

Abstract
To test the deterioration of endothelial function during the progression of diabetes, shear stress-induced dilation (SSID; 10, 20, and 40 dyn/cm2) was determined in isolated mesenteric arteries (80–120 μm in diameter) of 6-wk (6W), 3-mo (3M), and 9-mo (9M)-old male db/db mice and their wild-type (WT) controls. Nitric oxide (NO)-mediated SSID was comparable in 6W WT and db/db mice, but the dilation was significantly reduced in 3M db/db mice and declined further in 9M db/db mice. Vascular superoxide production was progressively increased in 3M and 9M db/db mice, associated with an increased expression of NADPH oxidase. Inhibition of NADPH oxidase significantly improved NO-mediated SSID in arteries of 3M, but not in 9M, db/db mice. Although endothelial nitric oxide synthase (eNOS) expression was comparable in all groups, a progressive reduction in shear stress-induced eNOS phosphorylation existed in vessels of 3M and 9M db/db mice. Moreover, inducible NOS (iNOS) that was not detected in WT, nor in 6W and 3M db/db mice, was expressed in vessels of 9M db/db mice. A significantly increased expression of nitrotyrosine in total protein and immunoprecipitated eNOS was also found in vessels of 9M db/db mice. Thus, impaired NO bioavailability plays an essential role in the endothelial dysfunction of diabetic mice, which becomes aggravated when endothelial nitrosative stress is further activated via perhaps, an additional iNOS-mediated pathway during the progression of diabetes.
Keywords: diabetes, shear stress, endothelium, nitric oxide, nitrotyrosine
type 2 diabetes is characterized by increased plasma glucose in the context of insulin resistance. A key feature of diabetes/insulin resistance in the cardiovascular system is the impairment of the endothelial phosphatidyl inositol 3 kinase (PI3K)-dependent pathway, leading to a reduced endothelial nitric oxide synthase (eNOS)/nitric oxide (NO)-mediated dilation of resistance arteries (15, 37). In addition, diabetes is associated with increases in oxidative stress, such as an increased NADPH oxidase-derived superoxide formation, which inactivates NO and transforms it into peroxynitrite, a highly reactive species, to uncouple eNOS and induce nitrotyrosine formation of signaling molecules, further impairing the endothelial function in diabetes (5, 19, 22, 35, 36). Many models of diabetic animals have been developed, among which db/db mice (a point mutation in the gene for the leptin receptor) have been accepted to be a specific model of type 2 diabetes. Consistent with the phenotypic changes of type 2 diabetes in db/db mice, the underlying mechanisms for the cardiovascular dysfunction in response to the mutation of leptin receptor remain unclear. Nevertheless, studies have shown that in the context of obesity, peripheral insulin resistance contributes to the endothelial dysfunction in db/db mice (1). Depletion of protein tyrosine phosphatase 1B, an insulin-desensitizing enzyme, improves acetylcholine-induced dilation in mesenteric arteries of db/db mice via a mechanism of increasing NO bioavailability. Similarly, we have also demonstrated that flow-induced dilation in coronary arteries of db/db mice is remarkably reduced, resulting mainly from an increased NADPH oxidase-derived superoxide that diminishes NO-mediated responses (2, 3).
Despite the fact that some underlying mechanisms for endothelial dysfunction in db/db mice have been explored, studies that investigate the progression of endothelial dysfunction in the advanced stage of diabetes are still sparse. To this end, the present study aimed to test the hypothesis that endothelial dysfunction is exacerbated during the progression of diabetes. Shear stress-induced dilation (SSID) in mesenteric arteries of 6-wk (6W), 3-mo (3M), and 9-mo (9M)-old db/db and wild-type (WT) control mice was assessed. We demonstrated that physiological maturation per se is not a major contributor to the exacerbation of endothelial dysfunction; rather, the pathological progression of oxidative stress and malfunction of eNOS are the major contributors to the exacerbation of endothelial dysfunction. As observed, db/db mice develop obesity, hyperglycemia, and hyperinsulinemia after their first month and do not survive longer than 10 mo. In this context, 3M and 9M db/db mice used in the present study could be defined as early and advanced diabetic, respectively, whereas 6W db/db mice could be defined as being in a prediabetic condition.
MATERIALS AND METHODS
Animals.
6W, 3M, and 9M male db/db mice (homozygous for the mutation of Leprdb, a model of type 2 diabetes; Jackson Laboratory) and WT controls (C57BL/J) were used. Experimental protocols were approved by the Institutional Animal Care and Use Committee of New York Medical College and conformed to the current guidelines of the National Institutes of Health and the American Physiological Society for the care and use of laboratory animals.
