by d-AP5 application in cells from control and naive animals (Fig. 2E), but not in cells from sleep-deprived animals; however, this difference was not significant. Examination of isolated AMPAR-mediated sEPSCs (measured after application of d-AP5) revealed no differences among the three treatment conditions in half-width (Fig. 2B), amplitude (Fig. 2D), or rise time (Fig. 2E). We also examined cumulative probability distributions for sEPSC amplitude and interevent interval (not shown), but found no differences among the groups for these measures. These findings indicate that AMPAR synaptic function was not affected by sleep deprivation.
Fig. 2.
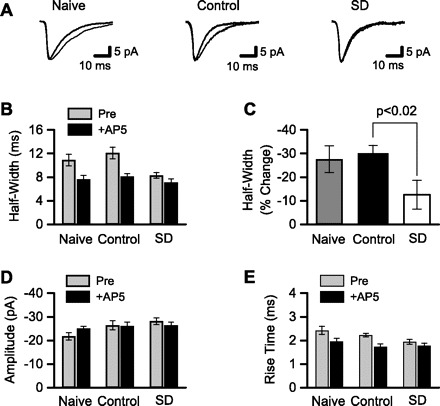
NMDAR-mediated, but not α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR)-mediated, synaptic currents were reduced after sleep deprivation. Whole cell patch-clamp was used to record sEPSCs. A: averaged sEPSCs from 3 different CA1 pyramidal neurons. sEPSCs were recorded in low-Mg2+ ACSF with GABA receptors blocked (thin line) and after addition of the NMDAR antagonist d-AP5 (thick line). Recordings are from single neurons in slices from (left to right): naive animal housed in standard animal cage, control animal kept on large platform over water, and sleep-deprived (SD) animal kept on small platform over water. The d-AP5-sensitive portion of each sEPSC reflects the NMDAR-mediated component of synaptic current. B: NMDAR-mediated synaptic currents were quantified by measuring EPSC half-width before and after application of d-AP5. sEPSC half-widths were averaged across all cells from the same treatment condition. d-AP5 substantially reduced sEPSC half-width in cells (n = 11, 9) from naive and control animals, but not in cells (n = 9) from SD animals. C: mean change in sEPSC half-width after d-AP5 application was calculated from the data shown in B. The change in half-width was significantly smaller in the SD group compared with control. sEPSC amplitudes (D) and rise times (E) are shown. After NMDARs were blocked by d-AP5, there were no differences among the 3 groups in sEPSC half-width (B), amplitude (D), or rise time (E), indicating that there was no effect of treatment on AMPAR function.
To assess NMDAR current stability over time, we attempted to wash out d-AP5 during all recordings. Because complete washout required a relatively long time (20 min), we achieved washout in a total of only eight recordings. As shown in Fig. 3, sEPSC half-width recovered completely following washout, indicating that NMDAR function remained stable over the entire duration of whole cell recording under our recording conditions.
Fig. 3.
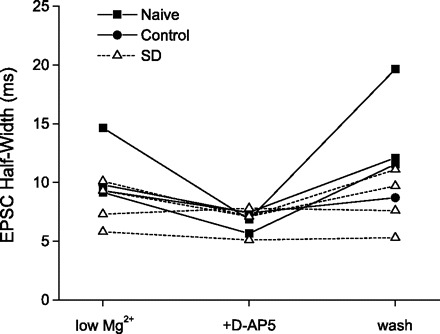
In cells that could be held long enough for complete washout of d-AP5, NMDAR function recovered to baseline levels. sEPSC half-width is plotted for each cell that was held long enough to complete washout of d-AP5. In cells from naive and control animals, sEPSC half-widths were consistently and substantially reduced from baseline (low Mg2+) during d-AP5 application as shown earlier (see Fig. 2), but fully recovered following washout of d-AP5 (wash). Even in this reduced sample size, it is evident that sEPSC half-widths in cells from SD animals were less sensitive to d-AP5 application, as shown above in Fig. 2; however, when half-width was reduced by d-AP5 in these cells, it recovered completely after washout.
Synaptic NMDAR function was impaired by sleep deprivation and rescued by growth hormone treatment.
