

Abstract
Many chronic pulmonary diseases are associated with pulmonary hypertension (PH) and pulmonary vascular remodeling, which is a term that continues to be used to describe a wide spectrum of vascular abnormalities. Pulmonary vascular structural changes frequently increase pulmonary vascular resistance, causing PH and right heart failure. Although rat models had been standard models of PH research, in more recent years the availability of genetically engineered mice has made this species attractive for many investigators. Here we review a large amount of data derived from experimental PH reports published since 1996. These studies using wild-type and genetically designed mice illustrate the challenges and opportunities provided by these models. Hemodynamic measurements are difficult to obtain in mice, and right heart failure has not been investigated in mice. Anatomical, cellular, and genetic differences distinguish mice and rats, and pharmacogenomics may explain the degree of PH and the particular mode of pulmonary vascular adaptation and also the response of the right ventricle.
Keywords: right ventricle failure, transgenic, vascular remodeling, Sugen, SU5416
“The mice, which helplessly find themselves between the cat's teeth, acquire no merit from their enforced sacrifice.”
—Mahatma Gandhi (1869–1948)
the discovery of hypoxic pulmonary vasoconstriction by von Euler and Liljestrand (24) brought attention to the “lesser circulation” and triggered the first series of systematic studies of the lung circulation and of pulmonary hypertension in both animals and normal people, at sea level and at high altitude (37, 38, 105, 119, 121). Later on, pulmonary hypertension (PH) was described in patients with a wide variety of diseases, ranging from chronic lung diseases (chronic obstructive pulmonary disease, collagen vascular diseases), infections (human immunodeficiency virus/acquired immunodeficiency syndrome, schistosomiasis) and heart diseases (congenital and acquired), to idiopathic and hereditary forms of PH. For recent reviews of the classification of PH and diseases associated with PH, see Refs. 3, 22, 26, 62, 87, 88. The epidemic of severe PH in three European countries associated with the intake of the amphetamine-related anorexigen aminorex fumarate generated an even greater interest in the pathogenesis of human PH diseases (2). This epidemic prompted the first World Health Organization meeting on PH and promoted intense research activities aimed to build animal models of drug-induced PH (65). Early studies conducted at the Cardiovascular Pulmonary Research Laboratory in Denver, Colorado, described species-specific differences in the response of the lung circulation to chronic hypoxia and presented the concept of “responders and nonresponders.” For example: cattle develop very severe PH when exposed to chronic hypoxia whereas sheep do not (38). Conversely, commercially bred laboratory rats, when not afflicted by bacterial or viral infections (36), develop moderate PH associated with significant pulmonary vascular remodeling (mainly media muscularization) and right ventricular hypertrophy (RVH) when exposed to chronic hypoxia (41, 45, 114). As a consequence, the species rattus, and several strains of this species (23, 41, 48, 49, 77), became the preferred animal for mechanistic studies of PH and preclinical drug trials (78, 84, 93, 100, 131, 132) for more than 50 years (95).
The advent and development of genetically modified mice, as tools to dissect cellular and molecular signaling pathways, created an understandable desire to generate mouse models for PH studies. Possibly the first experiments reporting the use of transgenic mice for PH research were conducted in 5-lipoxygenase (5-LO) knockout (KO) mice (5-LO−/−) exposed to chronic hypoxia (112). The 5-LO KO mice were introduced into the study protocol to test whether arachidonic acid metabolites, via the 5-lipoxygenase pathway, contributed to the development of chronic hypoxia-induced PH, and supported the pharmacological experimental use of the 5-LO inhibitor A866. These early studies demonstrated two main points: 1) the degree of hypoxia-induced PH and pulmonary vascular remodeling was significantly less in mice compared with rats; and 2) the relatively small degree of PH (compared with rats) and the extent of lung vessel structure changes were even less prominent in the mice lacking 5-LO, making a case for inflammation in the pathogenesis of chronic hypoxia-induced PH (112). The observed robust differences in the response to chronic hypoxia between mice and rats motivated a comparative mouse/rat study, evaluating the gene expression changes induced in the lung tissues following exposure to an identical degree and duration of chronic hypoxia (45). The microarray results showed a categorically different gene expression pattern between mouse and rat lung tissues. The expression of a number of genes was increased in the rat lungs but decreased in the mouse lungs (45). Chronic hypoxia finds the mouse lung circulation relatively unresponsive.
In recent years a considerable number of transgenic mice have been generated to investigate mechanisms of pulmonary hypertension (Table 1). In particular, the report of associations between mutations in the bone morphogenetic protein receptor 2 (BMPR2) gene with both familiar and idiopathic forms of pulmonary arterial hypertension (PAH) (46, 59, 70, 97) led to multiple attempts to generate novel animal models of PAH. In addition to BMPR2 KO mice, researchers have targeted other genes suspected to be involved in the pathogenesis of PAH (Table 1). Indeed, the transgenic mouse studies have strongly influenced the way we think of the pathogenesis of PAH and helped shift the early mechanical concepts of pressure (and flow) dependency toward disease models built on cell-to-cell interactions, immune dysregulation, metabolic changes, and abnormal cell phenotypes (19, 67, 75, 76, 101, 115, 123).
Table 1.
Pulmonary hypertension and pulmonary vasculature remodeling in mouse models published from 1996 to 2011
| RVSP, mmHg |
RV/LV+S |
Histology |
||||||
|---|---|---|---|---|---|---|---|---|
| Mouse Strain | Genetic Modification | Normoxia | Hypoxia | Normoxia | Hypoxia | Normoxia | Hypoxia | Author |
| C57BL6/J | Wild type/Normal | 13 | 18 | N/A | N/A | N/A | ↑ musc. | Vanderpool et al. (108) |
| C57BL6/J | Wild type/Normal | 10–20 | 14–26 | 0.24 | 0.31 | N/A | N/A | Tabima et al. (98) |
| C57BL6/J | 5-LO KO | N/A | N/A | 0.25 | 0.32 | Normal | ↓ musc. | Voelkel et al. (112) |
| C57BL6/J | PGI2 synthase OE | 30 | 30 | N/A | N/A | Normal | ↓ musc. | Geraci et al. (33) |
| C57BL6/J | HO-1 KO | 12 | 19 | N/A | N/A | N/A | modest musc. | Yu et al. (129) |
| FVB/N | SMC BMPR2 dom. neg | 44 | N/A | 0.315 | N/A | ↑ musc. | N/A | West et al. (116) |
| C57BL6/J | BMPR2± | 12 | N/A | N/A | N/A | N/A | N/A | Song et al. (91) |
| BMPR2± with Ad5LO OE | 23 | N/A | N/A | N/A | N/A | ↑ musc. | ||
| C57BL6/J | 5-HTT OE | 33 | N/A | 0.37 | N/A | ↑ musc. | N/A | Guignabert et al. (39) |
| C57BL6/J | VIP KO | 29.5 | N/A | 0.34 | N/A | ↑ musc. | ↑ musc. | Said et al. (80) |
| R26R | PVEC BMPR−/− KO | 27 | N/A | 0.27 | N/A | ↑ musc. Occlusion | N/A | Hong et al. (42) |
| C57BL6/J | SMC PPAR-γ −/− KO | 29 | N/A | 0.45 | N/A | ↑ musc. | N/A | Hansmann et al. (40) |
| C57BL6/J | SMC BMPR2± | 24 | N/A | N/A | N/A | ↑ musc. | N/A | Song et al. (90) |
| SMC BMPR2± with MCT + Ad5LO | 38 | N/A | N/A | N/A | ↑ musc. Inflamm. | N/A | ||
| C57BL/6 | Eln−/− | 80 | N/A | ↑ RV hyp. | N/A | ↑ musc. | N/A | Shifren et al. (86) |
| FVB/N | SMC BMPR2 R899X | 39 | N/A | N/A | N/A | ↑ musc. | N/A | West et al. (117) |
| C57BL/6 | Wildtype with OVA exposure | 25 | 32 | 0.20 | N/A | ↑↑↑ musc. | ↑↑↑ musc. | Daley et al. (19) |
| C57BL6/J | TPH1 −/− KO with dexfenflur. | 15 | N/A | 0.23 | N/A | Normal | N/A | Dempsie et al. (20) |
| HumanSERT+ with dexfenflur | 40 | 24 | 0.27 | 0.30 | ↑ musc. | ↑ musc. | ||
| Wild type with dexfenflur | 30 | N/A | 0.21 | N/A | ↑ musc. | N/A | ||
| C57BL6/J | Notch −/− KO | 20 | 20 | 0.20 | 0.22 | N/A | ↑ musc. | Li et al. (56) |
| C57BL6/J | IL-6 OE | 35 | 63 | 0.36 | 0.69 | N/A | ↑ musc. Occlusion Inflamm. | Steiner et al. (94) |
| C57BL6/J | SMC ID-1−/− KO | 20 | 28 | 0.258 | 0.36 | Normal | ↑ musc. | Lowery et al. (57) |
| C57BL6/J | Nogo−/− | 15* | 15* | 0.16 | 0.23 | N/A | ↓ musc. | Sutendra et al. (96) |
| C57BL6/J | A2AR−/− | 39 | 45 | 0.26 | N/A | ↑ musc. | N/A | Xu et al. (122) |
| C57BL6/J | Wild type with imatinib | 23 | 25 | 0.29 | 0.34 | ↓ musc. | Gambaryan et al. (31) | |
| N/A | CTGF OE | 22 | N/A | 0.42 | ↑ musc. | N/A | Chen et al. (15) | |
| C57BL6/J | HumanMMP9 OE | 29 | N/A | N/A | N/A | N/A | N/A | George et al. (32) |
| HumanMMP9 OE with MCT | 90 | N/A | N/A | N/A | ↑ musc. Inflamm | N/A | ||
| C57BL6/J | RKIP −/− | 25 | 35 | 0.28 | 0.35 | N/A | N/A | Morecroft et al. (66) |
| FVB/N | SMC BMPR2 R899X | 43 | N/A | 0.34 | N/A | ↑ musc. | N/A | Yasuda et al. (124) |
| SMC BMPR2 R899X with fasudil | 37 | N/A | 0.28 | N/A | ↓ musc. | N/A | ||
| C57BL6/J | COX-2 −/− KO | 13 | N/A | N/A | N/A | N/A | N/A | Seta et al. (85) |
| COX-2 −/− KO with MCT | 17 | N/A | N/A | N/A | Perivascular edema | N/A | ||
| FVB | HumanHO-1 OE | 20 | 21 | 0.21 | 0.22 | Normal | ↓ musc. | Vergadi et al. (109) |
| SV129 | Apelin KO | 25 | 33 | N/A | N/A | N/A | ↑ musc. | Chandra et al. (14) |
| C57BL6/J | Wild type/Normal | 22 | 35 | 0.20 | 0.45 | N/A | ↑ musc. | Yu et al. (130) |
| C57BL6/J | P53 KO | 18 | 35 | 0.21 | 0.35 | Normal | ↑ musc. | Mizuno et al. (64) |
| C57BL6/J | Wild type/Normal with SU5416 | 27 | 50 | 0.20 | 0.35 | Normal | ↑ musc. occlusion | Ciuclan et al. (16) |
| R26R | PVEC BMPR−/− KO | 37 | N/A | 0.23 | N/A | ↑ musc. | N/A | Burton et al. (13) |
| PVEC BMPR−/− KO with SCH527123 | 26 | N/A | 0.19 | N/A | ↓ musc. | N/A | ||
| CD1-B6.Cg 5Amc/J | Calpain-4 KO | 18 | 21 | 0.19 | 0.20 | Normal | ↓ musc. | Ma et al. (58) |
| C57BL6 | OX40L OE | 30 | N/A | 0.23 | N/A | ↑ musc of large vessels | N/A | Rabieyousefi et al. (74) |
| C57BL6 | S. Mansoni parasitic infection | 31 | N/A | 0.25 | N/A | ↑ musc. | N/A | Crosby et al. (18) |
RVSP, right ventricular systolic pressure; RV, right ventricle; LV, left ventricle; S, interventricular septum; N/A, not analyzed; KO, knockout; OE, overexpression; SMC, smooth muscle cell; PVEC, pulmonary vascular endothelial cell; 5-LO, 5-lipoxygenase; PGI2, prostacyclin; HO, heme oxygenase; BMPR2, bone morphogenetic protein receptor type 2; Ad5LO, adenoviral vector overexpressing 5-lipoxygenase; 5-HTT, 5-hydroxytryptamine transporter; VIP, vasoactive intestinal peptide; PPAR, peroxisome proliferator-activated receptor; MCT, monocrotaline; Eln, elastin; OVA, ovalbumin; TPH1, tryptophan hydroxylase-1; dexfenflur, dexfenfluramine; SERT, human serotonin receptor; IL-6, interleukin 6; ID-1, inhibitors of differentiation; A2aR, adenosine A2 receptor; CTGF, connective tissue growth factor; MMP, matrix metalloproteinase-9; RKIP, Raf-1 kinase inhibitor protein; COX-2, cyclooxygenase 2.