Measurements of serum glucose and insulin.
Serum glucose and insulin concentrations were measured with glucose assay kits (Sigma) and mouse ELISA kits (ALPCO Diagnostic), respectively (3).
Shear stress-induced dilation.
As described previously (36), mesenteric arteries (80–120 μm in diameter) were isolated from db/db and WT mice. Vessels were cannulated in a vessel chamber and perfused with MOPS-buffered physiological salt solution (PSS) at 37°C, pH 7.4. Intravascular pressure was maintained constant at 80 mmHg. After 1 h of stabilization, vessels developed spontaneous tone that reached to a basal tone of ∼65% maximal diameter. Wall shear stress (τ) was generated by increasing perfusate flow via a syringe pump (Harvard Apparatus). The flow rate (Q) was determined by the equation of τ = 4Qη/πr3, in which the radius (r) of vessels was measured before the onset of flow. 0.0069 poise was used for the viscosity (η) of PSS at 37°C. Changes in diameter in response to step increases in shear stress (10, 20, and 40 dyn/cm2) were recorded in the control condition and in the presence of nitro-l-arginine methyl ester (l-NAME, 300 μM) or 3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo(4,5-d)pyrimidine (VAS2870, 5 μM; Enzo Life Sciences), respectively. VAS2870 has been used to inhibit vascular NADPH oxidase in cultured cells and in isolated vessels and has been demonstrated to have higher specificity in the inhibition of vascular NADPH oxidase than that of diphenylene iodonium or apocynin (34). Dilation to an NO donor (acidified NaNO2 10−11 -10−6 M) was also assessed in the presence of l-NAME. Inhibitors were added into the vessel chamber directly at least 30 min prior to assessments. At the conclusion of the experiments, vessels were incubated in Ca2+-free MOPS-PSS with 1 mM EGTA for 10 min. The diameter recorded at this condition was defined as the passive diameter (PD).
Western blot analysis.
Equal amounts of total proteins isolated from mesenteric arteries were separated by SDS-PAGE. Proteins separated on the denaturing gels were transferred to nitrocellulose membranes, which were then probed with antibodies to eNOS, nNOS, inducible NOS (iNOS), Nox-2 (BD Transduction Laboratories), phospho-eNOS (Ser1177) (Cell Signaling), Nox-1 (Mox1), Nox-2 (Santa Cruz Biotechnology), and β-actin (Sigma-Aldrich). Prestained color protein markers (EZ-RUN, 170–10KD; Fisher Scientific) were used for monitoring protein separation and transfer efficiency. Immunoreactive bands were detected with an appropriate second antibody and visualized with a chemiluminescence kit (Pierce). Specific bands were normalized to β-actin. In separate experiments, the protein was extracted from shear stress (20 dyn/cm2 for 10 min)-stimulated mesenteric arteries. The ratio of phosphorylated eNOS (p-eNOS) and eNOS was then determined.
In experiments of immunoprecipitation of eNOS (36), the proteins isolated from mesenteric arteries of mice were incubated overnight with eNOS antibody and protein A/G beads. The immunocomplexes were then subjected to SDS-PAGE, followed by Western blot analysis with anti-nitrotyrosine antibody (Upstate).
Detection of superoxide production.
Superoxide production in isolated mesenteric arteries was measured from quantifying the chemiluminescence of 5 μM lucigenin in a liquid scintillation counter (LS6000IC; Beckman Instruments). Vessels of 3M and 9M WT and db/db mice were incubated in Krebs-HEPES buffer (pH 7.4) at 37°C in the control and in the presence of apocynin (10 μM) or VAS2870 (5 μM), inhibitors of NADPH oxidase, or l-NAME (300 μM) for 60 min. Vessels of 3M db/db mice were also incubated with ANG II (100 nM; a stimulus of NADPH oxidase) before and after administration of VAS2870. After incubation, vessels were transferred to scintillation vials containing 1 ml of Krebs-HEPES/lucigenin (5 μM) solution and counted in the scintillation counter over the next 10 min. Average chemiluminescence of each sample was subtracted with background signals determined from the vials without vessels. After that, every vessel was completely digested with 50 μl of 1 N NaOH, and the amount of total protein was determined. The final superoxide production was expressed as counts per minute per microgram of protein.
Statistical analysis.
