

Abstract
In lipoatrophic diabetes, a lack of fat is associated with insulin resistance and hyperglycemia. This is in striking contrast to the usual association of diabetes with obesity. To understand the underlying mechanisms, we transplanted adipose tissue into A-ZIP/F-1 mice, which have a severe form of lipoatrophic diabetes. Transplantation of wild-type fat reversed the hyperglycemia, dramatically lowered insulin levels, and improved muscle insulin sensitivity, demonstrating that the diabetes in A-ZIP/F-1 mice is caused by the lack of adipose tissue. All aspects of the A-ZIP/F-1 phenotype including hyperphagia, hepatic steatosis, and somatomegaly were either partially or completely reversed. However, the improvement in triglyceride and FFA levels was modest. Donor fat taken from parametrial and subcutaneous sites was equally effective in reversing the phenotype. The beneficial effects of transplantation were dose dependent and required near-physiological amounts of transplanted fat. Transplantation of genetically modified fat into A-ZIP/F-1 mice is a new and powerful technique for studying adipose physiology and the metabolic and endocrine communication between adipose tissue and the rest of the body.
Introduction
Insulin resistance and type 2 diabetes mellitus are frequent complications of obesity. Lipoatrophic (or lipodystrophic) diabetes is paradoxical, as this form of insulin-resistant diabetes is associated with a low fat mass (1, 2). The lipodystrophies are a heterogeneous group of disorders, with genetic, autoimmune, and probably other etiologies that exhibit a wide range in the extent of fat loss, from generalized to partial to focal. The age of onset ranges from congenital through adulthood and is often characteristic of the particular subtype of the disease.
The causes of lipoatrophic diabetes are not known. Mechanisms both intrinsic and extrinsic to the adipocyte have been proposed, including destruction or inadequate production of adipocytes, abnormal nonadipocyte signals affecting metabolism, and intrinsic adipocyte abnormalities that alter their metabolic response (3). A number of candidate genes have been excluded (4). The location of the causative mutations have been mapped for the Dunnigan variety of partial lipodystrophy (5, 6) and for a subset of patients with congenital generalized lipodystrophy (7). However, despite the recent identification of a mutation in the lamin A/C gene in Dunnigan lipodystrophy patients (8), the mechanism(s) and tissue(s) of action remain a mystery.
Recently, we developed a transgenic mouse, named A-ZIP/F-1, which has virtually no white adipose tissue (WAT) (9). These mice express, selectively in adipose tissue, a dominant negative protein that heterodimerizes with and inactivates members of the C/EBP and JUN families of B-ZIP transcription factors. The A-ZIP/F-1 phenotype strikingly resembles that of humans with severe lipoatrophic diabetes, including the lack of fat, insulin resistance and hyperglycemia, hyperlipidemia, fatty liver, and organomegaly. Transgenic mice with similar, but less severe, phenotypes resulted from adipose expression of a modified diphtheria toxin (10, 11) or a constitutively active SREBP-1c transcription factor (12).
The phenotypes of the transgenic mice are consistent with the hypothesis that the metabolic abnormalities in lipoatrophy are due to the lack of adipose tissue. However, the A-ZIP/F-1 mice provide a way to test rigorously the role of WAT in the etiology of lipoatrophic diabetes. If the lack of fat is causing the diabetes, reconstitution with normal adipose tissue should make the mice normoglycemic. In this study, we show that transplantation of wild-type fat into A-ZIP/F-1 mice reversed the diabetes. This proves that the lack of WAT is causing the metabolic abnormalities. These results establish fat transplantation as an important new technique for studying adipose tissue physiology and the mechanisms of insulin resistance and type 2 diabetes.
Methods
Mice.
The A-ZIP/F-1 animals were hemizygous on the FVB/N background, produced by breeding hemizygous males with wild-type females. Unless noted otherwise, female mice were used. Wild-type controls were littermates. Mice were kept on a 12-hour light/dark cycle and fed NIH-07 rodent chow (Zeigler Brothers Inc., Gardners, Pennsylvania, USA) and water ad libitum.
Fat transplantation.
To avoid rejection, donor fat was taken from littermates. Five-week-old A-ZIP/F-1 females (∼20 g) were anesthetized with pentobarbital (0.01 mL/g body weight of 5 mg/mL in 5% ethanol/PBS, intraperitoneally). Donor fat pads (parametrial unless noted otherwise) from wild-type littermates euthanized by cervical dislocation were placed into sterile PBS and cut into 100- to 150-mg pieces. These grafts were implanted subcutaneously through small incisions in the shaved skin of the back, with 1 piece per incision. When 900 mg of fat per mouse was transplanted, one graft was placed ventrally. Incisions were closed using 4-0 silk sutures. After surgery, the mice were housed individually for a week, and then, at 2–3 mice per cage. Tail venous blood (∼200 μL) was withdrawn at ∼1000 hours. Week 0 bleeds were from the day before surgery. Upon sacrifice the grafts were evaluated visually, weighed, and fixed for histological analysis. This article includes 6 independent series totaling 47 transplanted mice. Exclusive of 4 perioperative deaths (presumably anesthesia related), no transplanted mice died during the study. One mouse was excluded because its single 100-mg graft was necrotic at sacrifice.