, mean pulmonary artery pressure.
Here we review many of the published studies of mouse models of PH and provide an overview of the hemodynamic- and tissue analysis-derived data for normal and hypoxia-challenged wild-type (WT) and transgenic mice. In addition, we report our own data derived from experiments designed to generate a mouse model of predictable and severe PH, guided by our own experience with the rat model of SU5416-induced VEGF receptor blockade and chronic hypoxia (100). Our experimental data obtained with different mouse strains and genetically engineered mice illustrate both the problems and opportunities associated with mouse models of chronic PH. Here, we evaluate the published data, identify several potential problems related to data acquisition and interpretation, and point out gaps that might explain divergent results. With this review, we also intend to contrast mouse models of PH with informative data derived from rat PH models, and we try to put the murine models in a perspective with the clinical and translational reality of severe human PAH.
Overview of Mouse Models of Pulmonary Hypertension
In the early studies of PH in mice, the animals were challenged with chronic hypoxia because monocrotaline had been found to be ineffective. This circumstance had been explained by the inability of mice to metabolize monocrotaline to its active metabolite (dehydromonocrotaline), which requires a CYP3A isoenzyme lacking in the mouse liver (35, 50, 120). Although the degree of hypoxia-induced PH developed in mice is rather small (98), precise measurements of cardiac output and right ventricular and central pulmonary arterial elastance can be made (104, 108). In 2004, West et al. (116) tested the hypothesis that the loss of BMPR2 signaling in smooth muscle cells was sufficient to cause PH in transgenic mice. The authors constructed a smooth muscle-specific transgenic mouse expressing a dominant-negative BMPRII variant, under the control of a tetracycline-gene switch. These mice were studied under anesthesia (heart rates 260 ± 40 bpm), and right ventricular systolic pressure (RSVP) was measured with a Millar transducer-tipped catheter. The RVSP ranged from 40 to 70 mmHg at Denver altitude. However, although the RVSP was increased, these transgenic mice mainly developed muscularization of the media but not severe intimal cell proliferation, which is the hallmark of human plexogenic PAH (106). In 2005, Song et al. (91) hypothesized that BMPRII haploinsufficiency required an additional hit to trigger vascular remodeling: in this particular case, an inflammatory hit (“two-hit hypothesis”). By means of an adenoviral vector, Song et al. overexpressed 5-lipoxygenase in the lungs of heterozygous mutant BMPR+/− mice and demonstrated a small increase in RVSP with modest pulmonary vascular changes. In contrast to the findings reported by West et al., Song's study reported that mice heterozygous for a BMPR2-null allele, which have a genetic defect similar to that of some patients with familial and idiopathic PAH, do not develop PH spontaneously (91).
It was not until 2008 that another group of researchers reported a BMPR2-KO mouse that developed spontaneous PH with evidence of vascular obliteration. Hong et al. (42) conditionally ablated the BMPRII gene specifically in pulmonary vascular endothelial cells, utilizing a novel endothelial Alk1-Cre transgenic mouse. This report was relevant given the fact that complete deletion of the BMPR2 gene had not been reported before, because BMPR2-null mice are embryonically lethal (BMPR2 is required for septation of the outflow tract of the mammalian heart and embryonic gastrulation) (8, 21). Hong et al. demonstrated that complete loss of BMPR2 in endothelial cells was sufficient to generate pulmonary vascular remodeling and spontaneous PH. However, there were two important findings: 1) There was a high variability within the transgenic mice (RVSP ranged from 20 to 56 mmHg), with only a third of the animals developing PH. 2) Even after organizing the mice into groups with and without PH, PH mice developed only a modest degree of right ventricular (RV) hypertrophy, even when the RVSP was elevated (Table 1). Although it has been reported that human plexiform lesions are characterized by a significant decrease in the expression of BMPR2 (in idiopathic, familiar, and some associated PAH forms) (4), Hong et al.'s study (42) exemplifies how, even after complete ablation, BMPR2 dysfunction is neither sufficient nor required for the development of complex vascular lesions or for the development of right ventricular failure (RVF).
The large variability in the RVSP and pulmonary vascular remodeling in BMPR2-KO mouse models complicates the evaluation of preclinical drug trials. To our knowledge there are only two studies that attempted an interventional preclinical trial in genetically engineered mice. Yasuda, West, and colleagues (124) treated mice expressing a dominant-negative BMPRII gene (with a termination mutation at amino acid 899) specific for smooth muscle cells (BMPR2R899X), with fasudil, a Rho kinase inhibitor. These mice developed spontaneous PH (RVSP of 43 mmHg) with mild RV hypertrophy. Fourteen days of treatment with fasudil ameliorated PH. However, the authors could not show a decrease in Rho kinase activation nor modified Smad protein phosphorylation (BMPR2 downstream signaling proteins) in treated animals compared with vehicle controls. Furthermore, the drug was not administered after PH had been established, which somewhat diminished the translational potential. The second study by Burton and collaborators (13) utilized the same endothelial cell restricted BMPR−/− mice published by Hong, Oh, and colleagues (42). Mice were treated with the CXCR1/2 antagonist SCH527123, with the rationale that CXCR1–2-mediated leukocyte migration plays a role in the development of pulmonary hypertension. SCH527123 treatment resulted in decreased muscularization of pulmonary arteries, decreased RVSP, and increased (systemic) cardiac output (13). Interestingly, in contrast to what had been published originally by Hong et al., Burton and collaborators did not report pulmonary angioobliteration.
The role of inflammation and immune dysfunction in the pathogenesis of pulmonary hypertension has been increasingly discussed in recent years, but whether inflammation is a cause or consequence of pulmonary vascular remodeling continues to be debated (71, 101, 111). It has now been 15 years since the report that patients with idiopathic PAH have increased plasma levels of interleukins 1β and 6 (47), and it has been recently reported that high levels of IL-6 predict mortality in idiopathic PAH patients (92). Other proinflammatory conditions associated with PAH are also characterized by increased levels of IL-6, such as systemic lupus erythematosus (128) and Castleman's disease (73). In 2009, Steiner et al. (94) generated a transgenic mouse using the Clara cell 10-kDa promoter to constitutively drive lung-specific expression of IL-6 (IL6-OE). They tested the hypothesis that IL-6 overexpression would be sufficient to generate pulmonary hypertension in mice. Under baseline conditions IL6-OE mice did not develop pulmonary hypertension. However, upon chronic hypoxia exposure, the IL6-OE mice developed severe pulmonary hypertension (average RVSP of 60 mmHg), neointimal proliferation, and robust RV hypertrophy. Furthermore, Steiner and collaborators demonstrated a significantly higher number of fully and partially occluded vessels compared with controls. The vascular lesions present in this transgenic model reproduced some molecular features of human plexiform lesions (76, 106), including increased expression of the angiogenic factors VEGF and PDGF, increased expression of prosurvival proteins such as survivin and Bcl-2, and increased phosphorylation of ERK. Most importantly, Steiner's study was the first to quantify the number of occluded vessels in a transgenic mouse model. Unfortunately, the authors did not evaluate whether this model of angiooblierative pulmonary hypertension was reversible upon return to normoxia. Reversibility is particularly important for the design of preclinical drug trials, since most patients with PAH with specific therapy do not experience a large drop in the pulmonary artery pressure (30) and the complex vascular lesions do not regress. Spontaneous regression of pulmonary hypertension becomes an impediment for PAH animal models and should be investigated. For example, Ciuclan et al. (16) reported a mouse model of PH utilizing a VEGF receptor blocker (SU5416) and chronic hypoxia in WT C57BL6 mice (Fig. 1A), based on the rat protocol originally described by Taraseviciene-Stewart in 2001 (100). This mouse model is particularly attractive because it does not require genetic manipulations. After three doses of SU5416 (as opposed to a single dose in the rat model) and 3 wk of exposure to 10% hypoxia, these mice develop PH (average RVSP 49 mmHg) with modest RV hypertrophy (RV/LV+S 0.35, where LV+S is left ventricle plus septum), at least, compared with the IL6-OE mouse model (RV/LV+S 0.69)(94) or the original SU5416/hypoxia rat model (RV/LV+S 0.77) (9, 11) (Fig. 1, B and D). The SU5416/hypoxia mouse model develops muscularization and, although not quantified, various degrees of concentric neointimal thickening. The main disadvantage of this model is that, upon return to normoxia, the vascular remodeling, pulmonary hypertension, and RV hypertrophy return to normal. In contrast, the relevance of the SU5416/hypoxia rat model is illustrated mainly by two aspects: 1) the high reproducibility of severe PAH is accompanied by angioobliterative lesions and RV failure (1, 100); and 2) akin to human disease, PAH in the SU5416/hypoxia rat model is progressive, nonreversible, fatal, and unresponsive to conventional PAH drug treatment (12).