Previous investigations have established that growth hormone is released during sleep, and prevention of sleep, in turn, suppresses circulating growth hormone. If growth hormone is a mediator of sleep effects on hippocampal synaptic function, then experimental restoration of growth hormone to sleep-deprived animals should reverse effects of sleep loss. In our first test of this prediction, we injected sleep-deprived or control animals with growth hormone (1 mg/kg) or an equivalent volume of saline vehicle (1 ml/kg) once on each of three consecutive days of treatment, and we measured NMDAR-mediated synaptic currents as described above (Figs. 1 and 2). In cells from vehicle-injected control animals, sEPSC half-width was consistently reduced from a mean 9.0 ± 0.6 ms to 6.7 ± 0.5 ms after the addition of d-AP5 (−25.0 ± 2.8%, n = 13; see Fig. 4). In contrast, sEPSCs in cells from vehicle-injected, sleep-deprived animals were less sensitive to d-AP5; half-widths averaged 8.0 ± 0.3 ms prior to and 6.8 ± 0.3 ms after d-AP5 application (−15.1 ± 2.4%, n = 13). This difference in d-AP5 sensitivity indicates a decrease in NMDAR-mediated synaptic currents as a consequence of sleep deprivation, in agreement with results shown above (Fig. 2) and in a prior report (37). This loss of NMDAR synaptic function was rescued by growth hormone treatment. In cells from growth hormone-injected, sleep-deprived animals, sEPSC half-widths averaged 10.1 ± 0.6 ms and were reduced to 7.7 ± 0.5 ms by d-AP5 (decrease of −24.3 ± 2.7%, n = 15). This change in sEPSC half-width was virtually identical to that observed in cells from growth hormone-injected control animals (9.6 ± 0.9 ms before d-AP5, 7.2 ± 0.4 ms after d-AP5, −23.1 ± 4.0%, n = 11). ANOVA indicated a significant interaction (P < 0.05) between sleep condition (control vs. sleep-deprived) and injection treatment (vehicle vs. growth hormone). The basis of this interaction was a significant decrease in sEPSC half-width in the vehicle sleep-deprived group compared with the vehicle-injected control group (P < 0.05), with no other pairwise comparisons being significant. This pattern of results indicates that growth hormone selectively rescued NMDAR-mediated synaptic currents in sleep-deprived animals. sEPSC peak amplitudes, either before or after d-AP5 application, were not different among the four groups (Fig. 4D), nor were there differences in sEPSC half-width or rise time in d-AP5 (Fig. 4, B and E), indicating that AMPAR function was not affected by either sleep deprivation, as reported previously (37), or by growth hormone treatment.
Fig. 4.
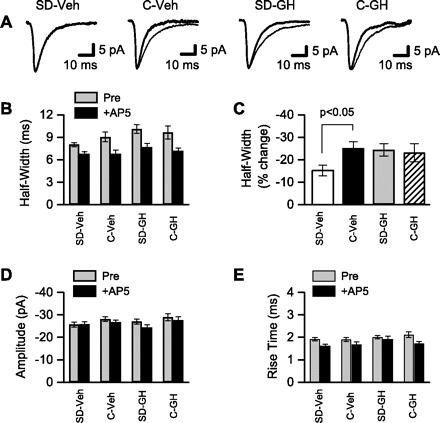
NMDAR-mediated synaptic currents were reduced after sleep deprivation, but were restored by growth hormone (GH) treatment. Whole cell patch-clamp was used to record sEPSCs. A: averaged sEPSCs from 4 different CA1 pyramidal neurons. sEPSCs were recorded in low-Mg2+ ACSF with GABA receptors blocked (thin line) and after addition of the NMDAR antagonist, d-AP5 (thick line). Recordings are from single neurons in slices from (left to right): SD animals that received daily injections of saline vehicle (SD-Veh), control animals that received daily injections of Veh (C-Veh), SD animals that received daily injections of GH (SD-GH), and control animals that received daily injections of GH (C-GH). The d-AP5-sensitive portion of each sEPSC reflects the NMDAR-mediated component of synaptic current. B: NMDAR-mediated synaptic currents were quantified by measuring sEPSC half-width before and after application of d-AP5. sEPSC half-widths were averaged across all cells from the same treatment condition. sEPSC half-width was minimally affected by d-AP5 in cells (n = 13) from SD-Veh animals, but was substantially reduced in cells (n = 13, 15, 11) from the remaining 3 treatment conditions. C: mean change in sEPSC half-width after d-AP5 application was calculated from the data shown in B. The change in half-width was significantly smaller in the SD-Veh group compared with C-Veh. In contrast, sEPSC half-width was restored to control levels in SD-GH animals. sEPSC amplitudes (D) and rise times (E) are shown. Following block of NMDARs with d-AP5, there were no differences among the 4 groups in sEPSC half-width (B), amplitude (D), or rise time (E), indicating no effect of sleep deprivation or GH on AMPAR function.
LTP was impaired by sleep deprivation but rescued by growth hormone treatment.
