

Abstract
Activation of NFAT (nuclear factor of activated T cells)-mediated hypertrophic signaling is a major regulatory response to hypertrophic stimuli. A recent study unveiled potential regulatory roles for microRNA-133a (miR-133a) in cardiac hypertrophy. To date, however, no connection has been made between miR-133a and NFAT signaling. In this study, we determined that NFATc4, a hypertrophy-associated mediator, is negatively regulated by miR-133a. Two conserved base-pairing sites between the NFATc4 3′-untranslated region (UTR) and miR-133a were verified. Mutation of these sites in the NFATc4 3′-UTR completely blocked the negative effect of miR-133a on NFATc4, suggesting that NFATc4 is a direct target for miR-133a regulation. Using a gain-of-function approach, we demonstrate that miR-133 significantly reduces the endogenous level of, as well as the hypertrophic stimulus-mediated increase in, NFATc4 gene expression. This latter effect of miR-133a on NFATc4 gene expression was coincided with an attenuated cardiomyocyte hypertrophy induced by an α-adrenergic receptor agonist. Conversely, cells treated with miR-133a inhibitor resulted in an increase in NFATc4 expression level. Application of miR-133a had no apparent effect on NFATc4 nuclear localization. We conclude that the negative regulation of NFATc4 expression contributes to miR-133a-mediated hypertrophic repression.
Keywords: micro-ribonucleic acid-133a, cardiomyocyte, hypertrophy, fibrosis, nuclear factor of activated T cells
cardiac hypertrophy reflects a remodeling of the myocardium in response to hemodynamic stress and various hypertrophic stimuli. Defining features of hypertrophy for cardiomyocytes are increases in cell size, protein synthesis, and heightened organization of the sarcomere. At the molecular level, these phenotypes are accompanied by reactivation of the expression of fetal cardiac genes. Calcineurin-dependent NFAT (nuclear factor of activated T cells) activation is part of an important signaling cascade in response to hypertrophic signaling. NFATc4, one of five NFAT family members, was demonstrated to be sufficient in promoting cardiac hypertrophy in vivo (35, 46). NFATc4 functions as a calcineurin effector, trafficking between the cytosol and the nucleus to regulate the cardiac hypertrophic response. Heart-specific expression of a constitutively active NFATc4 lacking the calcineurin-binding domain also promotes cardiac hypertrophy in transgenic mice (35), suggesting pro-hypertrophic roles for NFATc4 via both calcineurin-dependent and calcineurin-independent pathways.
MicroRNAs (miRs) are a class of ∼22 nucleotide noncoding RNA molecules. MiRs regulate gene expression by binding to partially or completely complementary sequence in the 3′-UTR (3′-untranslated region) of mRNAs, resulting in translational repression or mRNA degradation (1, 3, 16). Several miRs, including miR-133a, have been implicated as regulators of cardiomyocyte hypertrophy (6, 10, 20, 21, 38, 40–43). The miR-133 family consists of three miRs: miR-133a-1, miR-133a-2, and miR-133b. miR-133a-1 and -2 are identical in sequence and are expressed specifically in cardiac and skeletal muscles, whereas miR-133b differs from miR-133a by two nucleotides at the 3′ terminus and has enriched expression in skeletal muscle (9, 15, 26, 39).
Various cellular functions have been ascribed to miR-133a (4, 6, 9, 13, 30, 32, 47). The importance of miR-133a in cardiac hypertrophy was highlighted in a recent study, in which a significant decrease in miR-133a expression was observed in human and animal hypertrophic myocardium (6). In addition, this study revealed that several growth-related genes, including RhoA and Cdc42, were verified as miR-133a target genes, suggesting the potential regulatory role of miRs in cardiac hypertrophy (6). Given that individual miRs regulate potentially dozens of mRNAs, further verification of target genes in hypertrophic signaling pathways is crucial for fully understanding of the function of miR-133a in cardiac hypertrophy.
In this study, we explored the relationship between miR-133a and the NFAT-dependent signaling pathway. We demonstrate that NFATc4 levels are negatively regulated by miR-133a. Treatment with miR-133a resulted in striking decreases in NFATc4 mRNA and protein expression, coincident with attenuation of cardiomyocyte hypertrophy, which had been induced by application of an α-adrenergic receptor agonist. We suggest that silencing of NFATc4 may be one of the miR-133a-mediated mechanisms in repressing cardiac hypertrophy.
MATERIALS AND METHODS
Generation of plasmid constructs and synthesis of adenoviruses.