Fig. 1.
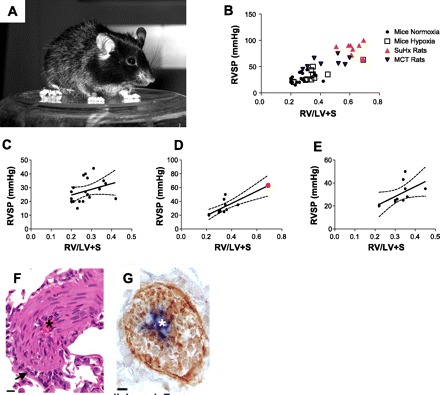
A: C57BL6 mouse. B: correlation between right ventricular systolic pressure (RVSP) and right ventricle (RV) to left ventricle (LV) plus septum (S) ratio (RV/LV+S) in mouse pulmonary hypertension (PH) models exposed to normoxia and hypoxia, compared with the combination of SU5416 and chronic hypoxia (SuHx) and monocrotaline (MCT)-injured rat models. C: correlation between RVSP and RV/LV+S in mouse models exposed to normoxia. Pearson coefficient 0.32, P = 0.150. D: correlation between RVSP and RV/LV+S in mouse models exposed to hypoxia. If all mouse models are included, the Pearson coefficient is 0.82, P = 0.0008. However, the correlation seems to be driven by an outlier, the IL6-OE mice (red dot). Thus, if the latter is not included in the statistical calculations, the coefficient becomes nonsignificant (r = 0.58, P = 0.057) (E). F: hematoxylin and eosin (HE)-stained lung section of a pulmonary vessel obtained from wild-type C57BL6 mice chronically exposed to ovalbumin. The mice develop severe pulmonary arterial muscularization without pulmonary hypertension. The vascular remodeling does not involve endothelial cell proliferation (G; brown staining is smooth muscle actin and blue is von Willebrand factor). F and G are reprinted from Daley et al. (2008), doi:10.1084/jem.20071008.
As severe human pulmonary arterial hypertension is characterized by complex pulmonary vascular lesions, it is surprising that vascular obliteration has not been the focus of experimental studies. Surprisingly, relatively few of the reported studies have systematically quantified the pulmonary vascular changes, with only three (16, 42, 94) studies reporting vasoocclusive changes and only one (94) measuring the percentage of fully and partially occluded lesions (Table 1).
As shown in Table 1, baseline RVSP measurements in unchallenged WT mice can vary from 10–20 mmHg up to 22 mmHg (98, 130), and up to 35 after chronic hypoxia exposure (16). From the 40 studies selected for this review, 30 of these reported PH, 10 of 30 studies reported an average RVSP higher than 30 mmHg, and only in 8 studies had mice been exposed to chronic hypoxia. Challenging the animals with chronic hypoxia can unmask pulmonary vascular hyperreactivity (57, 66, 122), trigger neointimal formation (16, 94, 100), or even render a paradoxical decrease in pressure. For example, Dempsie et al. (20) described that mice overexpressing a human variant of the serotonin transporter treated with the anorexigen dexfenfluramine developed PH (RVSP 40 mmHg). However, upon exposure to chronic hypoxia, the pulmonary artery pressure decreased resembling that of the unchallenged WT values (mean RVSP 24 mmHg).
There is one mouse model that perfectly exemplifies the complexity of vascular remodeling and PH. Daley and collaborators (19) described in 2008 a mouse model of PH induced by repeated antigen exposure, via a Th2 immune response. Wild-type C57BL6 mice were immunized with an intermittent exposure to intranasal Aspergillus fumigatus antigen. These mice developed severe thickening of the walls of small- and medium-caliber pulmonary arteries. Histologically, smooth muscle cells multilayered within the thickened wall of the remodeled pulmonary arteries, whereas von Willebrand factor-positive endothelial cells remained in a single-cell layer (Fig. 1, E and F). Strikingly, although pulmonary vascular remodeling was ubiquitous and severe, the RVSP was not elevated (range 21–29 mmHg) and there were no signs of right heart hypertrophy. Thus, at least in the mouse, severe pulmonary vascular remodeling does not necessarily cause severe pulmonary arterial hypertension.
Right Ventricular Failure in Transgenic Mouse Models of Pulmonary Hypertension
RVF is the leading cause of death and the main determinant of survival in patients with PAH (7, 83). Intriguingly, whereas the mechanisms of pulmonary vascular remodeling have been extensively studied in both mouse and rat models of PAH, the mechanisms underlying RVF remain largely elusive (9, 110). RV hypertrophy is easy to measure by separating the RV free wall from the rest of the heart (28). In rats, RVSP strongly correlates with the RV/LV+S (72), suggesting that the RV responds to an increased afterload with a corresponding degree of hypertrophy. Rat models of PAH, such as the SU5416/hypoxia and monocrotaline-injury models, generate robust RV hypertrophy (mean RV/LV+S 0.67–0.76) (11, 35). In contrast to rat models, only the IL6-OE mice seem to develop a comparable degree of hypertrophy (average RV/LV+S 0.69) and PH (94). When plotting data extracted from the reviewed mouse PH studies (Fig. 1, B–D), there seems to be an “uncoupling” between RVSP and RV/LV+S for the majority of PH mouse studies, even after hypoxia exposure. For example, Xu et al. (122) demonstrated that mice lacking the adenosine A2A receptor develop spontaneous PH (RVSP 39 mmHg); however, the RV/LV+S was only 0.26, a value that is within normal limits for C57BL6/J WT mice (98). In contrast, Chen and collaborators (15) reported that mice overexpressing connective tissue growth factor generate modest PH (average RVSP 22 mmHg vs. 10 mmHg in WT controls) but develop significant RV hypertrophy (RV/LV+S 0.42).
Another important point to make is that right ventricular hypertrophy does not necessarily imply RVF. For instance, patients with Eisenmenger's syndrome (PAH associated with a congenital systemic-to-pulmonary shunt) develop severe PH and right ventricular hypertrophy but do not develop signs or symptoms of overt RVF (at least not until late in the disease) (43, 44). Similarly, pulmonary artery banding in rats (a model of pure mechanical pressure overload) generates significant RV hypertrophy (mean RV/LV+S 0.52) with preserved RV function (11). Despite the fact that some investigators do not report mouse RV/LV+S data at all, it is surprising that only two studies attempted to evaluate cardiac function by means of echocardiography (13, 16) and both studies reported systemic cardiac output rather than RV cardiac output or contractility parameters [i.e., tricuspid annular plane systolic excursion (TAPSE)]. Although informative, systemic cardiac output does not characterize RV function.
It is true that the presence of RV hypertrophy supports the diagnosis of PH; however, it does not imply that the RV of these animals is failing. As such, RV function should be evaluated invasively by RV catheterization (cardiac output/index) or noninvasively by echocardiogram (TAPSE or RV fractional area change) or cardiac magnetic resonance (10, 12, 104, 107). Another useful echocardiographic finding to demonstrate RVF is the presence of paradoxical septal movement (Supplemental Video S1). When multiple echocardiographic signals such as RV dilatation, decreased TAPSE, and paradoxical septal movement are present, RVF is likely. Because severe chronic PAH is a cardiopulmonary disease, the study of PH in animal models should not only include assessment of RV hypertrophy but also evaluate whether the increase in PH pressure is affecting the function of the right ventricle.
Assessing the presence of PH by echocardiogram should also be part of the protocol when studying animal models of PAH. This is particularly important because, although RV catheterization is relatively simple in rats, the small hearts of mice impose a bigger challenge (79). Figure 2A illustrates how two different positions of the catheter tip, in a C57BL6/J W mouse heart, only 5 mm apart from each other, can significantly modify the measurement of RVSP. Thus a precatheterization echocardiographic assessment of PH, as well as an assessment of the RV/LV+S, complement the hemodynamic findings.
Fig. 2.
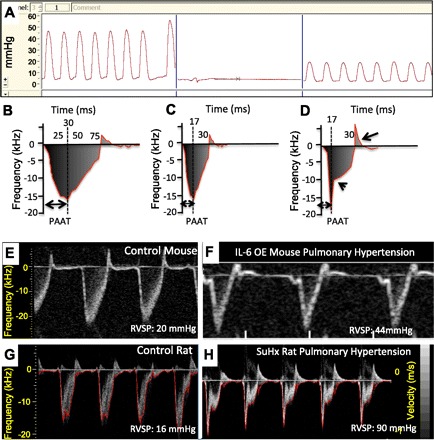
A: hemodynamic measurements in a normal C57BL6 mouse assessed in 2 different locations in the right ventricle. We can appreciate that a slight difference in the site of catheter insertion can cause dramatic changes in pressure recordings, even if the shape of the curve looks apparently normal. B–D: schematic representation of a pulsed-wave Doppler recording of the pulmonary artery. Animals with pulmonary hypertension show a pattern consistent with decreased pulmonary artery acceleration time (PAAT) with or without midsystolic notching (arrowhead). A bidirectional flow pattern appears when pulmonary valve insufficiency is present (D, arrow). E–H: pulsed-wave Doppler recordings from normal mouse (E), mouse overexpressing IL-6 with pulmonary hypertension (F, reproduced from Thibault HB, Kurtz B, Raher MJ, Shaik RS, Waxman A, Derumeaux G, Halpern EF, Bloch KD, Scherrer-Crosbie M. Noninvasive assessment of murine pulmonary arterial pressure: validation and application to models of pulmonary hypertension. Circ Cardiovasc Imaging 3: 157–163, 2010), normal Sprague-Dawley rat (G), and a SU5416/hypoxia exposed Sprague-Dawley rat (H). Compared with mice, rats develop a pulsed-wave Doppler pattern consisting of decreased PAAT, midsystolic notching, and bidirectional flow.
Two echocardiographic parameters obtained by pulsed-wave Doppler of the main pulmonary artery, can be used to address the presence of PH: 1) pulmonary artery acceleration time (PAAT) and 2) midsystolic notching. In humans, the pulmonary artery systolic pressure is usually estimated by measuring the tricuspid regurgitation peak flow velocity (Bernoulli's equation). However, tricuspid regurgitation is uncommon in rodents, and a proper apical/subcostal view, for adequate measurement of tricuspid regurgitation, is particularly challenging in mice (104). RVSP can be estimated by measuring the PAAT, which is the time from the onset of pulmonary flow to peak velocity by pulsed-wave Doppler recording (Fig. 2B). In response to an increase in pulmonary artery pressure, the pulmonary valve tends to close prematurely (118) and a peak flow velocity is reached earlier in systole (Fig. 2C). This is usually the pattern seen in mouse models of PH (Fig. 2, E and F). The PAAT inversely correlates with the mean pulmonary artery pressure in humans and RVSP in rats and mice (53, 104, 107).