In hippocampal area CA1, induction of LTP occurs in response to calcium influx into postsynaptic neurons through NMDARs. Because growth hormone treatment during sleep deprivation was able to prevent the loss of NMDAR function, which otherwise occurs as a consequence of sleep deprivation, we next asked whether growth hormone treatment could rescue the LTP deficit that also follows sleep deprivation. We made recordings of evoked field excitatory postsynaptic potentials (EPSPs) in stratum radiatum of area CA1 and used theta burst stimulation (TBS) of Schaffer collateral/commissural afferents to assess LTP. Following a stable 15-min baseline recording period, each slice received TBS consisting of 20 bursts of four stimuli (at 100 Hz) with bursts repeated at 5 Hz. Slices from vehicle-injected control animals consistently showed an LTP that was maintained throughout a 60-min post-TBS recording period (25.3 ± 5.5% increase in EPSP slope at 50–60 min post-TBS, n = 10; see Fig. 5A). As shown previously (6, 12, 20, 37), LTP in slices from sleep-deprived (vehicle-injected) animals was significantly reduced compared with control slices (Fig. 5A). In these slices, EPSP slopes completely returned to the baseline level after ∼45 min, and at 50–60 min post-TBS, were slightly reduced (−2.6 ± 5.5%, n = 7) compared with baseline. This loss of LTP following sleep deprivation is consistent with the significant loss of NMDAR function shown above. Just as growth hormone treatment of sleep-deprived animals rescued normal NMDAR function, growth hormone treatment also rescued LTP (see Fig. 5B). Slices from sleep-deprived animals that received daily injections of growth hormone demonstrated robust LTP after TBS (24.4 ± 8.8% increase in EPSP slope, n = 10) similar to that obtained in slices from growth hormone-injected control animals (38.8 ± 10.4% increase in EPSP slope, n = 8, P > 0.25). ANOVA showed significant main effects for both sleep condition (control vs. sleep-deprived) and injection treatment (vehicle vs. growth hormone); however, the only significant pairwise comparison was between the vehicle-injected, sleep-deprived, and vehicle-injected control groups (P < 0.05).
Fig. 5.
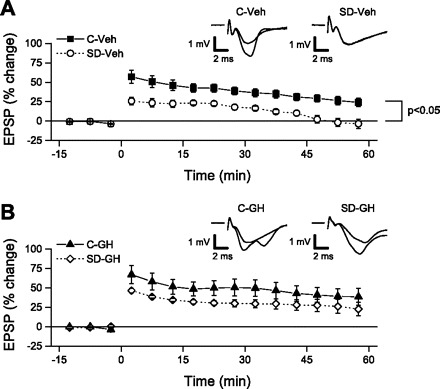
Long-term potentiation (LTP) was significantly impaired after sleep deprivation, but was rescued by GH treatment. For each slice, field excitatory postsynaptic potential (EPSP) slopes were normalized relative to baseline, averaged over consecutive 5-min periods, and then averaged across all slices in the same group. Theta burst stimulation (TBS; at time = 0 min) was used to test for LTP. A: LTP was greatly reduced in slices from the SD-Veh group compared with slices from C-Veh animals. By 45 min following TBS, LTP in the SD-Veh group had completely decayed, but LTP remained throughout the 60-min post-TBS recording period in slices from the C-Veh group. At 50–60 min post-TBS, the change in EPSPs was significantly greater in the C-Veh group compared with the SD-Veh group. Insets show averaged EPSPs from representative slices in C-Veh and SD-Veh groups obtained over the final 5 min of the baseline period and the final 5 min of the recording. B: GH treatment restored LTP in slices from SD animals to control levels. LTP was maintained at equivalent levels throughout the 60-min post-TBS recording in both SD-GH and C-GH slices. At 50–60 min post-TBS, there was no significant difference between the 2 groups. Insets show averaged EPSPs from representative slices in C-GH and SD-GH groups obtained over the final 5 min of the baseline period and the final 5 min of the recording.
Synaptic NMDAR 2B subunit protein level was decreased by sleep deprivation but restored by growth hormone treatment.
To determine whether the subunit composition of NMDARs in hippocampal synapses might be altered by sleep deprivation, we prepared synaptosomal membranes from whole hippocampus and quantified subunit protein levels using immunoblotting (see Fig. 6). There were no differences among the four groups (all n =5) for subunits NR1 or NR2A, nor were there differences in PSD-95 levels. For subunit NR2B protein, however, there was a significant interaction (P < 0.05) between sleep condition (control vs. sleep-deprived) and injection treatment (vehicle vs. growth hormone), with NR2B protein significantly decreased in the vehicle-injected, sleep-deprived group compared with the vehicle-injected control group (P < 0.01). Critically, growth hormone treatment of sleep-deprived animals restored NR2B protein to control levels, with no difference between growth hormone-injected, sleep-deprived and growth hormone-injected control animals.
Fig. 6.
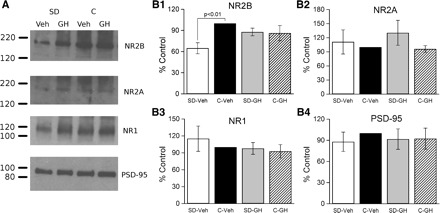
Hippocampal synaptic NMDAR subunit 2B (NR2B) expression was decreased after sleep deprivation but restored by GH injection. Proteins were isolated from synaptosomal membranes, separated by gel electrophoresis, transferred to nitrocellulose membranes, and probed using antibodies specific for NMDAR subunits (NR2B, NR2A, NR1) and the postsynaptic density protein-95 (PSD-95). A: results from 1 replication of this experiment. Each replication included 4 animals, 1 from each group (SD-Veh, SD-GH, C-Veh, C-GH). In this replication, NR2B expression (top) was substantially reduced in SD-Veh animals, but was restored to control level by GH injection, whereas NR2A and NR1 subunits (middle rows) were less affected. Subunit expression did not differ between control groups (GH or Veh). There were no differences in PSD-95. B, 1–4: this experiment was repeated a total of 5 times (a total of 5 animals/treatment condition). Protein expression was quantified by film densitometry and normalized within each blot to C-Veh. Normalized values were averaged across blots (animals). The only consistent change in protein expression was for the NR2B subunit (B1), which was significantly reduced in the SD-Veh group compared with the C-Veh group, and that was restored by GH injection (no significant difference between SD-GH and C-GH). B, 2–4: there were no differences between the SD-Veh and C-Veh or SD-GH and C-GH groups for NR2A, NR1, and PSD-95.