To determine whether NFATc4 is regulated by miR-133a, a 524-bp long duplex of NFATc4 exon 10 (9109–9632) containing two putative miR-133a targeting sites (Fig. 1A) was inserted into the 3′-UTR of the pGL3-promoter luciferase reporter vector (Promega). To generate the mutant version of the luciferase reporter, the “seed” sequences of the two miR-133a targeting sites were mutated with adenines by the QuickChange site-directed mutagenesis kit (Stratagene). A pSV-β-galacosidase vector (Promega) was cotransfected in all luciferase assessment experiments as an internal control. To induce miR-133a expression in neonatal rat cardiomyocytes, an adenovirus expressing miR-133a (v-miR-133a) was synthesized using the Adeno-X-virTrak Expression system 2 (Clontech) (31). miR-133a precursor cDNA was subcloned into the donor vector pDNR-CMV, followed by a cre-loxP-mediated homologous recombination with the adenoviral vector pLP-Adeno-X-ViraTrak containing a red fluorescent protein (RFP) cassette. The recombinant adenoviral vector was linearized by PacI and transfected into the human embryonic kidney-293 cells. For the control virus, the vector contained only the RFP cassette. Cells were harvested 7–10 days after transfection, and the virus was released by repeated freeze-thaw. The concentrations of v-miR-133a and v-control (expressing RFP only) were 1.14 × 107 and 2.0 × 107 plaque-forming unit/ml, respectively. An MOI (multiplicity of infection) of 5 was used to infect neonatal rat ventricular cardiomyocytes in all experiments. To determine the dosage effect of miR-133a on NFATc4 expression, multiple MOIs in the range of 1–5 were applied.
Fig. 1.
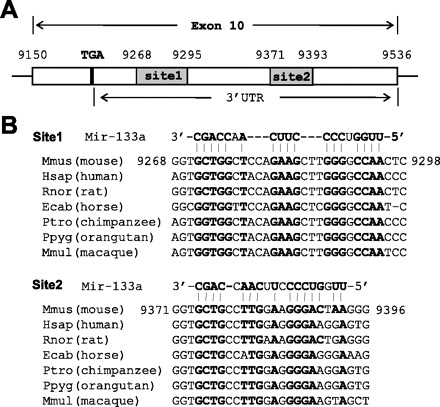
Prediction of two putative targeting sites of micro-RNA (miR)-133a in the 3′-untranslated region (UTR) of nuclear factor of activated T cell (NFAT) c4. A: schematic of exon 10 of NFATc4, including 3′-UTR. B: alignment of mouse miR-133a sequence with the 3′-UTRs of NFATc4 among different species. Matched nucleotides were highlighted in bold. Two highly conserved 3′-UTR sites were recognized.
Neonatal rat ventricular cardiomyocyte isolation and cell cultures.
The usage of isolated Sprague-Dawley rat cardiomyocytes was approved by the Institutional Animal Care and Use Committee of the Texas A&M Heath Science Center-Houston. Ventricles from 1- to 2-day-old Sprague-Dawley rats were extracted followed by collagenase (75 U/ml; Worthington) and pancreatin (0.6 mg/ml; Sigma) digestion. After multiple digestions, undigested tissue pieces were removed by passing the sample through a cell strainer (BD Biosciences), and cells were isolated by Percoll gradient centrifugation. Cells were then plated and incubated for 1 h, nonadherent cardiomyocytes were collected using a gentle wash, and rapidly attaching noncardiomyocytes were left in the culture dish and discarded. The desired cardiomyocytes in the collected washing medium were spun down and resuspended in culture medium. Cardiomyocytes were cultured in DMEM/F-12 50/50 (Cellgro) with 2 mM l-glutamine and 10% fetal bovine serum (FBS) for 24 h and then switched to the same medium without FBS for 24 h before the experiments. The purity of cardiomyocytes was evaluated by flow cytometry (LSRII, BD Biosciences) after the cells were stained with anti-striated sarcomeric myosin antibody MF-20. Mouse myoblast C2C12 cells were cultured in DMEM medium with 10% FBS.
Transient transfection for plasmids, miR mimic and inhibitor, luciferase assay, and cardiomyocyte hypertrophy experiments.
All transient transfections were conducted in C2C12 cells (1 × 105 cells/well in a 12-well plate for a confluent cell layer) using Lipofectamine 2000 in the Opti-MEM reduced serum medium (Invitrogen). The miR mimic, the miR inhibitor (antagomir), and the negative controls (chemically modified and cholesterol-conjugated synthetic miRs) were purchased from Dharmacon. Throughout the study, individual miRs were used at 20 nM. Two negative controls were used in each experiment. The Luciferase assay was conducted 36 h posttransfection. Lysis buffer was purchased from Promega (E3971), and 20 of 60 μl of the lysis supernatant were used for the measurement of luciferase activity using Monolight 3010 (Pharmingen). Each sample was measured three times. All results were adjusted by β-galactosidase activities (MRX Revelation, DYNEX Technologies). Cardiomyocyte hypertrophy was induced with the application of 20–100 μM phenylephrine (PE) for 48 h. When used in combination with the adenoviral experiments, PE was added 5 h after virus infection. Cell surface area was assessed using Image Tool software (UTHSCSA, version 3.0, San Antonio, TX).
Quantitative polymerase chain reaction analysis, Western blot analysis, and immunostaining.