Data are expressed as means ± SE. Changes in the diameter of vessels in response to increases in shear stress, as well as to NaNO2, were normalized to their PD and were expressed as % of PD. All raw data were included and grouped for statistical analysis. Statistical significance was calculated by Student's t-test and by repeated-measures, two-way ANOVA, followed by the Tukey-Kramer multiple-comparison test. Significance level was taken at P < 0.05.
RESULTS
Hyperglycemia and hyperinsulinemia in db/db mice.
Table 1 shows serum glucose and insulin concentrations in db/db and WT mice. In db/db mice, serum glucose increased significantly in 3M and further increased in 9M of age. Serum insulin level was three-fold higher in 3M db/db than in age-matched WT mice. It declined in 9M db/db mice, but it was still significantly higher than that of 9M WT controls. Serum levels of glucose and insulin were slightly increased in 6W db/db compared with age-matched WT mice, but the increments did not reach statistical significance.
Table 1.
Serum glucose and insulin in WT and db/db mice
| 6 wk |
3 mo |
9 mo |
||||
|---|---|---|---|---|---|---|
| WT | db/db | WT | db/db | WT | db/db | |
| Glucose, mM | 5.5 ± 0.6 | 6.6 ± 0.9 | 5.4 ± 0.4 | 15.6 ± 0.3* | 7.9 ± 0.3 | 17.6 ± 0.5*# |
| Insulin, ng/ml | 5.1 ± 1.8 | 6.7 ± 1.5 | 5.6 ± 1.6 | 16.5 ± 2.6* | 3.5 ± 1.2 | 7.7 ± 3.1*# |
(n = 4 for each group).
Significant difference from age-matched WT mice.
Significant difference from 3-mo-old db/db mice.
Impaired endothelial function during progression of diabetes.
To determine endothelial dysfunction at different stages of the disease, changes in diameter of mesenteric arteries in response to step increases in shear stress were assessed in WT and db/db mice. As shown in Fig. 1, SSID was comparable in vessels of 6W WT and db/db mice, suggesting a yet unchanged endothelial function in prediabetic condition. Inhibition of NO synthesis initiated an equal reduction of SSID in both groups of vessels. In contrast, SSID was steadily attenuated in arteries of 3M and 9M db/db compared with their age-matched WT mice. This attenuation was greater in 9M than in 3M db/db mice. In the presence of l-NAME, however, the difference in SSID, between WT and db/db mice, disappeared, indicating that the reduced SSID in vessels of diabetic mice was mainly due to an impaired NO-mediated dilation. The endothelium-independent dilation to acidified NaNO2 (Fig. 2) was comparable in all six groups of mice. Of note, there was no significant difference in SSID between vessels of 3M and 9M WT mice, implying that the process of natural maturity per se is not a major contributor to the deterioration of endothelial function during the development of diabetes.
Fig. 1.
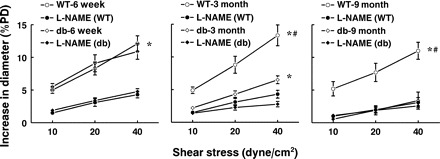
Shear stress-induced dilation in mesenteric arteries of 6-wk, 3-mo, and 9-mo-old db/db and wild-type (WT) mice in control conditions and after inhibition of nitric oxide (NO) synthesis with nitro-l-arginine methyl ester (l-NAME) (n = 12 for each group). *Significant difference between the curves of control and l-NAME, P < 0.05. #Significant difference between the curves of WT and db/db, P < 0.05.
Fig. 2.
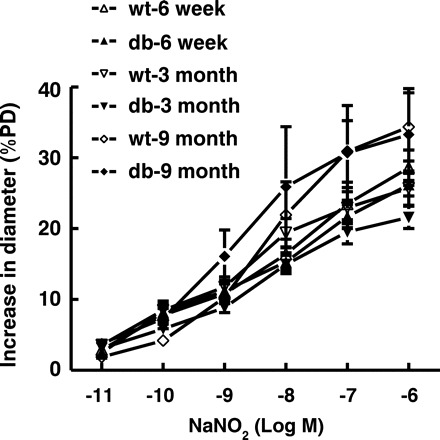
NaNO2 (NO donor)-induced dilation in mesenteric arteries of 6-wk, 3-mo, and 9-mo-old db/db and WT mice (n = 12 for each group).
Elevated superoxide production in db/db mice.