Indirect calorimetry.
Oxygen consumption and carbon dioxide production were measured using a 4-chamber Oxymax system (Columbus Instruments, Columbus, Ohio, USA), with 1 mouse per chamber, testing transplanted mice simultaneously with controls. Motor activity (total and ambulating) was determined by infrared beam interruption (Opto-Varimex mini; Columbus Instruments). Resting oxygen consumption was calculated as the average of the points with less than 6 ambulating beam breaks per minute. The respiratory exchange ratio (RER; the ratio of carbon dioxide produced to oxygen consumed) was calculated from the resting data points. Oxidation of carbohydrate produces an RER of 1.00, whereas fatty acid oxidation results in an RER of approximately 0.70 (13).
Biochemical assays.
Glucose (nonfasting) was measured using a Glucometer Elite (Bayer Corp., Elkhart, Indiana, USA). Triglycerides (#339-11; Sigma Chemical Co., St. Louis, Missouri, USA), FFA (#1383175; Boehringer Mannheim, Indianapolis, Indiana, USA), β-hydroxybutyrate (#907979; Boehringer Mannheim), and blood urea nitrogen (BUN; #640; Sigma Chemical Co.) were assayed according to the suppliers’ procedures. Insulin and leptin were measured by RIA (#SRI-13K and #ML-82K; Linco Research Inc., St. Charles, Missouri, USA).
Assays of glucose homeostasis.
In vitro glucose uptake into isolated extensor digitorum longus muscle was measured using [3H]2-deoxyglucose, with [14C]mannitol to correct for extracellular fluid (14). Briefly, muscles were isolated and allowed to recover for 3 hours in Krebs Heinsleit buffer in a 95%O2, 5% CO2 atmosphere and were then incubated in the presence or absence of 10 mU/mL insulin (Humulin R; Eli Lilly, Indianapolis, Indiana, USA) for 20 minutes, followed by addition of 100 μM (2.4 μCi) [1,2-3H]2-deoxyglucose, and 0.7 μCi [1-14C]mannitol. After 20 minutes at 30°C, the tissue 3H and 14C were measured.
Glucose tolerance tests were performed essentially as described (15).
In vivo glucose uptake into muscle and adipose tissue was measured by intraperitoneal injection of [1-14C]2-deoxyglucose (10 μCi, 51 Ci/mmol; ICN Radiochemicals Inc., Irvine, California, USA) and insulin (0.75 mU/g). After 45 minutes, tissues were removed and the [14C]2-deoxyglucose 6-phosphate in muscle and fat was quantitated (16).
Statistical analysis.
Values are reported as means ± SEM. Statistical significance was determined using t test or ANOVA, with differences considered significant at P < 0.05 (2 tailed).
Results
Fat transplantation is feasible.
To test the role of WAT in reversing the metabolic derangement of the A-ZIP/F-1 mice, we introduced wild-type fat by surgical implantation. Previous fat transplantation studies in mice used small grafts (< 50 mg) (17, 18). We reasoned that a larger amount of adipose tissue might be needed. Pieces of parametrial fat, up to 150 mg each, were successfully implanted subcutaneously. These grafts had a healthy gross and microscopic appearance, including the presence of blood vessels and nerves (Figure 1, a and b). Larger grafts showed necrosis and poor viability, so multiple implantation sites were used for transplantation of more than 150 mg of fat.
Figure 1.