In a similar way, when PH is present, the pulsed-wave Doppler demonstrate an alternative ejection pattern consisting of an abnormally rapid rise in flow velocity to a peak level, preceded by a rapid deceleration, followed in turn by a secondary slower rise to form a notch (Fig. 2D). The midsystolic notching is observed in patients with PAH (53) and is usually the pattern found in the SU5416/hypoxia (Fig. 2, G and H) and monocrotaline rat models of PH (107). Both flow patterns have been reported in PH (53); nonetheless, the second pattern (with midsystolic notching) also presents with a bidirectional broadening spectrum of the Doppler frequency during early diastole, indicating pulmonary regurgitation (Fig. 2D), an indirect sign of increased pulmonary pressure.
Three of the published mouse studies provided unpredicted and puzzling results which ought to be followed up to better understand the integrated response of the lung circulation-heart axis, in the setting of PAH and chronic hemodynamic stress. For instance, Daley et al. (19) reported very pronounced pulmonary artery muscularization, but no RV hypertrophy. In contrast, the studies of Shifren et al. (86) demonstrate that mice lacking elastin develop impressive elevation of RVSP without RV hypertrophy. Finally, in hemeoxygenase-1 KO mice, Yen et al. demonstrated that, after chronic hypoxia exposure, mice did not develop RV hypertrophy, but developed RV dilatation. All together, these last three studies exemplify that, in mice, 1) severe muscularization does not necessarily indicate PH, 2) PH is not always translated into RV hypertrophy, and 3) RV remodeling could occur in the absence of PH. Whether increased pulmonary arterial pressure and RV remodeling are two mechanistically distinct processes that are usually coupled in PAH remains to be investigated.
The SU5416/Hypoxia Rat Model of Pulmonary Arterial Hypertension: A Brief Summary
The combined VEGF receptor 1 (Flt) and 2 (KDR) blocker, SU5416, was one of the first agents discovered by screening for growth inhibitory activity of cultured endothelial cells incubated with the potent angiogenic VEGF ligand (27). Kindly provided by Dr. Peter Hirth (Sugen, South San Francisco, CA), for preclinical studies, SU5416 was tested with the hypothesis that inhibition of VEGF signaling would result in pulmonary emphysema. Indeed, a single subcutaneous injection of 20 mg/kg of SU5416 caused air space enlargement and mild PH in adult rats (51). Unexpectedly, rats injected with SU5416 (20 mg/kg) and exposed to chronic hypoxia in a hypobaric chamber (hereafter SUHx), as an attempt to worsen the air space enlargement, surprisingly developed severe PAH, which was not reversible when the animals were returned to Denver altitude (100) or in later experiments to sea level (11). The PH was associated with angioobliterative pulmonary lesions that were preventable by treating the animals concomitantly with a pan-caspase inhibitor, indicating that apoptosis was necessary for the development of the pulmonary vascular lesions (100). This rat model has served as a model for preclinical drug studies designed to examine whether the PH and pulmonary vascular disease in the SUHx rats could be reversed once established. Simvastatin (a 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor) and a bradykinin agonist had a small effect on established PH and RVH, whereas a Ca2+ channel blocker (nifedipine), angiotensin converting enzyme inhibitor (lisinopril), angiotensin receptor blocker (ibersartan) and bosentan (a nonselective endothelin receptor blocker) were all not effective (102, 103). Interestingly, when treated with infused prostacyclin, the animals died acutely, presumably because prostacyclin caused overwhelming peripheral vasodilation, dropping RV perfusion pressure and RV preload (72). Because in this rat model the PH is severe, is accompanied by plexiform-like lesions, and is largely refractory to treatment, it was concluded that the SUHx rat model of angioobliterative severe PH had a number of features that resembled the human disease. Moreover, Abe et al. (1) have shown the histopathological similarities of the SUHx lung vascular lesions compared with the plexiform lesions in human disease. In addition to severe PH, rats treated with SU5416 and exposed to chronic hypoxia also develop severe RV failure characterized by a cardiac output reduction, decreased TAPSE, increased RV diastolic diameter, capillary rarefaction, RV fibrosis, and cardiomyocyte apoptosis (11, 12, 72).
The SUHx model has been modified to uncover immunological mechanisms in the pathobiology of severe PAH. We have shown that treatment of athymic rats with SU5416 is sufficient to cause severe angioobliterative PAH, indicating that hypoxia is not necessary (101) and that early immune reconstitution with rat regulatory T lymphocytes prevents the development of severe PAH (99, 101). For further information regarding the SUHx model the reader is referred to Refs. 11, 81, 82, 100.
Attempts To Generate a SU5416/Hypoxia Mouse Model of Pulmonary Hypertension
Given the wealth of mechanistic insights gained from genetically engineered mice (Table 1), it was only intuitive to combine the SUHx model with gene knockout technology. Subsequent to the first publication describing the SUHx rat model (100), multiple attempts to generate a SUHx mouse model of severe PH have been pursued but, until now, none have been fruitful. Here we present some of our data utilizing different mouse strains, genetic manipulations, and multiple SU5416 dosing as we tried to generate a SU5416-based mouse model. All attempts to generate a SUHx mouse model are summarized in Table 2.
Table 2.
Right ventricular systolic pressure measurements and pulmonary vasculature remodeling in mice treated with SU5416 with or without hypoxia
| RVSP, mmHg |
RV/LV+S |
Histology |
|||||
|---|---|---|---|---|---|---|---|
| Mouse Strain | Genetic Modification | Normoxia | Hypoxia | Normoxia | Hypoxia | Normoxia | Hypoxia |
| C57BL6/J | Wild type/normal | N/A | 25 | N/A | N/A | N/A | ↑ musc. |
| with single SU5416 dose | |||||||
| C57BL6/J | Wild type/normal | N/A | 33 | N/A | N/A | N/A | ↑ musc. |
| with single SU5416 + 2 wk Denver altitude | |||||||
| C57BL6/J | ADAM 10TG | 20 | N/A | 0.26 | N/A | No changes | N/A |
| Wild type | |||||||
| C57BL6/J | ADAM 10TG OE | 41 | N/A | 0.28 | N/A | No changes | N/A |
| C57BL6/J | ADAM 10TG | 22 | 18 | 0.25 | 0.23 | No changes | ↑ musc. |
| Wild type | |||||||
| with SU5416 | |||||||
| C57BL6/J | ADAM 10TG OE with SU5416 | 25 | 20 | 0.27 | 0.22 | No changes | ↑ musc. |
| R26R | PVEC BMPR−/− KO | 21 | N/A | 0.28 | N/A | No changes | N/A |
| R26R | PVEC BMPR−/− KO | N/A | 23 | N/A | 0.26 | N/A | ↑ musc. |
| with SU5416 | |||||||
| NU/NU | Nude mice Foxn1nu | 21 | N/A | 0.21 | N/A | No changes | N/A |
| NU/NU | Nude mice Foxn1nu | 22 | N/A | 0.25 | N/A | No changes | N/A |
| with SU5416 | |||||||
| 3 doses once a week | |||||||
| NU/NU | Nude mice Foxn1nu | 22 | N/A | 0.23 | N/A | No changes | N/A |
| with SU5416 | |||||||
| 4 doses once a week | |||||||
ADAM10, A Disintegrin And Metalloproteinase 10.
The first attempt goes back 10 years ago, shortly after the first report of Taraseviciene-Stewart in 2001 (100). We injected 8–12 wk old adult C57BL6 mice with either 20 or 50 mg/kg of SU5416 at Denver (altitude 5,600 ft above sea level) and then exposed the mice to a simulated altitude of 17,000 ft. Littermates injected with saline instead of SU5416 served as controls. No significant differences in the RV/LV+S or the lung vascular morphology were detected between control and SU5416 groups. In the aggregate, the data indicated that, at least C57BL6, mice subjected to the same experimental protocol that had been used to generate severe PAH in WT rats did not develop PAH, nor did they develop pulmonary vascular remodeling or RVH (Table 2). Because the protocol used to generate severe PAH in rats did not generate in mice PH beyond the degree observed with chronic hypoxia exposure alone, we postulated that mouse-specific genetic background factors played an important role and did not publish these results.
On the basis of the findings that athymic nude rats lacking T cells exposed to SU5416 generated PH and vascular remodeling without hypoxia (101), we next attempted to induce PH in nude (immune-insufficient) mice (SCID). Given that a single dose of SU5416 did not induce the expected changes in WT mice, we decided to expose the SCID mice to one injection per week for 3 wk (3 doses total) and for 4 wk (4 doses total). Table 2 shows that there was no significant change in the mean RVSP when compared with nude mice without SU5416. SU5416 did not induce PH in nude mice.
As a further attempt to explore the role of abnormal immune regulation in the pathobiology of PAH, we investigated transgenic mice overexpressing the A Disintegrin And Metalloproteinase 10 (ADAM10). Saleem, Conrad, and colleagues (34) had reported that overexpression of ADAM10 abrogated B cell development, delayed T cell development, and strikingly induced systemic expansion of CD11b+Gr-1+ myeloid-derived suppressor cells. MDSCs were first characterized in tumor-bearing mice and in patients with cancer; however, there are other numerous pathological conditions that markedly induce MDSC expansion (29). MDSCs are characterized by a remarkable ability to suppress T cell responses and regulate innate immune responses by modulating cytokine production by macrophages (29, 89). Furthermore, circulating MDSCs can migrate to tumor sites and rapidly differentiate into tumor-associated macrophages (TAMs) (54). TAMs acquire the ability to produce several cytokines, such as IL-1β, IL-6, and TGF-β (60). Because 1) circulating MDSCs have been described in patients with PAH (127), 2) MDSCs/TAMs can produce large amounts of IL-6, 3) IL-6 is increased in patients with PAH (47) and overexpression of IL-6 generates PH (94), and finally, 4) abnormal T-cell function is associated with the development of plexiform-like lesions in rats exposed to SU5416 (99, 101), we postulated that MDSCs in ADAM10 transgenic mice (ADAM10Tg) would facilitate the development of pulmonary arterial hypertension and vascular remodeling in mice. Figure 3 shows the data obtained from the first ADAM10Tg mouse studied. The baseline echocardiogram showed RV dilatation (Fig. 3A) and hemodynamic measurements demonstrated a RVSP of 57 mmHg (Fig. 3B). Histological analysis revealed the presence of multiple occluded vessels (Fig. 3, C and D); however, immunohistochemistry for Von Willebrand factor revealed that none of the cells occluding the vessels were endothelial cells (Fig. 3, E and F). When additional animals were evaluated, we observed that, whereas some of the mice exhibited a high RVSP (range 40–60 mmHg) (Fig. 3H), the majority of the ADAM10Tg mice did not spontaneously develop PH. Given that few ADAM 10 mice (on the C57BL6 background) exhibited high RVSP values, we treated WT and ADAM10Tg animals with SU5416, on the basis of the double-hit hypothesis of PAH. Mice were injected with 20 mg/kg of SU5416 subcutaneously, once a week for 4 consecutive wk; however, we did not find an increase in the RVSP compared with WT mice (Table 2).