No differences in corticosterone levels.
Corticosterone is released in stressful situations, and sleep deprivation may be associated with stress (10, 20). To determine whether differences in stress level might have contributed to the differences we reported above, we measured serum corticosterone, using ELISA, in each of our four experimental groups (see Fig. 7A). Serum corticosterone averaged 65.7 ± 20.3 ng/ml in vehicle-injected, sleep-deprived animals (n = 14), 31.7 ± 9.0 ng/ml in vehicle-injected control animals (n = 10), 33.8 ± 13.6 ng/ml in growth hormone-injected, sleep-deprived animals (n = 8), and 26.4 ± 10.2 ng/ml in growth hormone-injected control animals (n = 6). ANOVA failed to find a significant difference in corticosterone concentration among these four groups. The absence of any significant differences in serum corticosterone demonstrates that differences in stress levels cannot explain our findings.
Fig. 7.
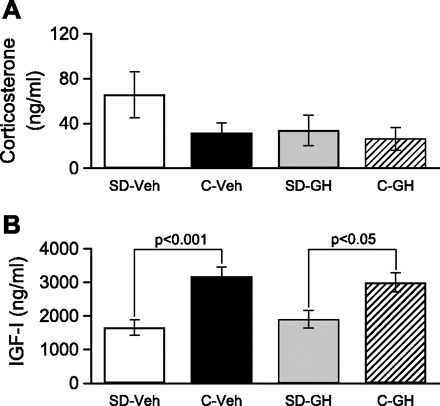
Serum corticosterone was not altered by SD or GH treatment; however, serum IGF-I was reduced by sleep deprivation but was not restored by GH treatment. Serum hormone concentrations were determined by ELISA. A: serum corticosterone concentration was not significantly different among the 4 groups examined. B: serum IGF-I concentration was significantly lower in SD-Veh animals compared with C-Veh animals. A significant difference remained between SD-GH and C-GH animals, indicating that GH treatment did not restore serum IGF-I following SD.
Reduced IGF-I in sleep-deprived animals.
IGF-I is produced by liver and target tissues in response to growth hormone stimulation, and IGF-I, in turn, mediates some, but not all, effects of growth hormone (28). In agreement with previous findings of reduced growth hormone during sleep deprivation (5, 15, 21, 52), a reduction in circulating IGF-1 has also been observed during sleep deprivation (15). To verify that growth hormone/IGF-1 signals were reduced in our sleep-deprived animals, and to investigate whether growth hormone treatment during sleep deprivation restored IGF-1 levels, we used ELISA to measure serum IGF-1 level in animals from each of our four experimental conditions (see Fig. 7B). As expected, IGF-1 was reduced in vehicle-injected, sleep-deprived animals (1,659.0 ± 228.4 ng/ml, n = 10) compared with vehicle-injected controls (3,171.7 ± 293.0 ng/ml, n = 11). Surprisingly, growth hormone treatment did not restore IGF-1 in sleep-deprived animals, with IGF-1 remaining low (1,909.5 ± 259.3 ng/ml, n = 8) compared with growth hormone-injected control animals (3,000.5 ± 284.03 ng/ml, n = 7). ANOVA revealed a significant main effect for sleep condition (control vs. sleep-deprived) only, with no effect of growth hormone injection and no interaction. Pairwise comparisons showed reduced IGF-I in sleep-deprived animals compared with controls regardless of whether they received vehicle (P < 0.001) or growth hormone (P < 0.05) injections.
DISCUSSION
Growth hormone is released in a major surge during sleep, and sleep deprivation substantially reduces circulating growth hormone level (5, 15, 21, 52). We hypothesized that loss of growth hormone as a consequence of sleep deprivation was responsible for the altered synaptic function that has been reported in the hippocampus after sleep deprivation (6, 12, 20, 36, 37). Our findings confirmed a critical role for growth hormone in maintaining hippocampal synaptic function. Hippocampal CA1 neurons from sleep-deprived animals had reduced NMDAR-mediated synaptic currents, reduced NMDA receptor-dependent LTP, and reduced synaptic NR2B subunit expression. Restoring growth hormone to sleep-deprived animals caused recovery of NMDAR function, NMDA receptor-dependent synaptic plasticity, and synaptic NMDAR subunit expression.