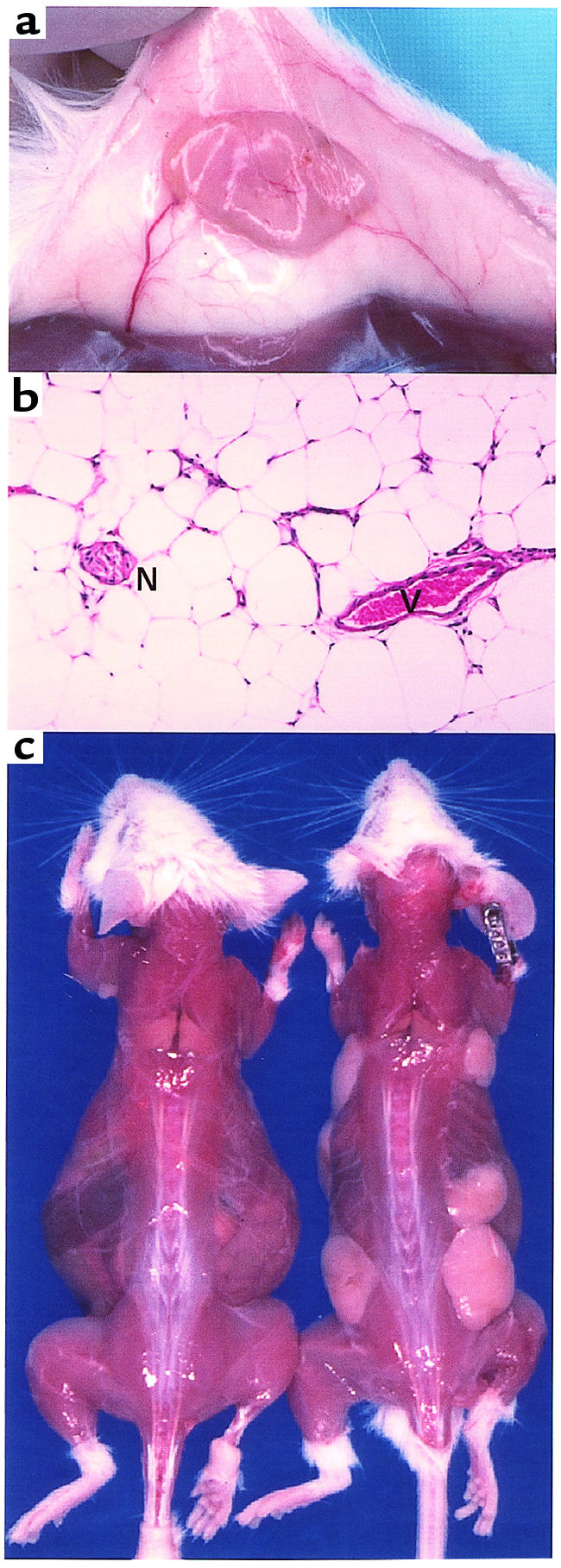
(a) Adipose tissue graft 3 weeks after transplantation. The graft (yellow, at center, originally 100 mg), attached to the skin (white), was dissected from the muscle (brown at bottom). Note the blood vessels supplying the graft. (b) Adipose tissue graft 13 weeks after transplantation. Hematoxylin and eosin stain. Original magnification, ×200. Note the blood vessel (V) and nerve (N). (c) A-ZIP/F-1 mice 13 weeks after transplantation. Skin was dissected from a sham-operated mouse (left) and from a mouse that received 900 mg of parametrial fat (right) in seven grafts (a ventral graft is not visible).
Fat transplantation reverses the A-ZIP/F-1 phenotype.
In the first experimental series, 900 mg of parametrial fat was transplanted into 5-week-old A-ZIP/F-1 mice. At 13 weeks after transplantation, 40 of 42 grafts appeared healthy (Figure 1c), although an occasional scarred area was visible. The total weight of transplanted fat increased 1.44 ± 0.23 fold (n = 23), with the greatest enlargement being 4.7-fold.
The A-ZIP/F-1 mice were already diabetic at transplantation, with serum glucose approximately 2-fold elevated and insulin approximately 50-fold elevated, compared with wild-type mice (Figure 2). Fat transplantation reversed the diabetes, improving both the glucose and insulin levels. By 3 weeks after transplantation, the glucose was nearly normal and remained so for the next 10 weeks (Figure 2). Insulin levels increased in sham-operated mice (from 45 ± 4 to 188 ± 63 ng/mL), whereas in transplanted mice (initially 53 ± 10 ng/mL) levels decreased through week 8 (2.1 ± 0.3 ng/mL), before increasing by week 13 (to 9.7 ± 3.7 ng/mL). These results suggest that transplantation improves insulin sensitivity.
Figure 2.

Fat transplantation improves plasma glucose and insulin levels and reduces postweaning growth in A-ZIP/F-1 mice. The sham-operated (filled circles [n = 5, except n = 3 at 13 weeks]) and transplanted animals (open circles [n = 6], 900 mg of parametrial fat) were significantly different beginning 5 weeks after transplantation for glucose, 2 weeks for insulin, and 3 weeks for body weight. The shaded region is the normal range for plasma/serum glucose for fed FVB/N mice (mean ± 2 SD; 150–306 mg/dL; n = 84).