Expression of NADPH oxidase, one of the primary sources of superoxide production in the vasculature, was assessed by Western blot analysis. As shown in Fig. 3, A and B, the expression of Nox-2 (gp91) was increased significantly in 3M db/db vessels and increased additionally in that of 9M db/db mice. The expression of Nox-1 was unchanged. Congruent with the upregulation of Nox-2, superoxide production in arteries of 3M and 9M db/db mice was significantly increased (Fig. 3C). The increment was greater in 9M than that of 3M db/db mice. Inhibition of NADPH oxidase with apocynin or VAS2870 decreased vascular superoxide production in db/db mice to a level similar to that in WT mice. Inhibition of nitric oxide synthase with l-NAME did not affect superoxide production in db/db mice, indicating that eNOS uncoupling is not the major source responsible for the increased superoxide production in mesenteric arteries of db/db mice. We also found that VAS2870 inhibited ANG II-induced superoxide production in 3M db/db mice (Fig. 3D), confirming further the effectiveness of VAS2870 on the inhibition of vascular NADPH oxidase. The role of NADPH oxidase-derived superoxide in the interruption of NO-mediated dilation was further revealed, as shown in Fig. 4 that blockage of NADPH oxidase with VAS2870 restored significantly the attenuated SSID in arteries of 3M db/db mice. This restoration was sensitive to l-NAME, suggesting an NO-mediated response. However, inhibition of NADPH oxidase did not improve SSID in vessels of 9M db/db mice.
Fig. 3.
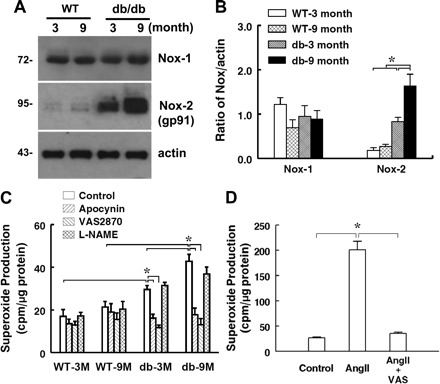
Protein expression of Nox-1, Nox-2, and β-actin (A and B; n = 3 blots) and superoxide production in the control and in the presence of apocynin, VAS2870, and l-NAME (C; n = 6–9 for each group) in mesenteric arteries of 3- and 9-mo-old WT and db/db mice. D: effects of VAS2870 (VAS) on ANG II-induced superoxide production in mesenteric arteries of 3-mo-old db/db mice (n = 4). *Significant difference between groups, P < 0.05.
Fig. 4.
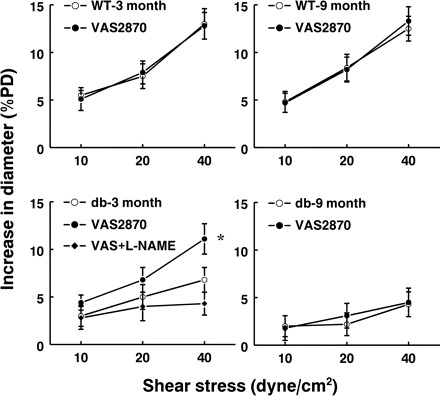
Shear stress-induced dilation in mesenteric arteries of 3- and 9-mo-old db/db and WT mice in control and after inhibition of NADPH oxidase with VAS2870 (n = 8 for each group). In the group of 3-mo-old db/db mice, additional l-NAME was administered in the presence of VAS2870. *Significant difference from control, P < 0.05.
Continuously altered NOS signaling during the process of diabetes.
To determine whether, in addition to increased superoxide production, an altered expression of eNOS also plays a role in the impaired SSID of db/db mice, Western blot analysis was used to detect vascular expressions of eNOS and shear stress-stimulated p-eNOS. Fig. 5 shows that eNOS expression was comparable in vessels of all six groups (Fig. 5A), but shear stress-induced p-eNOS, an index of eNOS activation, was significantly decreased in 3M and 9M db/db mice compared with their age-matched WT controls (Fig. 5, B and C). The reduction in p-eNOS was more severe in 9M compared with 3M db/db mice, indicating a progressive impairment of eNOS activation as the progression of diabetes. Natural aging does not significantly affect shear stress-induced eNOS activation, as indicated by the comparable expression of p-eNOS in both ages of WT mice. In addition to the unchanged eNOS expression, nNOS expression was not detected (data not shown) in any of the vessels. However, a predominant expression of iNOS was only observed in vessels of 9M db/db mice (Fig. 5A). Of six 9M db/db mice, five showed positive iNOS expression. The upregulated iNOS in age-advanced diabetic vessels (Fig. 5A) coexisted with a significant reduction in p-eNOS expression (Fig. 5B) and augmentation of nitrotyrosine level for immunoprecipitated eNOS (Fig. 5D) and total protein as well (Fig. 5E). These results indicate that increased protein nitration, specifically eNOS nitration, which most likely is a result from the increased expression of iNOS and superoxide production, exacerbates endothelial function during the progression of diabetes.