Fig. 3.
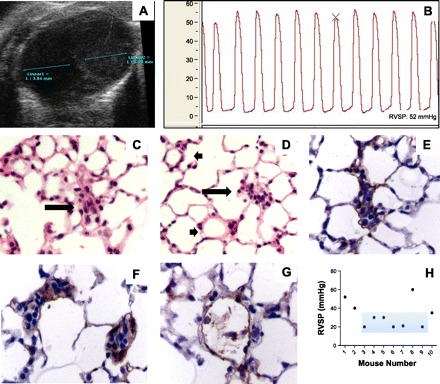
A: short-axis view obtained from a mouse overexpressing ADAM10 illustrates significant right ventricular dilatation compared with the left ventricle. B: hemodynamic measurement illustrating an increase in the right ventricular systolic pressure. C and D: HE-stained lung section illustrating occluded vessel (arrows) and normal vessels (arrowheads). E–F: immunohistochemistry of lung sections illustrating that the occluded vessels are not occluded by von Willebrand-positive endothelial cells. G: normal vessel. H: RVSP measurements in ADAM10 transgenic mouse. Light blue area marks mice that had a normal pulmonary artery pressure. Although some mice demonstrated high RVSP, the measurements were highly variable.
Mizuno et al. (64) had demonstrated that mice lacking the p53 gene developed severe pulmonary vascular muscularization and mild PH upon exposure to chronic hypoxia (Table 2). Thus we examined the impact of SU5416 in combination with hypoxia in p53 mice (FVB/B6-Trp53 tm1tyj/J, Jackson Laboratory, Sacramento, CA), hypothesizing that the lack of “the guardian of the genome” p53 would perhaps be permissive and lead to a more severe form of PH. Mice were injected with a single subcutaneous dose of SU5416 and exposed for 4 wk to 10% hypoxia, in keeping with the rat SUHx protocol. We did not find differences between SUHx p53−/− mice and p53−/− mice exposed to hypoxia alone (data not published).
In a recent experiment, we injected C57Bl6 WT mice with 20 mg/kg of SU5416 subcutaneously, once a week for 3 consecutive wk, and exposed the animals to nitrogen dilution hypoxia (10% oxygen). We then compared the data to those obtained with a group of C57BL6 mice exposed to hypoxia alone. The animals were killed following echocardiography and measurement of RVSP, and the lungs were inflated according to our standard protocol. The data derived from these studies are displayed in Fig. 4.
Fig. 4.
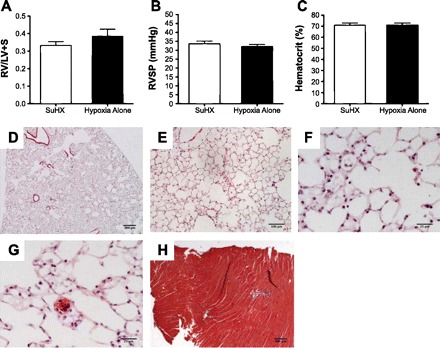
Mice administered SU5416 (20 mg/kg sc) once a week for 3 consecutive wk and exposed to nitrogen dilution hypoxia for 3 wk (n = 5) are compared with mice exposed only to chronic hypoxia (n = 5). Neither right ventricular hypertrophy nor RVSP were different when the 2 groups were compared (A and B); there was no difference in the hematocrit (C). Lung histology (HE staining) shows some degree of air space enlargement (D–F). Rare small vessel thrombi were observed (G). As evidenced by trichrome staining, RV fibrosis was absent (H).
Mice Are Not Small Rats
There are several factors that could explain the differences between the data obtained and published in mice following the implementation of SU/Hx protocols and the data obtained with rats subjected to the SU/Hx protocols. In the study by Ciuclan et al. (16), more than one dose of SU5416 was apparently required to generate PAH, strongly suggesting that mice are less responsive to SU5416 and that the compound is differently or more rapidly metabolized and inactivated in mice compared with rats. Possibly the accumulation of SU5416 in the lung tissues is less in mice when compared with rats. Ye et al. (125) compared the distribution, metabolism, and excretion of SU5416 (semaxinib) between rats and mice and found that the systemic and renal clearance of SU5416 was much greater in mice and that SU5416 was hepatically metabolized by CYP1A, a cytochrome differentially expressed between animal species (61, 69). Of interest, repeated dosing with SU5416 induced an increase in the activity of several cytochrome P-450 enzymes. It is likely that pharmacogenomic differences between rats and mice can explain the difference in pulmonary vascular responses observed after administration of the alkaloid monocrotaline (35). This hypothesis of a significantly different performance and action pattern of SU5416 between the mouse and rat species is also supported by the observation that, at least in the SUHx mouse model, PH is not sustained after the hypoxic exposure had been discontinued (16). This is in clear contrast to the response of SUHx rats in which PH is maintained for many weeks after the hypoxic exposure had been terminated, and in which lung lesion maturation continues for at least 13 wk after the initial and single dose of SU5416 had been administered (1).
The spontaneous reversal of PH is the hallmark of most (or all) “single-hit” chronic hypoxia models and is unfortunately not a feature of human forms of idiopathic PAH (113). Thus it appears that the recently reported mouse model of SUHx exposure (16) is most likely a variant of chronic hypoxia-related PH models in that the reported pulmonary vascular changes are predominantly arteriolar muscularization (16). Importantly, as documented by the studies by Daley et al. (19), very impressive degrees of muscularization of the pulmonary arterioles can occur in mice without the development of PH and RV hypertrophy.
Because PH researchers are increasingly investigating mice (Table 1), consideration should be given to the known important differences between the rat and the mouse species (5, 6, 17, 25, 55). Such differences, which may categorically determine the mechanisms leading to the development of pulmonary vascular diseases, can be found on anatomical, cellular, and gene expression levels and also in pharmacogenomics. For instance, rat lungs have less alveolar and more blood vessel walls and higher mean alveolar diameters than mouse lungs (25), which translate into distinct tissue mechanical properties. In mice, the systemic blood vessels that supply the trachea and mainstem bronchi do not penetrate into the intraparenchymal airways, as they do in other species (63). Although we do not know whether the alveolar space design influences the development of PAH, the presence or absence of a fully developed bronchial circulation is likely important for the pathobiology of PAH. Precursor cells, derived from the bone marrow, can be transported into the pulmonary vascular adventitia via vasa vasorum (via bronchial circulation) (126) and affect vascular remodeling.
In the heart the proportion of fibroblasts and myocardiocytes differs between the rat and mouse (5) and the cytochrome P-450 gene expression pattern, critically important for drug and steroid hormone metabolism both in the liver and the lung, differs between the two species (17, 61) (see above). As mentioned before, Hoshikawa et al. (45) demonstrated differences in lung tissue gene expression between WT mice and Sprague-Dawley rats. Some relevant differences included a sixfold upregulation of the expression of the gene encoding the integrin-α chain, a sixfold increase in the expression of the gene encoding MHC class II antigen and a sevenfold upregulation of the osteopontin gene in chronically hypoxic rat lungs compared with mice. In contrast, the gene encoding the CYP1A1 isozyme was more than fivefold downregulated in the lungs from hypoxic mice. Whether any of these gene expression differences can explain differences in the degree of lung vascular remodeling remains unclear but can be investigated.
Decoding the complete genome of the C57BL/6J mouse (Mouse Genome Sequencing Consortium) improved our ability to relate sequence to function and allowed, in an unprecedented way, the task of creating null alleles for all genes (68). However, genetic catalogs remain incomplete, and we are still largely ignorant of the molecular basis of the majority of genetically influenced phenotypes (52). For instance, Keane and collaborators (52) examined tissues in a single cross (C57BL/6J × DBA/2J) and were able to detect high levels of allelic bias. Interestingly, they showed divergent allele-specific patterns between tissues. For example, an allele that was relatively highly expressed in one tissue was underexpressed in a different one. Furthermore, they demonstrated the nature of sequence variants and how their relative position to other genes, influences function. Thus it is necessary to be circumspect when extrapolating data from a certain knockout mouse strain to other strains, as the phenotype could also be the result of a combination of sequence variants and not only the result of a specific genetic manipulation. For example, Rabieyousefi et al. (74) reported that mice overexpressing OX40L, a tumor necrosis factor family molecule, develop vascular remodeling of large pulmonary arteries in C57BL6 mice but not in BALB/c mice, supporting the importance of genetic background (strain differences).
Mouse Models of Pulmonary Hypertension: Translational Potential
Having reviewed many of the published data on mouse models of PAH and having failed to establish a mouse model of severe angioproliferative PAH in our own laboratory, we feel obligated to ask 1) To what extent can the information derived from the mouse PAH models be related to human forms of severe PAH? 2) Are mouse models really productive when it comes to preclinical drug testing and the exploration of new treatment strategies? 3) What are the criteria we should apply to recommend a particular animal model of PAH? These questions certainly depend on the disease aspect and pathobiological mechanisms of interest. For instance, if the investigational focus is on vasoconstriction or mechanisms of pulmonary arteriolar muscularization, many of the PAH mouse models will likely be very informative, especially if the model produces highly reproducible data without large variability. In contrast, if the investigational focus is a model of severe angioobliterative PAH and RVF, the animal model ought to display at least some of the critical features of human forms of PAH (Table 3). One critical feature of any new animal models of pulmonary arterial hypertension should be the development of RVF, not just RV hypertrophy.
Table 3.
Salient features of human pulmonary arterial hypertension that ought to be considered in animal models
| Hemodynamics |
|---|
| Mean pulmonary artery pressure >25 mmHg or RV systolic pressure >60 mmHg |
| Decreased cardiac output |
| Histology |
| Small pulmonary artery lumen obliteration |
| Muscularization of the media |
| Plexiform-like lesions |
| RV hypertrophy (RV/LV/S >0.45) |
| RV fibrosis and capillary rarefaction |
| Echocardiography |
| Dilatation of the right heart chambers |
| Hypertrophy of the RV |
| Paradoxical septal movement (or at least rectification of the interventricular septum) |
| Decreased tricuspid annulus plane systolic excursion (∼ <1.5 mm) |
Highly specific molecular targets or the opportunity to block certain signaling pathways are pleasing ideas to many researchers. However, without the context of the human phenotype, gene deletion may just produce another descriptive association. Killing one bee is unlikely to modify the complex interactions occurring in the colony (unless, of course, it is the queen!). This example may be simplistic, but the message is clear: Ablating one gene, in one particular cell type, is likely insufficient to explain a complex disease such as PAH. Taking our own data as an example (Table 2), knocking out the p53 gene in mice certainly caused enhanced pulmonary arteriolar muscularization after chronic hypoxic exposure (64) but did not cause a severe angioobliterative pathology, even after SU5416/hypoxia exposure. Does this result mean that p53 is uninvolved in the mechanisms of pulmonary angioobliteration? Or do we have to consider that mice are, perhaps, not suitable to explore this question, because the combination of SU5416 and hypoxia causes highly reproducible, progressive, and irreversible severe angioobliteration in the rat, but not in mice?