Quantification of miR-133a was conducted (Stratagene Mx3000p qPCR System) by quantitative polymerase chain reaction (qPCR) analysis using the TaqMan miR assay (Applied Biosystems, catalog no. 4373142) and normalized with mmu-miR-16 (catalog no. 4427975). The SYBR green method with MasterMix buffer system containing Taq polymerase (Stratagene) was used to assess non-miR gene mRNA levels. MirVana miR isolation kit (Ambion) was used for RNA extraction. RNA quality was evaluated using the ratio of 260-to-280-nm optical density. Cloned AMV Reverse Transcriptase kit (Invitrogen) was used for reverse transcription to generate cDNA using an oligo(dT) primer with the RT buffer system. The PCR primers used are as follows (5′ to 3′): NFATc4 (NM_023699) gggggctgtcaaggctgctc/gcgcccgatgtctgtctcacc; Acta1 (NM_009606, skeletal muscle α1-actin) ggcggtgctgtccctctatgct/cgggcaacggaaacgctcatt; Actb (NM_007393, β-actin) agcggttccgatgccctgag/aggggccggactcatcgtactc; myh7 (NM_080728, cardiac β-myosin heavy chain) gccgcgccagtacttcataggtg/tggccttggggaacatgcact; brain natriuretic peptide (nm_008726, natriuretic peptide precursor type B) gcggcatggatctcctgaaggtg/agcccaaacgactgacggatcc. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression levels were used for qPCR normalization. Expression levels were determined by the 2−ΔΔCt method. Primary antibodies used were NFATc4 (sc-13036), NFATc2 (sc-7295), NFATc3 (sc-8321), cardiac-specific troponin I (sc-31655), GAPDH (sc-20357) (Santa Cruz Biotechnology), and MF-20 (DSHB, University of Iowa). Secondary antibody was Alexa Fluor 488 anti-goat IgG (Invitrogen, A11055). Protein samples for Western blot analysis were extracted and separated as described previously (8). Protein even loading was evaluated by the expression of GAPDH.
Statistical analysis.
All experiments were conducted three times. For a comparison of two groups, the data were analyzed by the t-test (SigmaStat, SPSS, version 2.03). For an analysis of multiple groups, data were analyzed by one-way ANOVA. A P < 0.05 was considered significant. Data are presented as means ± SE.
RESULTS
Bioinformatics analysis reveals NFATc4 as a potential miR-133 target.
Using the search engine of the miRBase Targets in silico database (http://www.mirbase.org), we examined the 3′-UTR of NFATc4 and identified two putative binding sites for miR-133a 76 nucleotides apart with free energies of −24.4 and −21.7 cal/mol, respectively (Fig. 1). The site with low free energy implicates a high possibility as an actual targeted sequence (25, 36). The miR-133a seed-matched sequences are highly conserved among species. Collectively, analyses of these suggest that the two sites in the 3′-UTR of NFATc4 are potential miR-133a targets.
miR-133a targets 3′-UTR of NFATc4.
To validate the two putative miR-133a target sites, a 524-bp-long duplex of exon 10 of the NFATc4 gene containing these sites was subcloned into the 3′-UTR of a luciferase reporter vector (Fig. 2A). The chimeric vector, luciferase-NFATc4 3′-UTR, carries a constitutively activated promoter, which can express luciferase as a reporter. The effect of miR-133a on luciferase expression can be represented by the changes in luciferase activity. We cotransfected this vector with the miR-133a mimic in myogenic C2C12 cells. As indicated in Fig. 2B, the introduction of miR-133a mimic decreased luciferase activity 40% compared with the cells treated with a control mimic (P < 0.05). In a parallel experiment, the inhibitory effect of miR-133a in cells transfected with the mutant reporter vector (the two putative targeting sites were mutated) was completely abolished, as evidenced by high luciferase activity (P < 0.05). We also observed increased baseline luciferase activity in this mutant reporter vector group due to the elimination of the response to the endogenous miR-133a. Thus these results confirm the bioinformatics prediction that the 3′-UTR of NFATc4 is targeted by miR-133a.
Fig. 2.
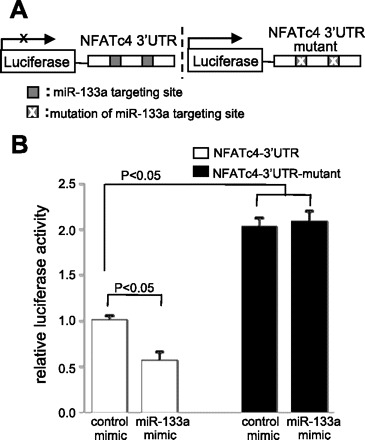
Analysis of the NFATc4–3′-UTR by luciferase activity assay. A: schematic presentation of the luciferase assay. The NFATc4–3′-UTR containing two miR-133a targeting sites was fused with the reporter gene luciferase. B: addition of the miR-133a mimic attenuated NFATc4–3′-UTR reporter gene activity. Mutation of miR-133a putative target sites blocked the repressive effect of miR-133a on the target, suggesting NFATc4 as a miR-133a target gene. The data in each group represent the average of nine measurements.