Fat transplantation also reversed other aspects of the A-ZIP/F-1 phenotype, including the polyuria, polydypsia, polyphagia, and glucosuria. The transplanted mice had less enlarged livers (1.9 ± 0.1 g in transplanted versus 3.1 ± 0.5 g in shams); histological examination revealed less hepatic steatosis (Figure 3), and hepatic triglyceride levels were reduced but not normalized (76 ± 9 μmol/g in transplanted versus 199 ± 29 μmol/g in shams and 28 ± 2 μmol/g in wild-type mice). Adult A-ZIP/F-1 mice have larger bodies and organs than wild-type mice, presumably due to the IGF-like effects of very high insulin levels (9). Fat transplantation (but not sham operation) prevented the continued adult growth of the A-ZIP/F-1 mice (Figure 2), and these mice had smaller spleens and kidneys (data not shown).
Figure 3.

Histology of the liver 13 weeks after transplantation of 900 mg of parametrial fat. Sections from sham operated (left) and transplanted (right) A-ZIP/F-1 mice were stained with hematoxylin and eosin. Original magnification, ×50. Note the large number of vacuolated hepatocytes (due to lipid deposition) located predominantly in the centrilobular zone of the liver in the sham-operated but not the transplanted mice.
At the time of transplantation at 5 weeks, A-ZIP/F-1 mice had enlarged pancreatic islets (data not shown). Despite the dramatic decrease in insulin levels with transplantation, the islets were not noticeably smaller. Transplantation did improve the histological appearance of the β cells, as evidenced by a more uniform shape of the cells and nuclei, as well as a more uniform pattern of insulin immunostaining (not shown).
Glucose tolerance testing was performed on transplanted and control mice. Fasting normalizes glucose levels in A-ZIP/F-1 mice (19). Glucose intolerance, with delayed return of the glucose to basal, was present in sham-operated, but not transplanted, A-ZIP/F-1 mice (Figure 4a). Whole-body insulin resistance, as measured with an insulin tolerance test, was reduced by transplantation (Figure 4b). These data suggest that pancreatic β-cell function is relatively preserved but that there is insulin resistance in A-ZIP/F-1 mice, which is reversed by fat transplantation. Next, in vivo insulin-stimulated glucose uptake into specific tissues was measured using [14C]2-deoxyglucose (Figure 4c). Transplanted adipose tissue was functional, taking up glucose as effectively as native adipose tissue in wild-type mice. Skeletal muscle from transplanted fat took up 4 times more glucose than muscle in sham-operated mice. Thus the transplanted fat shows normal glucose uptake and reverses the in vivo skeletal muscle insulin resistance.
Figure 4.

Effect of fat transplantation on insulin sensitivity of A-ZIP/F-1 mice. (a) Female mice (n = 3–9/group; transplanted with 900 mg of adipose tissue 5 weeks earlier) were fasted for 8 hours, then glucose (2 mg/g, intraperitoneally) was injected and blood glucose was measured at the indicated times. *P < 0.001 WT versus sham; +P < 0.02 transplanted versus sham. (b) Male mice (n = 3 to 9/group; transplanted with 900 mg of adipose tissue 8 weeks earlier) were fasted for 15–21 hours and then injected with insulin (0.75 mU/g), and blood glucose was measured at the indicated times. *P < 0.03 transplanted versus sham. (c) Tissue uptake of [14C]2-deoxyglucose in mice (n = 3/group, a subset of those in b) was measured 45 minutes after a single intraperitoneal injection. Muscle uptake was measured in gastrocnemius muscle and in either epididymal (WT) or transplanted adipose tissue. +P = 0.07, sham A-ZIP/F-1 versus wild-type; *P = 0.004, transplanted versus sham A-ZIP/F-1. dpm, disintegrations per minute. (d) Female mice (n = 4/group; transplanted with 900 mg of adipose tissue 6 weeks earlier) were fasted for 13 hours, then [3H]2-deoxyglucose (2-DG) uptake into extensor digitorum longus muscle in the absence (basal) or presence of insulin was measured. *P < 0.001 versus basal WT; **P = 0.01 versus insulin-treated WT; +P = 0.01 versus insulin-treated sham A-ZIP/F-1.
In vitro insulin responsiveness was measured in skeletal muscle (Figure 4d). Reduced responsiveness was observed in A-ZIP/F-1 mice, and this was improved by fat transplantation. These results demonstrate that transplantation of 900 mg of parametrial fat into 5-week-old mice reverses much of the A-ZIP/F-1 phenotype, indicating that the lack of fat is the primary cause of these aspects of lipoatrophic diabetes.
Inguinal fat works as well as parametrial fat.
There is much evidence that different fat depots are not metabolically equivalent. For example, the risk of diabetes and lipid abnormalities is greater with increased visceral adiposity than with an excess of subcutaneous fat (20). We compared the antidiabetic effects of inguinal (subcutaneous) and parametrial fat (intraperitoneal) fat, transplanting 600 mg of each subcutaneously. The 2 types of fat were equally efficient in reversing the diabetes, decreasing glucose levels to near normal and insulin by 17- to 35-fold (Figure 5) and preventing weight gain (not shown).