The decrease in NR2B subunits in hippocampal synaptosomal membranes from sleep-deprived animals may explain our observation of reduced NMDAR contribution to EPSC half-width. The decay kinetics of NMDAR synaptic currents depend on NR2 subunit identity: receptors composed of NR1 and NR2A subunits have currents with faster decay times than receptors composed of NR1 and NR2B subunits (54). The decreased NR2B subunit abundance we saw after sleep deprivation would have increased the fraction of hippocampal synapses with faster decay time constants and therefore produced EPSCs with shorter half-widths after sleep deprivation. Restoration of normal NR2B subunit expression with growth hormone treatment can also explain the recovery of EPSC half-width that we observed in neurons from growth hormone-injected, sleep-deprived animals. The loss of NR2B subunits after sleep deprivation, and the recovery of NR2B levels with growth hormone treatment may also explain the changes in LTP that we report here. In our study, LTP was deficient in hippocampal slices from sleep-deprived animals, which had a lower NR2B subunit level. Growth hormone restoration to sleep-deprived animals rescued LTP and NR2B subunit expression in parallel. Previous studies suggest a mechanism for this association between NR2B subunits and LTP. Subunit NR2B of the NMDAR is critical for targeting activated calcium-calmodulin-dependent protein kinase II, a downstream target for Ca2+ during LTP induction (34, 46), to the synapse (3, 49), and loss or specific inhibition of NR2B containing NMDA receptors impairs LTP.
Our finding that hippocampal synaptic impairment caused by sleep deprivation was reversed by growth hormone treatment adds to a growing literature on the role of growth hormone in cognitive and memory functions. In both humans and animal models, growth hormone deficiency impairs cognitive functions, including memory (41), and growth hormone treatment of growth hormone-deficient humans and animals restores these functions (1, 13, 27). Changes seen during normal aging further emphasize the critical role of growth hormone/IGF-I in maintaining memory function. Circulating growth hormone and IGF-I are decreased during aging (11), and growth hormone or IGF-I treatment improves memory-dependent performance (35, 43). Aged rats show impaired hippocampal LTP and hippocampal-dependent learning and memory (2) that have been linked to decreased NR2B expression (8). In 2002, Le Grevès et al. (25) showed that chronic growth hormone treatment increased NR2B mRNA in adult rats, but increased NR1 and NR2A mRNAs in aged rats. These authors suggested that growth hormone facilitates hippocampal function and enhances LTP in young adult rats by upregulating NR2B gene transcripts; however, they showed no change in NR2B with growth hormone treatment in aged rats. In addition, mRNA levels of NR1, NR2A, and PSD-95 were increased by growth hormone treatment in hypophysectomized rats, but there was no change in NR2B subunit expression (27). Although these results indicate that growth hormone may regulate NMDAR composition differently depending on age, growth hormone regulation of hippocampal memory-related functions through altered NMDA receptor expression is of general and considerable significance.
Many growth hormone effects are mediated by IGF-1 produced by liver and target tissues. Reduced growth hormone release leads to a decrease in plasma IGF-1. Effects of growth hormone on hippocampal synaptic function or NMDA receptor expression could be mediated by IGF-I. In support of this possibility, repeated injections of IGF-1 increased NR2B mRNA levels in young (11 wk) adult rats (26). However, in our sleep-deprived animals, growth hormone treatment did not restore serum IGF-1 concentration, indicating that the growth hormone effects we report here were not mediated through increased circulating IGF-I. If the growth hormone effects that we obtained were not mediated by circulating IGF-I, then they may have resulted from either direct action of growth hormone on hippocampal neurons, or perhaps by growth hormone stimulation of IGF-I production within the hippocampus (or other central nervous system region), which then acted on hippocampal neurons as a growth hormone mediator. A direct role for growth hormone in altering hippocampal synaptic function is supported by our previous finding that direct application of growth hormone to in vitro hippocampal slices can enhance NMDA receptor-mediated EPSP/Cs (33). Alternatively, since the hippocampus is a site of IGF-I production (28), growth hormone entry into the central nervous system might be capable of stimulating hippocampal IGF-I production, with hippocampal-derived IGF-I regulating NMDARs in a paracrine or autocrine manner.
Growth hormone regulation of IGF-I production by the liver is well established (28). It was surprising, therefore, that growth hormone treatment of sleep-deprived animals did not restore circulating IGF-I levels. Several factors could explain this lack of effect. First, growth hormone is not the only hormone affected by sleep deprivation: circulating thyroid hormone, insulin, corticosterone, leptin, and ghrelin are all altered by sleep deprivation (48). Second, although IGF-I expression is regulated by growth hormone, circulating IGF-I concentration is dependent on the presence of IGF-binding proteins (IGF-BPs). Sleep deprivation has diverse effects on metabolic function and hormone levels (15, 22, 48), which in turn may alter production of IGF-I, IGF-BPS, or decouple IGF-I production from growth hormone (38, 45, 51). Restoration of growth hormone alone may not have been sufficient to restore IGF-I levels.