Highly complex, multicellular vascular lesions characterize the lungs of humans with severe PAH, and many layers of redundant control mechanisms prevent the formation of such lesions in healthy, not genetically susceptible people. Thus how likely it is that one single gene will explain the entire pathobiology of PAH? We have already learned that BMPR2 mutations are not sufficient to trigger PAH, but to interpret that the presence of BMPR2 gene mutations in humans with familial and idiopathic PAH is just an epiphenomenon, not relevant to the disease, would be a mistake.
Many questions remain, but one question should be highlighted: What from the PAH mouse models can be translated and applied to the human disease?
Conclusions
Here we have reviewed and compared a large number of representative mouse model studies of PAH and contrasted, in passing, the salient features of these models with those that are present in rat models. We addressed some of the potential problems that can arise when evaluating PH in mice and underlined the importance of evaluating right ventricular function. Stating the obvious: There is not a perfect or ideal animal model of PAH yet. However, we invite researchers interested in designing new models of PAH to consider, among others (Table 3), three critical features of the human disease: 1) obliteration of the lung arterioles, 2) nonreversibility of PAH, and 3) development of RVF.
Although there have been recent general reviews of animal models of PH (79, 95), here we have attempted to fill a gap by putting current mouse models of PAH into perspective, and tried to highlight some important innate murine characteristics, which partially explain why many of the mouse models could have been lost in translation.
Supplementary Material
REFERENCES
- 1. Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121: 2747–2754, 2010. [DOI] [PubMed] [Google Scholar]
- 2. Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J, Kurz X, Higenbottam T, Oakley C, Wouters E, Aubier M, Simonneau G, Bégaud B. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med 335: 609–616, 1996. [DOI] [PubMed] [Google Scholar]
- 3. Archer S, Rich S. Primary pulmonary hypertension: a vascular biology and translational research “Work in progress.” Circulation 102: 2781–2791, 2000. [DOI] [PubMed] [Google Scholar]
- 4. Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 105: 1672–1678, 2002. [DOI] [PubMed] [Google Scholar]
- 5. Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol 293: H1883–H1891, 2007. [DOI] [PubMed] [Google Scholar]
- 6. Bazykin GA, Kondrashov FA, Ogurtsov AY, Sunyaev S, Kondrashov AS. Positive selection at sites of multiple amino acid replacements since rat-mouse divergence. Nature 429: 558–562, 2004. [DOI] [PubMed] [Google Scholar]
- 7. Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 122: 164–172, 2010. [DOI] [PubMed] [Google Scholar]
- 8. Beppu H, Kawabata M, Hamamoto T, Chytil A, Minowa O, Noda T, Miyazono K. BMP type II receptor is required for gastrulation and early development of mouse embryos. Dev Biol 221: 249–258, 2000. [DOI] [PubMed] [Google Scholar]
- 9. Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 135: 794–804, 2009. [DOI] [PubMed] [Google Scholar]
- 10. Bogaard HJ, Mizuno S, Hussaini Al AA, Toldo S, Abbate A, Kraskauskas D, Kasper M, Natarajan R, Voelkel NF. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am J Respir Crit Care Med 183: 1402–1410, 2011. [DOI] [PubMed] [Google Scholar]
- 11. Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 120: 1951–1960, 2009. [DOI] [PubMed] [Google Scholar]
- 12. Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, Chau VQ, Hoke NN, Kraskauskas D, Kasper M, Salloum FN, Voelkel NF. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med 182: 652–660, 2010. [DOI] [PubMed] [Google Scholar]
- 13. Burton VJ, Holmes AM, Ciuclan LI, Robinson A, Roger JS, Jarai G, Pearce AC, Budd DC. Attenuation of leukocyte recruitment via CXCR1/2 inhibition stops the progression of PAH in mice with genetic ablation of endothelial BMPR-II. Blood 118: 4750–4758, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chandra SM, Razavi H, Kim J, Agrawal R, Kundu RK, de Jesus Perez V, Zamanian RT, Quertermous T, Chun HJ. Disruption of the apelin-APJ system worsens hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol 31: 814–820, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen S, Rong M, Platteau A, Hehre D, Smith H, Ruiz P, Whitsett J, Bancalari E, Wu S. CTGF disrupts alveolarization and induces pulmonary hypertension in neonatal mice: implication in the pathogenesis of severe bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 300: L330–L340, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, Stringer R, Jones P, Morrell NW, Jarai G, Walker C, Westwick J, Thomas M. A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med 184: 1171–1182, 2011. [DOI] [PubMed] [Google Scholar]
- 17. Craft ES, DeVito MJ, Crofton KM. Comparative responsiveness of hypothyroxinemia and hepatic enzyme induction in Long-Evans rats versus C57BL/6J mice exposed to TCDD-like and phenobarbital-like polychlorinated biphenyl congeners. Toxicol Sci 68: 372–380, 2002. [DOI] [PubMed] [Google Scholar]
- 18. Crosby A, Jones FM, Southwood M, Stewart S, Schermuly R, Butrous G, Dunne DW, Morrell NW. Pulmonary vascular remodeling correlates with lung eggs and cytokines in murine schistosomiasis. Am J Respir Crit Care Med 181: 279–288, 2010. [DOI] [PubMed] [Google Scholar]
- 19. Daley E, Emson C, Guignabert C, de Waal Malefyt R, Louten J, Kurup VP, Hogaboam C, Taraseviciene-Stewart L, Voelkel NF, Rabinovitch M, Grunig E, Grunig G. Pulmonary arterial remodeling induced by a Th2 immune response. J Exp Med 205: 361–372, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dempsie Y, Morecroft I, Welsh DJ, MacRitchie NA, Herold N, Loughlin L, Nilsen M, Peacock AJ, Harmar A, Bader M, MacLean MR. Converging evidence in support of the serotonin hypothesis of dexfenfluramine-induced pulmonary hypertension with novel transgenic mice. Circulation 117: 2928–2937, 2008. [DOI] [PubMed] [Google Scholar]
- 21. Délot EC, Bahamonde ME, Zhao M, Lyons KM. BMP signaling is required for septation of the outflow tract of the mammalian heart. Development 130: 209–220, 2003. [DOI] [PubMed] [Google Scholar]
- 22. Elliott CG, Glissmeyer EW, Havlena GT, Carlquist J, McKinney JT, Rich S, McGoon MD, Scholand MB, Kim M, Jensen RL, Schmidt JW, Ward K. Relationship of BMPR2 mutations to vasoreactivity in pulmonary arterial hypertension. Circulation 113: 2509–2515, 2006. [DOI] [PubMed] [Google Scholar]
- 23. Engebretsen BJ, Irwin D, Valdez ME, O'Donovan MK, Tucker A, van Patot MT. Acute hypobaric hypoxia (5486 m) induces greater pulmonary HIF-1 activation in hilltop compared with madison rats. High Alt Med Biol 8: 312–321, 2007. [DOI] [PubMed] [Google Scholar]
- 24. Euler von US, Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand 12: 301–320, 1946. [Google Scholar]
- 25. Faffe DS, Rocco PRM, Negri EM, Zin WA. Comparison of rat and mouse pulmonary tissue mechanical properties and histology. J Appl Physiol 92: 230–234, 2002. [DOI] [PubMed] [Google Scholar]
- 26. Farmakis D, Aessopos A. Pulmonary hypertension associated with hemoglobinopathies: prevalent but overlooked. Circulation 123: 1227–1232, 2011. [DOI] [PubMed] [Google Scholar]
- 27. Fong TA, Shawver LK, Sun L, Tang C, App H, Powell TJ, Kim YH, Schreck R, Wang X, Risau W, Ullrich A, Hirth KP, McMahon G. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res 59: 99–106, 1999. [PubMed] [Google Scholar]
- 28. Fulton RM, Hutchinson EC, Jones AM. Ventricular weight in cardiac hypertrophy. Br Heart J 14: 413–420, 1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9: 162–174, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Galiè N, Manes A, Negro L, Palazzini M, Bacchi-Reggiani ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J 30: 394–403, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gambaryan N, Perros F, Montani D, Cohen-Kaminsky S, Mazmanian GM, Humbert M. Imatinib inhibits bone marrow-derived c-kit+ cell mobilisation in hypoxic pulmonary hypertension. Eur Respir J 36: 1209–1211, 2010. [DOI] [PubMed] [Google Scholar]
- 32. George J, D'Armiento J. Transgenic expression of human matrix metalloproteinase-9 augments monocrotaline-induced pulmonary arterial hypertension in mice. J Hypertens 29: 299–308, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Geraci MW, Gao B, Shepherd DC, Moore MD, Westcott JY, Fagan KA, Alger LA, Tuder RM, Voelkel NF. Pulmonary prostacyclin synthase overexpression in transgenic mice protects against development of hypoxic pulmonary hypertension. J Clin Invest 103: 1509–1515, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gibb DR, Saleem SJ, Kang DJ, Subler MA, Conrad DH. ADAM10 overexpression shifts lympho- and myelopoiesis by dysregulating site 2/site 3 cleavage products of Notch. J Immunol 186: 4244–4252, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gomez-Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel NF, Bogaard HJ. The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol 302: L363–L369, 2012. [DOI] [PubMed] [Google Scholar]
- 36. Graham LM, Vasil A, Vasil ML, Voelkel NF, Stenmark KR. Decreased pulmonary vasoreactivity in an animal model of chronic Pseudomonas pneumonia. Am Rev Respir Dis 142: 221–229, 1990. [DOI] [PubMed] [Google Scholar]
- 37. Grover RF, Reeves JT. Exercise performance of athletes at sea level and 3100 meters altitude. Med Thorac 23: 129–143, 1966. [DOI] [PubMed] [Google Scholar]
- 38. Grover RF. Pulmonary circulation in animals and man at high altitude. Ann NY Acad Sci 127: 632–639, 1965. [DOI] [PubMed] [Google Scholar]
- 39. Guignabert C, Izikki M, Tu LI, Li Z, Zadigue P, Barlier-Mur AM, Hanoun N, Rodman D, Hamon M, Adnot S, Eddahibi S. Transgenic mice overexpressing the 5-hydroxytryptamine transporter gene in smooth muscle develop pulmonary hypertension. Circ Res 98: 1323–1330, 2006. [DOI] [PubMed] [Google Scholar]
- 40. Hansmann G, Perez de J VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, Rabinovitch M. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest 118: 1846–1857, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He LS, Chang SW, Voelkel NF. Pulmonary vascular reactivity in Fischer rats. J Appl Physiol 70: 1861–1866, 1991. [DOI] [PubMed] [Google Scholar]