NFATc4 expression is negatively regulated by miR-133a.
To determine whether miR-133a could silence NFATc4 expression, we applied gain- and loss-of-function approaches. C2C12 cells were transfected with either the miR-133a mimic or the miR-133a inhibitor. Total RNA and protein were isolated 36 and 48 h after transfection, respectively. The NFATc4 mRNA level was quantified by qPCR analysis. The results showed that the cells treated with the miR-133a mimic decreased NFATc4 mRNA levels 65% compared with cells treated with the control mimic (P < 0.05) (Fig. 3A). In contrast, an almost threefold increase in the NFATc4 mRNA level was observed in the cells treated with the miR-133a inhibitor compared with the control group (P < 0.05). Western blot analysis revealed that, while miR-133a mimic treatment decreased NFATc4 protein expression, the opposite result was observed by following miR-133a inhibitor treatment (Fig. 3B), consistent with the changes of NFATc4 mRNA. This result indicates that the expression level of NFATc4 can be negatively modulated by miR-133a, and that miR-133a may be a silencer of NFACTc4.
Fig. 3.
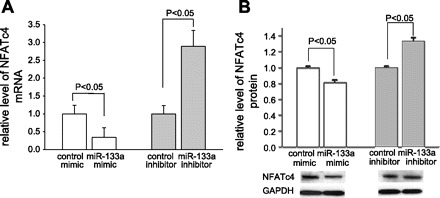
Effect of miR-133a mimic and inhibitor on the expression of NFATc4. Treatment of C2C12 cells with chemically modified miR-133a mimic reduced the levels of NFATc4 mRNA and protein. The converse results were observed when cells were treated with miR-133a inhibitor. A: quantitative polymerase chain reaction (qPCR) analysis. Each group contained six measurements. B: the expression level of NFATc4 protein was assessed by Western blot and semiquantified by densitometric analysis with six measurements in each group. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
miR-133a-mediated downregulation of NFAT contributes to the inhibition of cardiomyocyte hypertrophy induced by PE.
NFATc4 is one of the hypertrophy-associated transcription factors in the NFAT family. To examine whether the silencing of NFATc4 by miR-133a contributes to inhibition of cardiomyocyte hypertrophy, an adenovirus expressing miR-133a tagged RFP was generated (Fig. 4A). The virus was initially infected in C2C12 cells before its usage in cardiomyocytes. The expression of mature miR-133a in cells infected with v-miR-133a at MOI 5 was confirmed by qPCR (Fig. 4B). As expected, the control virus (v-control) expressed only RFP, with no effect on the endogenous miR-133a level (Fig. 4B). We then infected cells with different dosages of v-miR-133a (MOI from 1 to 5). Consistent with the miR-133a mimic study, decreases in NFATc4 mRNA and protein levels were observed after 36 and 48 h of infection, respectively (Fig. 4, C and D). The reduction of NFATc4 expression was dose dependent.
Fig. 4.
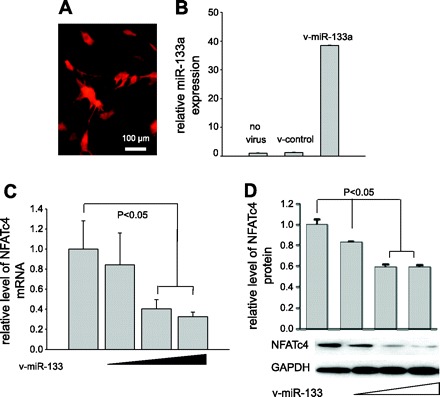
Generation of the adenovirus expressing miR-133a (v-miR-133a) and assessment of miR-133a-mediated NFATc4 silencing. A: red fluorescent protein was coexpressed as a marker along with the miR expression. B: qPCR analysis revealed a high level of miR-133a expressed by v-miR-133a. C and D: dose-dependent silencing of NFATc4 by v-miR-133a was assessed in C2C12 cells by qPCR and Western blot analysis, respectively. v-control, Control adenovirus expressing RFP. The data in each group represent the average of nine measurements, except for B and D, which contains four measurements.
We next applied the viruses to neonatal rat ventricle cardiomyocytes to see if the treatment of cells with miR-133a could directly block PE-induced cardiomyocyte hypertrophy. A 91.3% purity in cardiomyocytes was achieved (Fig. 5A). After 48-h PE treatment, cardiomyocytes exhibited robust hypertrophy (Fig. 5Bb), while the hypertrophy was completely blocked by the v-miR-133a (Fig. 5Bd), but not by the v-control infection (Fig. 5Bc). The alteration in cell surface area supported this morphological observation (Fig. 5C). This strongly suggests that application of miR-133a is sufficient to prevent PE-induced cardiomyocytes hypertrophy.
Fig. 5.