Sleep deprivation may be associated with stress (10, 20). We therefore measured circulating corticosterone to determine stress levels in each of our experimental conditions. We found no significant differences in corticosterone levels among our four treatment groups, indicating that differences in stress cannot explain the findings we obtained. This conclusion is in agreement with recent studies showing sleep deprivation effects on hippocampal function that are independent of differences in stress or stress hormones (40, 44).
Taken together, our data indicate that growth hormone is required for normal hippocampal synaptic function. When growth hormone signals are lost during sleep deprivation, NMDAR-mediated synaptic currents and NMDAR-dependent LTP are impaired. This deficit in NMDAR function may reflect a specific loss of NR2B subunits from synaptic membranes. Restoration of growth hormone to sleep-deprived animals rescued hippocampal NMDAR function, LTP, and NMDAR subunit expression.
Perspectives and Significance
While sleep has long been recognized as an essential regulator of biological function, including brain function, the signals linking sleep to brain function have not been known. In this study, we demonstrated a role for growth hormone as a mediator between sleep and hippocampal function, establishing for the first time that growth hormone is an essential link between sleep and normal brain function. A better understanding of the role of growth hormone in the critical maintenance functions of sleep will ultimately lead to improved understanding and development of countermeasures for the long-term, chronic, sleep deprivation that affects many people.
GRANTS
This work was supported by the National Aeronautics and Space Administration Grant NCC5–570 and the West Virginia IDeA Network of Biomedical Research Excellence Grant P20-RR-016477 from the National Center for Research Resources, National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial, or otherwise, are declared by the author(s).
ACKNOWLEDGMENTS
Present address of E. Kim: Molecular Neurophysiology and Biophysics Unit, Laboratory of Cellular and Synaptic Neurophysiology, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892.
REFERENCES
- 1. Arwert LI, Deijen JB, Müller M, Drent ML. Long-term growth hormone treatment preserves GH-induced memory and mood improvements: a 10-year follow-up study in GH-deficient adult men. Horm Behav 47: 343–349, 2005 [DOI] [PubMed] [Google Scholar]
- 2. Barnes CA. Long-term potentiation and the ageing brain. Philos Trans R Soc Lond B Biol Sci 358: 765–772, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 48: 289–301, 2005 [DOI] [PubMed] [Google Scholar]
- 4. Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31–39, 1993 [DOI] [PubMed] [Google Scholar]
- 5. Brandenberger G, Gronfier C, Chapotot F, Simon C, Piquard F. Effect of sleep deprivation on overall 24 h growth-hormone secretion. Lancet 356: 1408, 2000 [DOI] [PubMed] [Google Scholar]
- 6. Campbell IG, Guinan MJ, Horowitz JM. Sleep deprivation impairs long-term potentiation in rat hippocampal slices. J Neurophysiol 88: 1073–1076, 2002 [DOI] [PubMed] [Google Scholar]
- 7. Chen C, Hardy M, Zhang J, LaHoste GJ, Bazan NG. Altered NMDA receptor trafficking contributes to sleep deprivation-induced hippocampal synaptic and cognitive impairments. Biochem Biophys Res Commun 340: 435–440, 2006 [DOI] [PubMed] [Google Scholar]
- 8. Clayton DA, Mesches MH, Alvarez E, Bickford PC, Browning MD. A hippocampal NR2B deficit can mimic age-related changes in long-term potentiation and spatial learning in the Fischer 344 rat. J Neurosci 22: 3628–3637, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Coculescu M. Blood-brain barrier for human growth hormone and insulin-like growth factor-I. J Pediatr Endocrinol Metab 12: 113–124, 1999 [DOI] [PubMed] [Google Scholar]
- 10. Coenen AM, van Luijtelaar EL. Stress induced by three procedures of deprivation of paradoxical sleep. Physiol Behav 35: 501–504, 1985 [DOI] [PubMed] [Google Scholar]
- 11. Corpas E, Harman SM, Blackman MR. Human growth hormone and human aging. Endocr Rev 14: 20–39, 1993 [DOI] [PubMed] [Google Scholar]
- 12. Davis CJ, Harding JW, Wright JW. REM sleep deprivation-induced deficits in the latency-to-peak induction and maintenance of long-term potentiation within the CA1 region of the hippocampus. Brain Res 973: 293–297, 2003 [DOI] [PubMed] [Google Scholar]
- 13. Deijen JB, de Boer H, van der Veen EA. Cognitive changes during growth hormone replacement in adult men. Psychoneuroendocrinology 23: 45–55, 1998 [DOI] [PubMed] [Google Scholar]