Figure 5.

Inguinal and parametrial fat have similar effects on serum glucose, insulin, triglyceride, and FFA levels in the A-ZIP/F-1 mice. A-ZIP/F-1 were sham-operated (filled circles [n = 6]) or received 600 mg of parametrial (open circles [n = 7]) or inguinal (filled triangles [n = 6]) fat. Wild-type controls (open triangles [n = 5]) are age-matched mice from the experiment in Figure 8.
Fat transplantation has little effect on serum FFAs and triglycerides.
Characteristic features of the A-ZIP/F-1 phenotype include high serum triglycerides and FFAs (9). Increased FFAs may increase glucose levels via the glucose-fatty acid cycle, in which muscle burns FFA in preference to glucose (21). Interestingly, circulating FFAs were changed little by fat transplantation, being slightly lower in transplanted than sham mice at 3–5 weeks after surgery, but remaining higher than in the wild-type controls (Figure 5; similar results were obtained with 900 mg transplants). Similarly, serum triglyceride levels were only slightly decreased 3 weeks after fat transplantation, remaining 2- to 3-fold higher than in wild-type mice. Thus, fat transplantation was less effective in normalizing FFA and triglyceride than glucose and insulin levels.
Transplanted fat is a mobilizable, but insufficient source of energy.
We asked whether the triglyceride in the transplant was available for metabolism. The shift in fuel usage from carbohydrate to fatty acid is measurable as a decrease in the RER. Fat transplantation had no effect on the A-ZIP/F-1 RER in the fed state, but after a 4-hour fast, the transplanted mice decreased their RER more than the sham-operated mice (Figure 6), suggesting that the graft triglycerides were released and burned.
Figure 6.

Effect of fat transplantation on RER. Resting RER was measured 10 weeks after transplantation at 24°C in mice either sham-operated (n = 5) or transplanted with 900 mg of parametrial fat (n = 6). The animals were fasted starting at 0900 hours, and data were collected for 2 hours starting 3 hours later. Fed data were obtained using the same animals at the same time of the day a day before the fast. The difference between sham and transplanted mice was significant in the fasted state (P = 0.02). The drop in RER with fasting was significant in both groups (P < 0.01).
During a 24-hour fast, A-ZIP/F-1 mice show a drop in serum FFA, induction of protein catabolism, no increase in β-hydroxybutyrate levels, and they enter torpor (19). Despite the significant improvement in metabolic parameters, A-ZIP/F-1 mice transplanted with 600 mg of fat were similar to sham-operated controls in their response to a 24-hour fast. Both groups lost approximately 17% of body weight and entered torpor, as evidenced by a 7–10°C drop of body temperature. The mean FFA levels were not different between the sham and transplanted mice, but a few mice responded to fasting similarly to wild-type animals (Figure 7). Both the sham and transplanted groups also had an approximately 2-fold elevated fasting BUN, suggesting increased protein catabolism (Figure 7), and did not increase their β-hydroxybutyrate level with fasting (data not shown). Notably, the transplanted fat pads (originally 100 mg) were larger in the fed than the fasted mice (176 ± 11 mg [n = 12] versus 123 ± 7 mg [n = 30]; P < 0.001). These results demonstrate that the fat in the grafted tissue can be mobilized, but the grafts do not provide enough fuel to tolerate 24-hour fast.
Figure 7.

Effect of a 24-hour fast on serum FFA and BUN levels in A-ZIP/F-1 mice 8 weeks after transplantation of 600 mg of fat. The A-ZIP/F-1 mice (n = 6–7/group) are from the series shown in Figure 5. The wild-type controls (n = 3) are age- and sex-matched mice. The fed (filled circles) sera were obtained a week before fasting (open circles) in the A-ZIP/F-1 mice and a day before fasting in the wild-type mice.
Antidiabetic effect of adipose grafts is dose dependent.
We next investigated the effect of varying the amount of transplanted fat. Serum leptin concentration was measured as an indicator of adipose function. In A-ZIP/F-1 mice, free leptin levels are reduced 10-fold, compared with the wild-type mice (9). At these levels, the RIA is inaccurate, so we used serum from leptin-deficient ob/ob mice (22) to determine the assay background. The level in the ob/ob, sham, and 100-mg groups were not different, whereas the 300-mg and 900-mg groups had significantly increased the serum leptin levels (Figure 8a). Thus, the transplanted fat secretes leptin in proportion to the amount transplanted.
Figure 8.