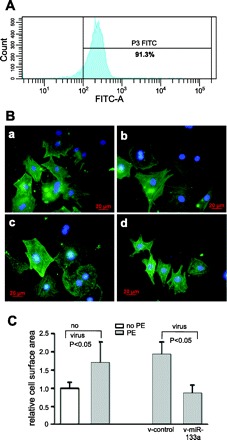
miR-133a treatment attenuates phenylephrine (PE)-induced cardiomyocyte hypertrophy. A: flow cytometry demonstrated 91.3% purity in neonatal rat cardiomyocytes. B: representative immunostaining of cardiomyocytes: control cardiomyocytes (a); PE-treated cardiomyocytes (b); PE-treated cardiomyocytes with control virus (c); PE-treated cardiomyocytes with v-miR-133a (d). C: morphometric analysis of cardiomyocytes. Size of cardiomyocytes, as evaluated by surface area, was significantly increased by PE treatment. This size increase was blocked by v-miR-133a. Cardiomyocytes were detected by cardiac-specific troponin I antibodies (green), followed by diamidino-2-phenylindole staining for nuclei (blue). At least 50 cardiomyocytes were randomly chosen for cell surface area measurement.
To further evaluate the repressive effect of miR-133a on the hypertrophic response of cardiomyocytes, we measured the levels of cardiac stress markers Acta1, Actb, Myh7, and brain natriuretic peptide via qPCR analysis. We found that expression levels of these genes were significantly upregulated after PE induction (Fig. 6). However, the increases were suppressed by the infection of v-miR-133a, suggesting an inhibitory effect of miR-133a on the hypertrophic response.
Fig. 6.
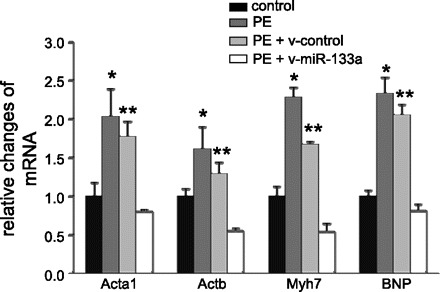
PE-mediated hypertrophic responses in cardiomyocytes were eliminated by miR-133a. The hypertrophic response was demonstrated in robust increases in various stress markers. The changes of the stress marker mRNAs were quantified by qPCR analysis. P < 0.05 compared with *controls or **PE + v-miR-133a groups. The data in each group represent the average of nine measurements. BNP, brain natriuretic peptide.
Finally, we assessed mRNA and protein levels of NFATc4 in PE-treated cardiomyocytes. Overt increases in NFATc4 mRNA (Fig. 7A) and protein (Fig. 7B) were detected in PE-treated cardiomyocytes, whereas these increases were significantly suppressed by the introduction of v-miR-133a, indicating that the expression of miR-133a eliminated PE-induced NFATc4 upregulation. No significant changes in expression of NFATc2 and NFATc3 (two other major isoforms of NFAT family in the heart) were detected with v-miR-133a treatment (Fig. 7B). Collectively, these data demonstrate that NFATc4 is the target of miR-133a, and that silencing of NFATc4 by miR-133a may contribute to miR-133a-mediated anti-hypertrophy.
Fig. 7.
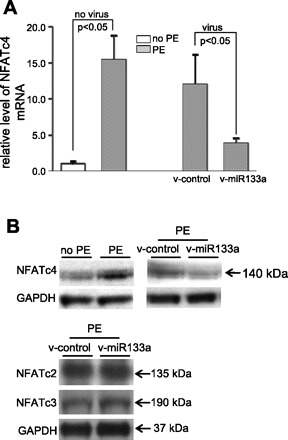
Silencing of NFATc4 gene expression by miR-133a. In response to PE and miR-133a treatments, expression of NFATc4 was assessed by qPCR (A) and Western blot analysis (B), respectively. Robust increases in NFATc4 mRNA and protein were observed after the PE treatment, whereas the increases were attenuated by infection of the v-miR-133a. Expression levels of NFATc2 and NFATc3 did not significantly change on v-miR-133a treatment. The data in each group in A represent the average of nine measurements.
It has been suggested that the activity of transcription factor NFAT is contingent on its nuclear import (11, 23). To address this possibility, we visualized the NFATc4 in the cellular compartment by immunostaining in cardiomyocytes (Fig. 8). No obvious change of nuclear localization of NFATc4 was observed after v-miR-133a treatment compared with the control group (Fig. 8, B and E). Hence, we conclude that miR-133a negatively regulates NFATc4 expression, but not the activity of NFATc4.
Fig. 8.
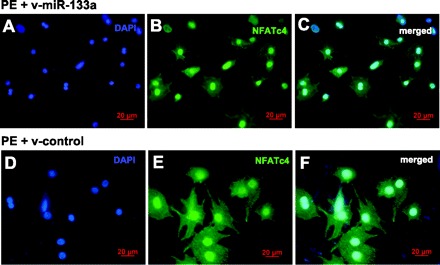
Application of miR-133a had no effect on NFATc4 nuclear localization. Immunostaining with antibody specific for NFATc4 was performed in neonatal rat ventricular myocytes. Expression of NFATc4 (green) was observed in both nucleus and cytoplasm. No significant changes of NFATc4 cellular distribution were found between the v-miR-133a (A–C) and v-control virus (D–F) treatment groups.