- 42. Hong KH, Lee YJ, Lee E, Park SO, Han C, Beppu H, Li E, Raizada MK, Bloch KD, Oh SP. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation 118: 722–730, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hopkins WE, Ochoa LL, Richardson GW, Trulock EP. Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or Eisenmenger syndrome. J Heart Lung Transplant 15: 100–105, 1996. [PubMed] [Google Scholar]
- 44. Hopkins WE, Waggoner AD. Severe pulmonary hypertension without right ventricular failure: the unique hearts of patients with Eisenmenger syndrome. Am J Cardiol 89: 34–38, 2002. [DOI] [PubMed] [Google Scholar]
- 45. Hoshikawa Y, Nana-Sinkam P, Moore MD, Sotto-Santiago S, Phang T, Keith RL, Morris KG, Kondo T, Tuder RM, Voelkel NF, Geraci MW. Hypoxia induces different genes in the lungs of rats compared with mice. Physiol Genomics 12: 209–219, 2003. [DOI] [PubMed] [Google Scholar]
- 46. Humbert M, Deng Z, Simonneau G, Barst RJ, Sitbon O, Wolf M, Cuervo N, Moore KJ, Hodge SE, Knowles JA, Morse JH. BMPR2 germline mutations in pulmonary hypertension associated with fenfluramine derivatives. Eur Respir J 20: 518–523, 2002. [DOI] [PubMed] [Google Scholar]
- 47. Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 151: 1628–1631, 1995. [DOI] [PubMed] [Google Scholar]
- 48. Imamura M, Luo B, Limbird J, Vitello A, Oka M, Ivy DD, McMurtry IF, Garat CV, Fallon MB, Carter EP. Hypoxic pulmonary hypertension is prevented in rats with common bile duct ligation. J Appl Physiol 98: 739–747, 2005. [DOI] [PubMed] [Google Scholar]
- 49. Ivy DD, Yanagisawa M, Gariepy CE, Gebb SA, Colvin KL, McMurtry IF. Exaggerated hypoxic pulmonary hypertension in endothelin B receptor-deficient rats. Am J Physiol Lung Cell Mol Physiol 282: L703–L712, 2002. [DOI] [PubMed] [Google Scholar]
- 50. Kasahara Y, Kiyatake K, Tatsumi K, Sugito K, Kakusaka I, Yamagata S, Ohmori S, Kitada M, Kuriyama T. Bioactivation of monocrotaline by P-450 3A in rat liver. J Cardiovasc Pharmacol 30: 124–129, 1997. [DOI] [PubMed] [Google Scholar]
- 51. Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 106: 1311–1319, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, Furlotte NA, Eskin E, Nellåker C, Whitley H, Cleak J, Janowitz D, Hernandez-Pliego P, Edwards A, Belgard TG, Oliver PL, McIntyre RE, Bhomra A, Nicod J, Gan X, Yuan W, van der Weyden L, Steward CA, Bala S, Stalker J, Mott R, Durbin R, Jackson IJ, Czechanski A, Guerra-Assunção JA, Donahue LR, Reinholdt LG, Payseur BA, Ponting CP, Birney E, Flint J, Adams DJ. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477: 289–294, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kitabatake A, Inoue M, Asao M, Masuyama T, Tanouchi J, Morita T, Mishima M, Uematsu M, Shimazu T, Hori M, Abe H. Noninvasive evaluation of pulmonary hypertension by a pulsed Doppler technique. Circulation 68: 302–309, 1983. [DOI] [PubMed] [Google Scholar]
- 54. Kusmartsev S, Gabrilovich DI. Inhibition of myeloid cell differentiation in cancer: the role of reactive oxygen species. J Leukoc Biol 74: 186–196, 2003. [DOI] [PubMed] [Google Scholar]
- 55. Lensch M, Lohr M, Russwurm R, Vidal M, Kaltner H, André S, Gabius HJ. Unique sequence and expression profiles of rat galectins-5 and -9 as a result of species-specific gene divergence. Int J Biochem Cell Biol 38: 1741–1758, 2006. [DOI] [PubMed] [Google Scholar]
- 56. Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P, Macias J, Yuan JXJ, Jamieson SW, Thistlethwaite PA. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med 15: 1289–1297, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lowery JW, Frump AL, Anderson L, DiCarlo GE, Jones MT, de Caestecker MP. ID family protein expression and regulation in hypoxic pulmonary hypertension. Am J Physiol Regul Integr Comp Physiol 299: R1463–R1477, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ma W, Han W, Greer PA, Tuder RM, Toque HA, Wang KKW, Caldwell RW, Su Y. Calpain mediates pulmonary vascular remodeling in rodent models of pulmonary hypertension, and its inhibition attenuates pathologic features of disease. J Clin Invest 121: 4548–4566, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, Phillips JA, Newman J, Williams D, Galiè N, Manes A, McNeil K, Yacoub M, Mikhail G, Rogers P, Corris P, Humbert M, Donnai D, Martensson G, Tranebjaerg L, Loyd JE, Trembath RC, Nichols WC. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet 68: 92–102, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23: 549–555, 2002. [DOI] [PubMed] [Google Scholar]
- 61. Martignoni M, Groothuis GMM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol 2: 875–894, 2006. [DOI] [PubMed] [Google Scholar]
- 62. Mathai SC, Hassoun PM. Pulmonary arterial hypertension associated with systemic sclerosis. Expert Rev Respir Med 5: 267–279, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mitzner W, Lee W, Georgakopoulos D, Wagner E. Angiogenesis in the mouse lung. Am J Pathol 157: 93–101, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mizuno S, Bogaard HJ, Kraskauskas D, Alhussaini A, Gomez-Arroyo J, Voelkel NF, Ishizaki T. p53 gene deficiency promotes hypoxia-induced pulmonary hypertension and vascular remodeling in mice. Am J Physiol Lung Cell Mol Physiol 300: L753–L761, 2011. [DOI] [PubMed] [Google Scholar]
- 65. Mlczoch J, Weir EK, Reeves JT, Grover RF. Long term effects of the anorectic agent fenfluramine alone and in combination with aminorex on pulmonary and systemic circulation in the pig. Basic Res Cardiol 74: 313–320, 1979. [DOI] [PubMed] [Google Scholar]
- 66. Morecroft I, Doyle B, Nilsen M, Kolch W, Mair K, Maclean MR. Mice lacking the Raf-1 kinase inhibitor protein exhibit exaggerated hypoxia-induced pulmonary hypertension. Br J Pharmacol 163: 948–963, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67. Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JX-J, Weir EK. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol 54, Suppl 1: S20–S31, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mouse Genome Sequencing Consortium Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigó R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O'Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Niederhausern Von AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang S-P, Zdobnov EM, Zody MC, Lander ES. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562, 2002. [DOI] [PubMed] [Google Scholar]
- 69. Nelson DR, Zeldin DC, Hoffman SMG, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 14: 1–18, 2004. [DOI] [PubMed] [Google Scholar]
- 70. Newman JH, Wheeler L, Lane KB, Loyd E, Gaddipati R, Phillips JA, Loyd JE. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med 345: 319–324, 2001. [DOI] [PubMed] [Google Scholar]
- 71. Nicolls MR, Taraseviciene-Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J 26: 1110–1118, 2005. [DOI] [PubMed] [Google Scholar]
- 72. Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 100: 923–929, 2007. [DOI] [PubMed] [Google Scholar]
- 73. Parravicini C, Corbellino M, Paulli M, Magrini U, Lazzarino M, Moore PS, Chang Y. Expression of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative Castleman's disease. Am J Pathol 151: 1517–1522, 1997. [PMC free article] [PubMed] [Google Scholar]
- 74. Rabieyousefi M, Soroosh P, Satoh K, Date F, Ishii N, Yamashita M, Oka M, McMurtry IF, Shimokawa H, Nose M, Sugamura K, Ono M. Indispensable roles of OX40L-derived signal and epistatic genetic effect in immune-mediated pathogenesis of spontaneous pulmonary hypertension. BMC Immunol 12: 67, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 118: 2372–2379, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rai PR, Cool CD, King JAC, Stevens T, Burns N, Winn RA, Kasper M, Voelkel NF. The cancer paradigm of severe pulmonary arterial hypertension. Am J Respir Crit Care Med 178: 558–564, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rehman J, Archer SL. A proposed mitochondrial-metabolic mechanism for initiation and maintenance of pulmonary arterial hypertension in fawn-hooded rats: the Warburg model of pulmonary arterial hypertension. Adv Exp Med Biol 661: 171–185, 2010. [DOI] [PubMed] [Google Scholar]
- 78. Resta TC, Chicoine LG, Omdahl JL, Walker BR. Maintained upregulation of pulmonary eNOS gene and protein expression during recovery from chronic hypoxia. Am J Physiol Heart Circ Physiol 276: H699–H708, 1999. [DOI] [PubMed] [Google Scholar]
- 79. Ryan J, Bloch K, Archer SL. Rodent models of pulmonary hypertension: harmonisation with the world health organisation's categorisation of human PH. Int J Clin Pract 65, Suppl 172: 15–34, 2011. [DOI] [PubMed] [Google Scholar]
- 80. Said SI, Hamidi SA, Dickman KG, Szema AM, Lyubsky S, Lin RZ, Jiang YP, Chen JJ, Waschek JA, Kort S. Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene. Circulation 115: 1260–1268, 2007. [DOI] [PubMed] [Google Scholar]
- 81. Sakao S, Taraseviciene-Stewart L, Lee JD, Wood K, Cool CD, Voelkel NF. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J 19: 1178–1180, 2005. [DOI] [PubMed] [Google Scholar]
- 82. Sakao S, Tatsumi K. The effects of antiangiogenic compound SU5416 in a rat model of pulmonary arterial hypertension. Respiration 81: 253–261, 2011. [DOI] [PubMed] [Google Scholar]
- 83. Sandoval J, Bauerle O, Palomar A, Gómez A, Martínez-Guerra ML, Beltrán M, Guerrero ML. Survival in primary pulmonary hypertension. Validation of a prognostic equation. Circulation 89: 1733–1744, 1994. [DOI] [PubMed] [Google Scholar]
- 84. Schermuly RT, Kreisselmeier KP, Ghofrani HA, Yilmaz H, Butrous G, Ermert L, Ermert M, Weissmann N, Rose F, Guenther A, Walmrath D, Seeger W, Grimminger F. Chronic sildenafil treatment inhibits monocrotaline-induced pulmonary hypertension in rats. Am J Respir Crit Care Med 169: 39–45, 2004. [DOI] [PubMed] [Google Scholar]
- 85. Seta F, Rahmani M, Turner PV, Funk CD. Pulmonary oxidative stress is increased in cyclooxygenase-2 knockdown mice with mild pulmonary hypertension induced by monocrotaline. PLoS ONE 6: e23439, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shifren A, Durmowicz AG, Knutsen RH, Faury G, Mecham RP. Elastin insufficiency predisposes to elevated pulmonary circulatory pressures through changes in elastic artery structure. J Appl Physiol 105: 1610–1619, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shlobin OA, Nathan SD. Pulmonary hypertension secondary to interstitial lung disease. Expert Rev Respir Med 5: 179–189, 2011. [DOI] [PubMed] [Google Scholar]