Fat transplantation increases serum leptin and improves insulin levels in a dose-dependent way. A-ZIP/F-1 mice (n = 5–7/group) were transplanted with 100, 300, or 900 mg of parametrial fat. (a) Serum leptin levels were measured 13 weeks after transplantation, with serum from ob/ob mice indicating the assay background. Leptin levels in the 900- and 300-mg groups were significantly different from the sham group. (b) Serum insulin, triglyceride, and FFA levels were measured at the indicated times in the sham-operated (filled circles), 100-mg (open circles), 300-mg (filled triangles), and 900-mg (open triangles) transplanted A-ZIP/F-1 mice and in wild-type controls (filled squares).
The improvement in the glucose and insulin levels was dose dependent, with 900 mg more effective than 300 or 100 mg (Figure 8b and data not shown). In all transplanted groups, the maximum effect on insulin levels was achieved at approximately 6 weeks, being 1.1, 3.4, and 8.9 ng/mL in the 900-, 300-, and 100-mg groups, compared with 113 ng/mL in the sham and 0.5 ng/mL in the wild-type controls. Beginning approximately 8 weeks after transplantation, the insulin levels gradually increased in all groups. The rapidity of the initial improvement was also dependent on the amount of adipose tissue transplanted, taking 2, 4, or 6 weeks to fall by a similar amount in the 900-, 300-, and 100-mg groups, respectively. The effects of fat transplantation on triglyceride and FFA levels were also dose dependent, but less obviously so due to the smaller difference between A-ZIP/F-1 and control mice and to biologic variation in the levels.
Discussion
We show that transplantation of wild-type fat into lipoatrophic mice reverses their diabetic phenotype. This demonstrates that a lack of fat can be the primary cause of the constellation of metabolic and biochemical findings known as lipoatrophic diabetes. These results were obtained in A-ZIP/F-1 mice, but it is likely that this conclusion applies to the other murine models and human forms of this disease. Adipose tissue transplantation has been used in humans with lipodystrophy, but typically small amounts of autologous tissue were transplanted, without increasing the body fat mass (23, 24).
Adipose tissue can exert its antidiabetic actions via endocrine or metabolic mechanisms. Possible endocrine mechanisms include secretion of leptin or TNF-α, both of which affect insulin sensitivity (25–28). Possible metabolic mechanisms include adipose uptake of glucose, triglyceride, and/or FFA. The transplanted adipose tissue was able to take up glucose as efficiently as in wild-type mice. We demonstrate that large amounts of transplanted tissue are needed to reverse the diabetes. This is consistent with metabolic mechanisms and those endocrine mechanisms in which hormone production is proportional to fat mass (e.g., leptin). Recently, leptin infusion was shown to reverse completely the diabetes of a less severely lipoatrophic mouse (29). However, we find that leptin is only minimally effective at reversing the diabetes of the A-ZIP/F-1 mice (O. Gavrilova, B. Marcus-Samuels, L.R. Leon, C. Vinson, and M.L. Reitman, manuscript submitted for publication). How, specifically, might the loss of adipose tissue lead to insulin resistance via a metabolic mechanism? We have shown that the improvement in insulin resistance is not due simply to massive glucose uptake by transplanted fat, but also to increased uptake into muscle. The lack of WAT causes increased triglyceride synthesis and storage by the liver and higher circulating FFA and triglyceride levels. However, it is not clear whether it is the lack of WAT per se, the high triglycerides or FFA, or some other factor, that causes the insulin resistance. One possibility, the Randle hypothesis, is that fatty acids are oxidized in preference to glucose by skeletal muscle via the glucose-fatty acid cycle, resulting in muscle resistant to insulin-stimulated glucose utilization (21). Indeed, in A-ZIP/F-1 mice, insulin-stimulated glucose uptake is severely impaired, both in vitro (this article) and under clamp conditions (J.K. Kim, O. Gavrilova, Y. Chen, M.L. Reitman, and G.I. Shulman, manuscript submitted for publication). However, arguing against the Randle hypothesis, the respiratory exchange ratio in both A-ZIP/F-1 mice (O. Gavrilova, unpublished observations) and lipoatrophic humans (E. Arioglu and S.I. Taylor, personal communication) is the same as or higher than controls, demonstrating that fatty acid oxidation is not increased. Another possible explanation for the insulin resistance is that the elevated FFA act via regulatory mechanisms, for example transcriptionally, by binding to PPAR-like transcription factors or biochemically, by inhibiting glucose uptake. FFA-mediated mechanisms are also leading candidates to explain the insulin resistance seen in obesity. Surprisingly, we observed that fat transplantation in the A-ZIP/F-1 mice lowered the glucose and insulin levels much more dramatically than the FFA and triglyceride levels. This does not rule out FFA as mediators of insulin resistance but suggests that the hypothesis should be carefully evaluated. Analysis of production rates and disposal rates, particularly of triglycerides and FFA within specific tissues should provide insight into this question.