DISCUSSION
Several features make miRs unique regulators of gene expression. First, a single miR can regulate a number of different mRNAs, as long as the UTRs carry a common targeting sequence. In addition, the same mRNA can be silenced by multiple miRs. Given these features, one of the challenges in any miR functional study is to identify and validate the multiple target genes of an individual miR. In this study, we identified NFATc4 as one of several genes negatively regulated by miR-133a. Two miR-133a hybridization sites in the NFATc4 3′-UTR were determined, and bioinformatics analysis revealed that they are highly conserved among species. Mutation of these sites completely blocked the negative effect of miR-133a on NFATc4, revealing NFATc4 as a direct target of miR-133. We further demonstrated that application of miR-133 significantly silenced the endogenous level of, as well as the hypertrophic stimulus-mediated increase in, NFATc4 gene expression. The decrease in expression of miR-133a resulted in an increase in the NFATc4 expression level. We found that manipulation of miR-133a had no overt effect on NFATc4 nuclear localization, as well as the expression levels of NFATc2 and NFATc3, the two major NFAT isoforms in the heart. We conclude that the negative regulation of NFATc4 expression contributes to miR-133a-mediated hypertrophic repression.
Given the high degree of similarity in the DNA-binding and the calcineurin-interaction domains among the NFAT isoforms (18, 22, 37), it is not surprising that functional redundancy has been demonstrated between NFATc3 and NFATc4. Hence, it was important to determine the complementary role of NFATc4 to that of NFATc3 in cardiac hypertrophy (5, 35, 45). NFATc4 was first identified as a downstream effector of the calcineurin-dependent hypertrophic signaling pathway (35). Phosphorylated NFATc4 localizes to the cytoplasm in an inactive state (18, 22, 37). Upon the calcium signaling, the NFATc4 is dephosphorylated by calcineurin, then translocates to the nucleus and associates with the zinc finger transcription factor GATA4, resulting in a synergistic activation of fetal cardiac genes and a hypertrophic response (35). The pro-hypertrophic effect of NFATc4 has also been demonstrated through the calcineurin-independent pathway. Pioneering work by Molkentin revealed that transgenic mice with cardiac-restricted expression of active NFATc4 (lacking the calcineurin binding domain) developed robust hypertrophic cardiomyopathy (35). The hypertrophic role of NFATc4 has been incessantly identified recently. Notably, much higher nuclear levels of NFATc4 protein were observed in the myocardium of heart failure patients than the myocardium from control noncardiac disease patients (2, 7, 12, 14, 19, 24, 27–29, 33, 44). Altogether, these studies demonstrate that activation of NFATc4 is sufficient for the development of cardiac hypertrophy.
Interestingly, the pathological significance of each individual NFAT member to the development of cardiac hypertrophy is varied. Using a loss-of-function approach, a major function of NFATc3 in cardiac hypertrophy has been demonstrated. Both calcineurin- and angiotensin II-induced cardiac hypertrophy can be partially attenuated by a loss of the NFATc3 gene, but not of the NFATc4 gene (45). On the other hand, the complementary role of NFATc4 has also been elucidated. Active NFATc4 is sufficient to rescue the NFATc3 NFATc4 double-knockout mouse phenotype (5), suggesting that redundant functions among different NFAT members are far more complicated than previously expected (5, 17, 45). Further clarification of the importance of NFATc4 in hypertrophy is required. Given the findings from these previous investigations, we believe that the upregulation of multiple hypertrophy-associated genes, such as RhoA, Cdc42, and NFATc4, collectively contribute to miR-133a-mediated hypertrophic repression.
Emerging studies have uncovered the anti-hypertrophy role of miR-133a in the heart. A profound reduction of miR-133a in human and animal hypertrophic ventricle myocardium was observed (6, 42), and the transient knockdown of miR-133a in vivo by an antagomir has resulted in cardiac hypertrophy (6). Interestingly, miR-133a-1 and -2 double knockout mice develop severe fibrotic but not hypertrophic cardiomyopathy (30). This discrepancy may be due to the loss of miR-133a expression in a different temporal pattern between the two mouse models. In the antagomir mouse model, a downregulation of miR-133a is induced in adult mouse hearts. In the knockout mice, however, genetic elimination of miR-133 occurs at the onset of embryogenesis. The disparity might also explain the dual roles of miR-133a in cardiac remodeling. The anti-fibrotic effect of miR-133a was characterized in a recent study, whereby miR-133a silences connective tissue growth factor, a strong profibrotic factor (13). Of note, mice with cardiac overexpression of NFATc4 develop overt cardiac fibrosis (35), the same phenotype observed in the miR-133a null mice (30). More recently, Matkovich et al. (34) reported that genetic overexpression of miR-133a failed to protect mouse hearts from pressure-overload-induced hypertrophy. The differing results between these studies may be explained by several factors; an obvious difference is that our study was conducted in cultured cardiomyocytes. Cardiomyocyte hypertrophy was induced by PE and not by mechanical stretch, consistent with an earlier in vitro study showing that forced miR-133a overexpression attenuated hypertrophy (6). Nevertheless, our finding supports the notion that the functions of an miR can be ascribed to several targets. The miR-133a-mediated NFATc4 silencing mechanism provides a new layer of regulatory complexity to the calcineurin-independent NFAT gene regulation program. Future identification of the new targets will extend our understanding of the biological functions of miR-133a in a variety of cardiac disorders.