- 14. DeMesquita S, Hale GA. Cardiopulmonary regulation after rapid-eye-movement sleep deprivation. J Appl Physiol 72: 970–976, 1992 [DOI] [PubMed] [Google Scholar]
- 15. Everson CA, Crowley WR. Reductions in circulating anabolic hormones induced by sustained sleep deprivation in rats. Am J Physiol Endocrinol Metab 286: E1060–E1070, 2004 [DOI] [PubMed] [Google Scholar]
- 16. Goebel SM, Alvestad RM, Coultrap SJ, Browning MD. Tyrosine phosphorylation of the N-methyl-d-aspartate receptor is enhanced in synaptic membrane fractions of the adult rat hippocampus. Brain Res Mol Brain Res 142: 65–79, 2005 [DOI] [PubMed] [Google Scholar]
- 17. Grahnstedt S, Ursin R. Platform sleep deprivation affects deep slow wave sleep in addition to REM sleep. Behav Brain Res 18: 233–239, 1985 [DOI] [PubMed] [Google Scholar]
- 18. Hartog M. Physiological and pathological roles of growth hormone. Postgrad Med J 40: 470–478, 1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johansson JO, Larson G, Andersson M, Elmgren A, Hynsjö L, Lindahl A, Lundberg PA, Isaksson OG, Lindstedt S, Bengtsson BA. Treatment of growth hormone-deficient adults with recombinant human growth hormone increases the concentration of growth hormone in the cerebrospinal fluid and affects neurotransmitters. Neuroendocrinology 61: 57–66, 1995 [DOI] [PubMed] [Google Scholar]
- 20. Kim EY, Mahmoud GS, Grover LM. REM sleep deprivation inhibits LTP in vivo in area CA1 of rat hippocampus. Neurosci Lett 388: 163–167, 2005 [DOI] [PubMed] [Google Scholar]
- 21. Kimura F, Tsai CW. Ultradian rhythm of growth hormone secretion and sleep in the adult male rat. J Physiol 353: 305–315, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Knutson KL, Spiegel K, Penev P, Van Cauter E. The metabolic consequences of sleep deprivation. Sleep Med Rev 11: 163–178, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kopp C, Longordo F, Nicholson JR, Lüthi A. Insufficient sleep reversibly alters bidirectional synaptic plasticity and NMDA receptor function. J Neurosci 26: 12456–12465, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lai ZN, Emtner M, Roos P, Nyberg F. Characterization of putative growth hormone receptors in human choroid plexus. Brain Res 546: 222–226, 1991 [DOI] [PubMed] [Google Scholar]
- 25. Le Grevès M, Steensland P, Le Grevès P, Nyberg F. Growth hormone induces age-dependent alteration in the expression of hippocampal growth hormone receptor and N-methyl-d-aspartate receptor subunits gene transcripts in male rats. Proc Natl Acad Sci USA 99: 7119–7123, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Le Grevès M, Le Grevès P, Nyberg F. Age-related effects of IGF-1 on the NMDA-, GH- and IGF-1-receptor mRNA transcripts in the rat hippocampus. Brain Res Bull 65: 369–374, 2005 [DOI] [PubMed] [Google Scholar]
- 27. Le Grevès M, Zhou Q, Berg M, Le Grevès P, Fhölenhag K, Meyerson B, Nyberg F. Growth hormone replacement in hypophysectomized rats affects spatial performance and hippocampal levels of NMDA receptor subunit and PSD-95 gene transcript levels. Exp Brain Res 173: 267–273, 2006 [DOI] [PubMed] [Google Scholar]
- 28. Le Roith D, Bondy C, Yakar S, Liu JL, Butler A. The somatomedin hypothesis: 2001. Endocr Rev 22: 53–74, 2001 [DOI] [PubMed] [Google Scholar]
- 29. Levin G, Gershonowitz A, Sacks H, Stern M, Sherman A, Rudaev S, Zivin I, Phillip M. Transdermal delivery of human growth hormone through RF-microchannels. Pharm Res 22: 550–555, 2005 [DOI] [PubMed] [Google Scholar]
- 30. Lobie PE, García-Aragón J, Lincoln DT, Barnard R, Wilcox JN, Waters MJ. Localization and ontogeny of growth hormone receptor gene expression in the central nervous system. Brain Res Dev Brain Res 74: 225–233, 1993 [DOI] [PubMed] [Google Scholar]
- 31. Lynch MA. Long-term potentiation and memory. Physiol Rev 84: 87–136, 2004 [DOI] [PubMed] [Google Scholar]
- 32. Machado RB, Hipólide DC, Benedito-Silva AA, Tufik S. Sleep deprivation induced by the modified multiple platform technique: quantification of sleep loss and recovery. Brain Res 1004: 45–51, 2004 [DOI] [PubMed] [Google Scholar]
- 33. Mahmoud GS, Grover LM. Growth hormone enhances excitatory synaptic transmission in area CA1 of rat hippocampus. J Neurophysiol 95: 2962–2974, 2006 [DOI] [PubMed] [Google Scholar]
- 34. Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 245: 862–866, 1989 [DOI] [PubMed] [Google Scholar]
- 35. Markowska AL, Mooney M, Sonntag WE. Insulin-like growth factor-1 ameliorates age-related behavioral deficits. Neuroscience 87: 559–569, 1998 [DOI] [PubMed] [Google Scholar]