Despite the dramatic improvement, the insulin levels and response to fasting were not completely normalized. It is possible that not enough fat was transplanted. Even at 900 mg, the grafts are less than 4% of body weight, less than the normal 5–23% body fat (30). Another possibility is that the transplanted fat cannot function properly. Normally lipolysis is stimulated via the sympathetic nervous system. The transplanted fat did contain nerves, but it is not known whether these nerves can stimulate lipolysis. Finally, we did not restore brown adipose tissue with transplantation, and this deficiency could account for the lack of total normalization of the phenotype.
After a 1-week lag (presumably required for graft vascularization), there is a biphasic response of the insulin levels to fat transplantation. From 1 to 6 weeks, there is continued improvement in the diabetic phenotype. After this time, there is a rise in the insulin levels, suggesting increasing insulin resistance. Because the fat grafts appeared healthy and vascularized at sacrifice, necrosis of adipose tissue does not appear to explain the rising insulin levels. The increased insulin levels may reflect an increase in the intrinsic insulin resistance of the recipients, either due to their aging and/or to progression of the disease (as the mice were already diabetic at transplantation). The rising insulin levels might also be explained if the transplanted adipocytes have a limited functional capacity. For example, the fat grafts are from young donors and contain small adipocytes, which are thought to be more effective in glucose disposal than bigger cells (31). Enlargement/engorgement of the adipocytes (or exhaustion of a preadipocyte precursor pool) during the course of the experiment might lead to their decreased efficiency. This hypothesis can be tested by comparing fat grafts differing in adipocyte size/triglyceride content.
Transplantation of mutant adipose tissue into A-ZIP/F-1 mice will enable investigation of the antidiabetic effects of fat. For example, the role of leptin can be examined by transplantation of fat from ob/ob mice. The contribution of adipose TNF-α can similarly be investigated using TNF-α–/– fat. Using the genetically appropriate donors, one can also ask the following questions: Are normal adipose insulin signaling and glucose disposal required for the antidiabetic action of fat? Similarly, are the abilities to take up FFA and store triglycerides needed? Fat transplantation into A-ZIP/F-1 mice is a powerful new technique for studying the communication, both endocrine and metabolic, between adipose tissue and the rest of the body.
Our data demonstrate that a lack of fat causes diabetes and that we can reverse the diabetes by surgical reconstitution with normal adipose tissue. Can a similar improvement be achieved in humans? If the requirement for a large graft amount can be met, patients with genetic lipoatrophy should benefit from adipose tissue transplantation.
Acknowledgments
We thank S. Taylor for continued encouragement and excellent advice, E. Arioglu for stimulating discussions, S. Cushman for support, and J. Owens for immunohistochemistry. S. Bi, D. Camerini-Otero, A. Ginsberg, D. LeRoith, J. Moitra, and L. Weinstein provided helpful comments on the manuscript.
References
- 1.Foster, D.W. 1994. The lipodystrophies and other rare disorders of adipose tissue. In Harrison’s principles of internal medicine. K.J. Isselbacher et al., editors. McGraw-Hill. New York, NY. 2131–2136.
- 2.Seip M, Trygstad O. Generalized lipodystrophy, congenital and acquired (lipoatrophy) Acta Paediatr Suppl. 1996;413:2–28. doi: 10.1111/j.1651-2227.1996.tb14262.x. [DOI] [PubMed] [Google Scholar]
- 3.Rossini, A.A., and Cahill, G.F.J. 1979. Lipoatrophic diabetes. In Endocrinology. Volume 2. L.J. DeGroot et al., editors. Grune & Stratton. New York, NY. 1093–1097.
- 4.Vigouroux C, et al. Genetic exclusion of 14 candidate genes in lipoatropic diabetes using linkage analysis in 10 consanguineous families. J Clin Endocrinol Metab. 1997;82:3438–3444. doi: 10.1210/jcem.82.10.4292. [DOI] [PubMed] [Google Scholar]
- 5.Peters JM, et al. Localization of the gene for familial partial lipodystrophy (Dunnigan variety) to chromosome 1q21-22. Nat Genet. 1998;18:292–295. doi: 10.1038/ng0398-292. [DOI] [PubMed] [Google Scholar]
- 6.Jackson SN, et al. A defect in the regional deposition of adipose tissue (partial lipodystrophy) is encoded by a gene at chromosome 1q. Am J Hum Genet. 1998;63:534–540. doi: 10.1086/301971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garg A, et al. A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. J Clin Endocrinol Metab. 1999;84:3390–3394. doi: 10.1210/jcem.84.9.6103. [DOI] [PubMed] [Google Scholar]
- 8.Cao H, Hegele RA. Nuclear lamin A/C#R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9:109–112. doi: 10.1093/hmg/9.1.109. [DOI] [PubMed] [Google Scholar]
- 9.Moitra J, et al. Life without white fat: a transgenic mouse. Genes Dev. 1998;12:3168–3181. doi: 10.1101/gad.12.20.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ross SR, Graves RA, Spiegelman BM. Targeted expression of a toxin gene to adipose tissue: transgenic mice resistant to obesity. Genes Dev. 1993;7:1318–1324. doi: 10.1101/gad.7.7b.1318. [DOI] [PubMed] [Google Scholar]
- 11.Burant CF, et al. Troglitazone action is independent of adipose tissue. J Clin Invest. 1997;100:2900–2908. doi: 10.1172/JCI119839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimomura I, et al. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: model for congenital generalized lipodystrophy. Genes Dev. 1998;12:3182–3194. doi: 10.1101/gad.12.20.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McLean, J.A., and Tobin, G. 1987. Animal and human calorimetry. Cambridge University Press. Cambridge, United Kingdom. 338 pp.