GRANTS
This work was supported by American Heart Association Grant-in-Aid 0855030F to J. Chang and National Natural Science Foundation of China Grant 30860103 to Q. Li and J. Chang.
DISCLOSURES
No conflicts of interest are declared by the author(s).
ACKNOWLEDGMENTS
The authors thank Dr. Robert J. Schwartz for critical suggestions on the project. We are grateful to Drs. Vladimir N. Potaman, Todd Heallen, John Ivy, and Guanghong Tan for editorial assistance on the manuscript, and to Yanlin Ma and Kong Ann for technical help.
REFERENCES
- 1. Ambros V. The functions of animal microRNAs. Nature 431: 350–355, 2004 [DOI] [PubMed] [Google Scholar]
- 2. Bao Y, Li R, Jiang J, Cai B, Gao J, Le K, Zhang F, Chen S, Liu P. Activation of peroxisome proliferator-activated receptor gamma inhibits endothelin-1-induced cardiac hypertrophy via the calcineurin/NFAT signaling pathway. Mol Cell Biochem 317: 189–196, 2008 [DOI] [PubMed] [Google Scholar]
- 3. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, function. Cell 116: 281–297, 2004 [DOI] [PubMed] [Google Scholar]
- 4. Boutz PL, Chawla G, Stoilov P, Black DL. MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev 21: 71–84, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bushdid PB, Osinska H, Waclaw RR, Molkentin JD, Yutzey KE. NFATc3 and NFATc4 are required for cardiac development and mitochondrial function. Circ Res 92: 1305–1313, 2003 [DOI] [PubMed] [Google Scholar]
- 6. Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW, 2nd, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat Med 13: 613–618, 2007 [DOI] [PubMed] [Google Scholar]
- 7. Cha H, Kim JM, Oh JG, Jeong MH, Park CS, Park J, Jeong HJ, Park BK, Lee YH, Jeong D, Yang DK, Bernecker OY, Kim do H, Hajjar RJ, Park WJ. PICOT is a critical regulator of cardiac hypertrophy and cardiomyocyte contractility. J Mol Cell Cardiol 45: 796–803, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chang J, Knowlton AA, Wasser JS. Expression of heat shock proteins in turtle and mammal hearts: relationship to anoxia tolerance. Am J Physiol Regul Integr Comp Physiol 278: R209–R214, 2000 [DOI] [PubMed] [Google Scholar]
- 9. Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 38: 228–233, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol 170: 1831–1840, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chow CW, Rincon M, Cavanagh J, Dickens M, Davis RJ. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science 278: 1638–1641, 1997 [DOI] [PubMed] [Google Scholar]
- 12. Diedrichs H, Chi M, Boelck B, Mehlhorn U, Schwinger RH. Increased regulatory activity of the calcineurin/NFAT pathway in human heart failure. Eur J Heart Fail 6: 3–9, 2004 [DOI] [PubMed] [Google Scholar]
- 13. Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, Herias V, van Leeuwen RE, Schellings MW, Barenbrug P, Maessen JG, Heymans S, Pinto YM, Creemers EE. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res 104: 170–178, 2009 [DOI] [PubMed] [Google Scholar]
- 14. Fan Y, Xie P, Zhang T, Zhang H, Gu D, She M, Li H. Regulation of the stability and transcriptional activity of NFATc4 by ubiquitination. FEBS Lett 582: 4008–4014, 2008 [DOI] [PubMed] [Google Scholar]
- 15. Feng J, Schaus BJ, Fallavollita JA, Lee TC, Canty JM., Jr Preload induces troponin I degradation independently of myocardial ischemia. Circulation 103: 2035–2037, 2001 [DOI] [PubMed] [Google Scholar]
- 16. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806–811, 1998 [DOI] [PubMed] [Google Scholar]
- 17. Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell 105: 863–875, 2001 [DOI] [PubMed] [Google Scholar]
- 18. Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17: 2205–2232, 2003 [DOI] [PubMed] [Google Scholar]
- 19. Ichida M, Finkel T. Ras regulates NFAT3 activity in cardiac myocytes. J Biol Chem 276: 3524–3530, 2001 [DOI] [PubMed] [Google Scholar]
- 20. Ikeda S, He A, Kong SW, Lu J, Bejar R, Bodyak N, Lee KH, Ma Q, Kang PM, Golub TR, Pu WT. microRNA-1 negatively regulates expression of the hypertrophy-associated genes calmodulin and Mef2a. Mol Cell Biol 29: 2193–2204, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, Golub TR, Pieske B, Pu WT. Altered microRNA expression in human heart disease. Physiol Genomics 31: 367–373, 2007 [DOI] [PubMed] [Google Scholar]