- 88. Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, Gaine SP, Gladwin MT, Jing ZC, Krowka MJ, Langleben D, Nakanishi N, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 54: S43–S54, 2009. [DOI] [PubMed] [Google Scholar]
- 89. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 179: 977–983, 2007. [DOI] [PubMed] [Google Scholar]
- 90. Song Y, Coleman L, Shi J, Beppu H, Sato K, Walsh K, Loscalzo J, Zhang YY. Inflammation, endothelial injury, and persistent pulmonary hypertension in heterozygous BMPR2-mutant mice. Am J Physiol Heart Circ Physiol 295: H677–H690, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Song Y, Jones JE, Beppu H, Keaney JF, Loscalzo J, Zhang YY. Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation 112: 553–562, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, Walker C, Budd DC, Pepke-Zaba J, Morrell NW. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 122: 920–927, 2010. [DOI] [PubMed] [Google Scholar]
- 93. Stanbrook HS, Morris KG, McMurtry IF. Prevention and reversal of hypoxic pulmonary hypertension by calcium antagonists. Am Rev Respir Dis 130: 81–85, 1984. [DOI] [PubMed] [Google Scholar]
- 94. Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res 104: 236–244, 28p following 244, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol 297: L1013–L1032, 2009. [DOI] [PubMed] [Google Scholar]
- 96. Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, McMurtry MS, Michalak M, Vance JE, Sessa WC, Michelakis ED. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med 3: 88ra55, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sztrymf B, Coulet F, Girerd B, Yaici A, Jais X, Sitbon O, Montani D, Souza R, Simonneau G, Soubrier F, Humbert M. Clinical outcomes of pulmonary arterial hypertension in carriers of BMPR2 mutation. Am J Respir Crit Care Med 177: 1377–1383, 2008. [DOI] [PubMed] [Google Scholar]
- 98. Tabima DM, Hacker TA, Chesler NC. Measuring right ventricular function in the normal and hypertensive mouse hearts using admittance-derived pressure-volume loops. Am J Physiol Heart Circ Physiol 299: H2069–H2075, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, Patterson AJ, Agrawal R, Rabinovitch M, Ambler K, Long CS, Voelkel NF, Nicolls MR. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res 109: 867–879, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 15: 427–438, 2001. [DOI] [PubMed] [Google Scholar]
- 101. Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C, Wood K, Parr JE, Boackle SA, Voelkel NF. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med 175: 1280–1289, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Taraseviciene-Stewart L, Scerbavicius R, Choe KH, Cool C, Wood K, Tuder RM, Burns N, Kasper M, Voelkel NF. Simvastatin causes endothelial cell apoptosis and attenuates severe pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 291: L668–L676, 2006. [DOI] [PubMed] [Google Scholar]
- 103. Taraseviciene-Stewart L, Slepikas L, Parr J, Kraskauskiene V, Kraskauskas D, Bogart H, Voelkel N. Bosentan fails to prevent right ventricular hypertrophy and heart failure in immune impaired animals exposed to chronic hypoxia (Abstract). Am J Respir Crit Care Med 179: A1822, 2009. [Google Scholar]
- 104. Thibault HB, Kurtz B, Raher MJ, Shaik RS, Waxman A, Derumeaux G, Halpern EF, Bloch KD, Scherrer-Crosbie M. Noninvasive assessment of murine pulmonary arterial pressure: validation and application to models of pulmonary hypertension. Circ Cardiovasc Imaging 3: 157–163, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tucker A, McMurtry IF, Alexander AF, Reeves JT, Grover RF. Lung mast cell density and distribution in chronically hypoxic animals. J Appl Physiol 42: 174–178, 1977. [DOI] [PubMed] [Google Scholar]
- 106. Tuder RM, Abman SH, Braun T, Capron F, Stevens T, Thistlethwaite PA, Haworth SG. Development and pathology of pulmonary hypertension. J Am Coll Cardiol 54: S3–S9, 2009. [DOI] [PubMed] [Google Scholar]
- 107. Urboniene D, Haber I, Fang YH, Thenappan T, Archer SL. Validation of high-resolution echocardiography and magnetic resonance imaging vs. high-fidelity catheterization in experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 299: L401–L412, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Vanderpool RR, Kim AR, Molthen R, Chesler NC. Effects of acute Rho kinase inhibition on chronic hypoxia-induced changes in proximal and distal pulmonary arterial structure and function. J Appl Physiol 110: 188–198, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Vergadi E, Chang MS, Lee C, Liang OD, Liu X, Fernandez-Gonzalez A, Mitsialis SA, Kourembanas S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 123: 1986–1995, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ, Gladwin M, Denholm EM, Gail DB, National Heart Lung, and Blood Institute Working Group on Cellular and Molecular Mechanisms of Right Heart Failure Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 114: 1883–1891, 2006. [DOI] [PubMed] [Google Scholar]
- 111. Voelkel NF, Tuder RM, Bridges J, Arend WP. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am J Respir Cell Mol Biol 11: 664–675, 1994. [DOI] [PubMed] [Google Scholar]
- 112. Voelkel NF, Tuder RM, Wade K, Höper M, Lepley RA, Goulet JL, Koller BH, Fitzpatrick F. Inhibition of 5-lipoxygenase-activating protein (FLAP) reduces pulmonary vascular reactivity and pulmonary hypertension in hypoxic rats. J Clin Invest 97: 2491–2498, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Voelkel NF, Tuder RM. Hypoxia-induced pulmonary vascular remodeling: a model for what human disease? J Clin Invest 106: 733–738, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Walker BR, Berend N, Voelkel NF. Comparison of muscular pulmonary arteries in low and high altitude hamsters and rats. Respir Physiol 56: 45–50, 1984. [DOI] [PubMed] [Google Scholar]
- 115. Wang L, Tamosiuniene R, Voelkel N, Gera L, Nicolls M. Restoration of regulatory T cells (Tregs) prevents autoimmune inflammation (AI) and pulmonary hypertension (PH) in rats (Abstract). Am J Respir Crit Care Med 179: A2355, 2009. [Google Scholar]
- 116. West J, Fagan K, Steudel W, Fouty B, Lane K, Harral J, Hoedt-Miller M, Tada Y, Ozimek J, Tuder R, Rodman DM. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res 94: 1109–1114, 2004. [DOI] [PubMed] [Google Scholar]
- 117. West J, Harral J, Lane K, Deng Y, Ickes B, Crona D, Albu S, Stewart D, Fagan K. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am J Physiol Lung Cell Mol Physiol 295: L744–L755, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Weyman AE, Dillon JC, Feigenbaum H, Chang S. Echocardiographic patterns of pulmonic valve motion with pulmonary hypertension. Circulation 50: 905–910, 1974. [DOI] [PubMed] [Google Scholar]
- 119. Will DH, Horrell JF, Reeves JT, Alexander AF. Influence of altitude and age on pulmonary arterial pressure in cattle. Proc Soc Exp Biol Med 150: 564–567, 1975. [DOI] [PubMed] [Google Scholar]
- 120. Winter CK, Segall HJ, Jones AD. Determination of pyrrolizidine alkaloid metabolites from mouse liver microsomes using tandem mass spectrometry and gas chromatography/mass spectrometry. Biomed Environ Mass Spectrom 15: 265–273, 1988. [DOI] [PubMed] [Google Scholar]
- 121. Wolfel EE, Selland MA, Mazzeo RS, Reeves JT. Systemic hypertension at 4,300 m is related to sympathoadrenal activity. J Appl Physiol 76: 1643–1650, 1994. [DOI] [PubMed] [Google Scholar]
- 122. Xu MH, Gong YS, Su MS, Dai ZY, Dai SS, Bao SZ, Li N, Zheng RY, He JC, Chen JF, Wang XT. Absence of the adenosine A2A receptor confers pulmonary arterial hypertension and increased pulmonary vascular remodeling in mice. J Vasc Res 48: 171–183, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, Dweik RA, Tuder RM, Stuehr DJ, Erzurum SC. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA 104: 1342–1347, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Yasuda T, Tada Y, Tanabe N, Tatsumi K, West J. Rho-kinase inhibition alleviates pulmonary hypertension in transgenic mice expressing a dominant-negative type II bone morphogenetic protein receptor gene. Am J Physiol Lung Cell Mol Physiol 301: L667–L674, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ye C, Sweeny D, Sukbuntherng J, Zhang Q, Tan W, Wong S, Madan A, Ogilvie B, Parkinson A, Antonian L. Distribution, metabolism, and excretion of the anti-angiogenic compound SU5416. Toxicol In Vitro 20: 154–162, 2006. [DOI] [PubMed] [Google Scholar]
- 126. Yeager ME, Frid MG, Stenmark KR. Progenitor cells in pulmonary vascular remodeling. Pulm Circ 1: 3–16, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Yeager ME, Nguyen CM, Belchenko DD, Colvin KL, Takatsuki S, Ivy DD, Stenmark KR. Circulating myeloid derived suppressor cells are increased and activated in pulmonary hypertension. Chest 141: 944–952, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127a. Yet SF, Perrella MA, Layne MD, Hsieh CM, Maemura K, Kobzik L, Christou H, Kourembanas S, Lee M. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J Clim Invest 103: R23–R29, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Yoshio T, Masuyama JI, Kohda N, Hirata D, Sato H, Iwamoto M, Mimori A, Takeda A, Minota S, Kano S. Association of interleukin 6 release from endothelial cells and pulmonary hypertension in SLE. J Rheumatol 24: 489–495, 1997. [PubMed] [Google Scholar]
- 129. Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest 103: 691–696, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Yu L, Hales CA. Hypoxia does neither stimulate pulmonary artery endothelial cell proliferation in mice and rats with pulmonary hypertension and vascular remodeling nor in human pulmonary artery endothelial cells. J Vasc Res 48: 465–475, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Yuki H, Sato S, Arisaka Y, Kato S, Tomoike H. Orally administered beraprost sodium inhibits pulmonary hypertension induced by monocrotaline in rats. Tohoku J Exp Med 173: 371–375, 1994. [DOI] [PubMed] [Google Scholar]
- 132. Yuyama H, Fujimori A, Sanagi M, Koakutsu A, Sudoh K, Sasamata M, Miyata K. The orally active nonpeptide selective endothelin ETA receptor antagonist YM598 prevents and reverses the development of pulmonary hypertension in monocrotaline-treated rats. Eur J Pharmacol 496: 129–139, 2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.