- 14.Lauro D, et al. Impaired glucose tolerance in mice with a targeted impairment of insulin action in muscle and adipose tissue. Nat Genet. 1998;20:294–298. doi: 10.1038/3112. [DOI] [PubMed] [Google Scholar]
- 15.Bruning JC, et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell. 1998;2:559–569. doi: 10.1016/s1097-2765(00)80155-0. [DOI] [PubMed] [Google Scholar]
- 16.Kim JK, Wi JK, Youn JH. Plasma free fatty acids decrease insulin-stimulated skeletal muscle glucose uptake by suppressing glycolysis in conscious rats. Diabetes. 1996;45:446–453. doi: 10.2337/diab.45.4.446. [DOI] [PubMed] [Google Scholar]
- 17.Bach-Mortensen N, Romert P, Ballegaard S. Transplantation of human adipose tissue to nude mice. Acta Pathol Microbiol Scand [C] 1976;84:283–289. doi: 10.1111/j.1699-0463.1976.tb00031.x. [DOI] [PubMed] [Google Scholar]
- 18.Ashwell M, Meade CJ, Medawar P, Sowter C. Adipose tissue: contributions of nature and nurture to the obesity of an obese mutant mouse (ob/ob) Proc R Soc Lond B Biol Sci. 1977;195:343–353. doi: 10.1098/rspb.1977.0014. [DOI] [PubMed] [Google Scholar]
- 19.Gavrilova O, et al. Torpor in mice is induced by both leptin-dependent and -independent mechanisms. Proc Natl Acad Sci USA. 1999;96:14623–14628. doi: 10.1073/pnas.96.25.14623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kissebah AH, Krakower GR. Regional adiposity and morbidity. Physiol Rev. 1994;74:761–811. doi: 10.1152/physrev.1994.74.4.761. [DOI] [PubMed] [Google Scholar]
- 21.Randle PJ, Garland PB, Hales CN, Hewsholme EA. The glucose fatty-acid cycle, its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 23.Langhof H, Zabel R. Zur lipodystrophia progressiva. Arch Klin Exp Dermatol. 1960;210:313–321. [PubMed] [Google Scholar]
- 24.Koshy CE, Evans J. Facial contour reconstruction in localised lipodystrophy using free radial forearm adipofascial flaps. Br J Plast Surg. 1998;51:499–502. doi: 10.1054/bjps.1998.0101. [DOI] [PubMed] [Google Scholar]
- 25.Kamohara S, Burcelin R, Halaas JL, Friedman JM, Charron MJ. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature. 1997;389:374–377. doi: 10.1038/38717. [DOI] [PubMed] [Google Scholar]
- 26.Rossetti L, et al. Short term effects of leptin on hepatic gluconeogenesis and in vivo insulin action. J Biol Chem. 1997;272:27758–27763. doi: 10.1074/jbc.272.44.27758. [DOI] [PubMed] [Google Scholar]
- 27.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610–614. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 28.Schreyer SA, Chua SC, Jr, LeBoeuf RC. Obesity and diabetes in TNF-alpha receptor-deficient mice. J Clin Invest. 1998;102:402–411. doi: 10.1172/JCI2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401:73–76. doi: 10.1038/43448. [DOI] [PubMed] [Google Scholar]
- 30.West DB, Boozer CN, Moody DL, Atkinson RL. Dietary obesity in nine inbred mouse strains. Am J Physiol. 1992;262:R1025–R1032. doi: 10.1152/ajpregu.1992.262.6.R1025. [DOI] [PubMed] [Google Scholar]
- 31.Okuno A, et al. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. J Clin Invest. 1998;101:1354–1361. doi: 10.1172/JCI1235. [DOI] [PMC free article] [PubMed] [Google Scholar]