- 36. McDermott CM, LaHoste GJ, Chen C, Musto A, Bazan NG, Magee JC. Sleep deprivation causes behavioral, synaptic, and membrane excitability alterations in hippocampal neurons. J Neurosci 23: 9687–9695, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McDermott CM, Hardy MN, Bazan NG, Magee JC. Sleep deprivation-induced alterations in excitatory synaptic transmission in the CA1 region of the rat hippocampus. J Physiol 570: 553–565, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miell JP, Taylor AM, Zini M, Maheshwari HG, Ross RJ, Valcavi R. Effects of hypothyroidism and hyperthyroidism on insulin-like growth factors (IGFs) and growth hormone- and IGF-binding proteins. J Clin Endocrinol Metab 76: 950–955, 1993 [DOI] [PubMed] [Google Scholar]
- 39. Morden B, Mitchell G, Dement W. Selective REM sleep deprivation and compensation phenomena in the rat. Brain Res 5: 339–349, 1967 [DOI] [PubMed] [Google Scholar]
- 40. Mueller AD, Pollock MS, Lieblich SE, Epp JR, Galea LAM, Mistlberger RE. Sleep deprivation can inhibit adult hippocampal neurogenesis independent of adrenal stress hormones. Am J Physiol Regul Integr Comp Physiol 294: R1693–R1703, 2008 [DOI] [PubMed] [Google Scholar]
- 41. Nyberg F. Growth hormone in the brain: characteristics of specific brain targets for the hormone and their functional significance. Front Neuroendocrinol 21: 330–348, 2000 [DOI] [PubMed] [Google Scholar]
- 42. Pan W, Yu Y, Cain CM, Nyberg F, Couraud PO, Kastin AJ. Permeation of growth hormone across the blood-brain barrier. Endocrinology 146: 4898–4904, 2005 [DOI] [PubMed] [Google Scholar]
- 43. Ramsey MM, Weiner JL, Moore TP, Carter CS, Sonntag WE. Growth hormone treatment attenuates age-related changes in hippocampal short-term plasticity and spatial learning. Neuroscience 129: 119–127, 2004 [DOI] [PubMed] [Google Scholar]
- 44. Ruskin DN, Dunn KE, Billiot I, Bazan NG, LaHoste GJ. Eliminating the adrenal stress response does not affect sleep deprivation-induced acquisition deficits in the water maze. Life Sci 78: 2833–2838, 2006 [DOI] [PubMed] [Google Scholar]
- 45. Schmid C, Brändle M, Zwimpfer C, Zapf J, Wiesli P. Effect of thyroxine replacement on creatinine, insulin-like growth factor 1, acid-labile subunit, and vascular endothelial growth factor. Clin Chem 50: 228–231, 2004 [DOI] [PubMed] [Google Scholar]
- 46. Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in α-calcium-calmodulin kinase II mutant mice. Science 257: 201–206, 1992 [DOI] [PubMed] [Google Scholar]
- 47. Smith C. Sleep states and memory processes. Behav Brain Res 69: 137–145, 1995 [DOI] [PubMed] [Google Scholar]
- 48. Steiger A. Neurochemical regulation of sleep. J Psychiatr Res 41: 537–552, 2007 [DOI] [PubMed] [Google Scholar]
- 49. Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem 273: 20689–20692, 1998 [DOI] [PubMed] [Google Scholar]
- 50. Tzanela M, Wagner C, Tannenbaum GS. Recombinant human growth hormone-binding protein fails to enhance the in vivo bioactivity of human growth hormone in normal rats. Endocrinology 138: 5316–5324, 1997 [DOI] [PubMed] [Google Scholar]
- 51. Thissen JP, Ketelslegers JM, Underwood LE. Nutritional regulation of the insulin-like growth factors. Endocr Rev 15: 80–101, 1994 [DOI] [PubMed] [Google Scholar]
- 52. Toppila J, Asikainen M, Alanko L, Turek FW, Stenberg D, Porkka-Heiskanen T. The effect of REM sleep deprivation on somatostatin and growth hormone-releasing hormone gene expression in the rat hypothalamus. J Sleep Res 5: 115–122, 1996 [DOI] [PubMed] [Google Scholar]
- 53. van Luijtelaar EL, Coenen AM. Electrophysiological evaluation of three paradoxical sleep deprivation techniques in rats. Physiol Behav 36: 603–609, 1986 [DOI] [PubMed] [Google Scholar]
- 54. Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J Neurophysiol 79: 555–566, 1998 [DOI] [PubMed] [Google Scholar]
- 55. Wilson MA. Hippocampal memory formation, plasticity, and the role of sleep. Neurobiol Learn Mem 78: 565–569, 2002 [DOI] [PubMed] [Google Scholar]
- 56. Youngblood BD, Zhou J, Smagin GN, Ryan DH, Harris RB. Sleep deprivation by the “flower pot” technique and spatial reference memory. Physiol Behav 61: 249–256, 1997 [DOI] [PubMed] [Google Scholar]
- 57. Zearfoss NR, Alarcon JM, Trifilieff P, Kandel E, Richter JD. A molecular circuit composed of CPEB-1 and c-Jun controls growth hormone-mediated synaptic plasticity in the mouse hippocampus. J Neurosci 28: 8502–8509, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]