- 22. Jain J, Burgeon E, Badalian TM, Hogan PG, Rao A. A similar DNA-binding motif in NFAT family proteins and the Rel homology region. J Biol Chem 270: 4138–4145, 1995 [PubMed] [Google Scholar]
- 23. Jain J, McCaffrey PG, Miner Z, Kerppola TK, Lambert JN, Verdine GL, Curran T, Rao A. The T-cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature 365: 352–355, 1993 [DOI] [PubMed] [Google Scholar]
- 24. Jeong D, Kim JM, Cha H, Oh JG, Park J, Yun SH, Ju ES, Jeon ES, Hajjar RJ, Park WJ. PICOT attenuates cardiac hypertrophy by disrupting calcineurin-NFAT signaling. Circ Res 102: 711–719, 2008 [DOI] [PubMed] [Google Scholar]
- 25. Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet 39: 1278–1284, 2007 [DOI] [PubMed] [Google Scholar]
- 26. Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol 12: 735–739, 2002 [DOI] [PubMed] [Google Scholar]
- 27. Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest 114: 1058–1071, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li R, Zheng W, Pi R, Gao J, Zhang H, Wang P, Le K, Liu P. Activation of peroxisome proliferator-activated receptor-alpha prevents glycogen synthase 3beta phosphorylation and inhibits cardiac hypertrophy. FEBS Lett 581: 3311–3316, 2007 [DOI] [PubMed] [Google Scholar]
- 29. Liao W, Wang S, Han C, Zhang Y. 14–3-3 proteins regulate glycogen synthase 3 beta phosphorylation and inhibit cardiomyocyte hypertrophy. FEBS J 272: 1845–1854, 2005 [DOI] [PubMed] [Google Scholar]
- 30. Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev 22: 3242–3254, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J, Sharff KA, Luu HH, Haydon RC, Kinzler KW, Vogelstein B, He TC. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc 2: 1236–1247, 2007 [DOI] [PubMed] [Google Scholar]
- 32. Luo X, Lin H, Pan Z, Xiao J, Zhang Y, Lu Y, Yang B, Wang Z. Down-regulation of miR-1/miR-133 contributes to re-expression of pacemaker channel genes HCN2 and HCN4 in hypertrophic heart. J Biol Chem 283: 20045–20052, 2008 [DOI] [PubMed] [Google Scholar]
- 33. Mathew S, Mascareno E, Siddiqui MA. A ternary complex of transcription factors, Nished and NFATc4, and co-activator p300 bound to an intronic sequence, intronic regulatory element, is pivotal for the up-regulation of myosin light chain-2v gene in cardiac hypertrophy. J Biol Chem 279: 41018–41027, 2004 [DOI] [PubMed] [Google Scholar]
- 34. Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM, Dorn GW., 2nd MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res 106: 166–175, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93: 215–228, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muckstein U, Tafer H, Hackermuller J, Bernhart SH, Stadler PF, Hofacker IL. Thermodynamics of RNA-RNA binding. Bioinformatics 22: 1177–1182, 2006 [DOI] [PubMed] [Google Scholar]
- 37. Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15: 707–747, 1997 [DOI] [PubMed] [Google Scholar]
- 38. Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res 100: 416–424, 2007 [DOI] [PubMed] [Google Scholar]
- 39. Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol 5: R13, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM, Wang DZ. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol 42: 1137–1141, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Van Rooij E, Olson EN. MicroRNAs put their signatures on the heart. Physiol Genomics 31: 365–366, 2007 [DOI] [PubMed] [Google Scholar]
- 42. van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A 103: 18255–18260, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316: 575–579, 2007 [DOI] [PubMed] [Google Scholar]
- 44. Vlasblom R, Muller A, Musters RJ, Zuidwijk MJ, Van Hardeveld C, Paulus WJ, Simonides WS. Contractile arrest reveals calcium-dependent stimulation of SERCA2a mRNA expression in cultured ventricular cardiomyocytes. Cardiovasc Res 63: 537–544, 2004 [DOI] [PubMed] [Google Scholar]
- 45. Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF, Molkentin JD. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol Cell Biol 22: 7603–7613, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun 322: 1178–1191, 2004 [DOI] [PubMed] [Google Scholar]
- 47. Xiao J, Luo X, Lin H, Zhang Y, Lu Y, Wang N, Yang B, Wang Z. MicroRNA miR-133 represses HERG K+ channel expression contributing to QT prolongation in diabetic hearts. J Biol Chem 282: 12363–12367, 2007. [DOI] [PubMed] [Google Scholar]