Fig. 5.
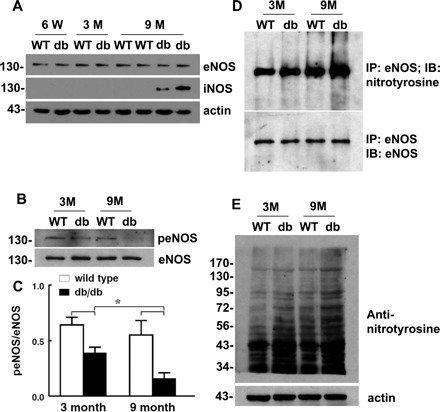
Western blots for mesenteric arteries of 6-wk, 3-mo-, and 9-mo-old db/db and WT mice. A: expressions of endothelial nitric oxide synthase (eNOS), inducible nitric oxide synthase (iNOS) and β-actin. B and C: shear stress (20 dyn/cm2, 10 min)-induced eNOS phosphorylation (peNOS). D: immunoprecipitation (IP) of eNOS and immunoblotting (IB) with anti-nitrotyrosine and eNOS antibodies. E: anti-nitrotyrosine in total proteins of mesenteric arteries is shown with 3 or 4 blots for each group. The darkness of protein bands in E reflects the level of nitrotyrosine formation. Actin was used for loading controls. *Significant difference between groups, P < 0.05.
DISCUSSION
The present study revealed a diabetic progression-dependent deterioration of endothelial function, characterized by a normal (prediabetes; 6W) to an impaired (early stage of diabetes; 3M), and finally a deficient (advanced stage; 9M) NO-mediated SSID during the process of the disease. The underlying mechanisms involve altered eNOS, NADPH oxidase, and iNOS signaling, which in concert, potentiate oxidative stress and exacerbate endothelial dysfunction.
Of note, compared with 3M WT mice, age-matched db/db mice have a three-fold increase in serum glucose that was associated with a parallel increase in serum insulin (hyperinsulinemia), a characteristic of insulin resistance. Further elevated glucose concentrations in 9M db/db mice were associated with greatly reduced insulin levels, a phenomenon that is also observed in advanced diabetic patients. This reduction in circulating insulin observed in 9M db/db mice does not necessarily mean any improvement in metabolic actions of insulin, but rather is indicative of a decompensation of pancreatic β-cells and an irretrievable loss of the metabolic function of insulin. This is evidenced by a dramatically high circulating glucose (Table 1), exacerbated endothelial dysfunction (Figs. 1, 3, and 4), and severely impaired eNOS activity, followed by activation of iNOS signaling (Fig. 5). These phenomena could also explain the observation that db/db mice became feeble at ∼10 mo of age, followed by a soaring rate of mortality.
Impaired NO mediation of shear stress-induced dilation in diabetes.
Shear stress initiated by increases in blood flow is one of the most important regulators of the cardiovascular system. Vascular endothelium responds rapidly and sensitively to shear stress, leading to vasodilation that is primarily mediated by NO (17). In this context, SSID is used as a reproducible and accessible parameter to probe endothelial function. Reduced SSID observed in many cardiovascular diseases, such as hypertension, heart failure, and aging as well, is an important index of endothelial dysfunction (16, 31, 32, 36). In the present study, we tested endothelial function of arteries at different stages of diabetes, and demonstrated that SSID was close to normal in 6W db/db mice, suggesting maintained vascular endothelial function before a prolonged exposure of the vessels to the hyperglycemia and hyperinsulinemia. SSID became significantly attenuated in the early (3M) stage, and further reduced in the advanced (9M) stage of diabetes (Fig. 1). This reduced endothelium-dependent dilation in resistant arteries may well contribute to the increased blood pressure in db/db mice (2, 30). Specific role of NO in the mediation of SSID was confirmed by inhibition of the responses with l-NAME in WT, as well as in prediabetic mice. The inhibitory effect of l-NAME was significantly reduced in 3M db/db vessels, and became deficient in those of 9M db/db mice, indicating a steady deterioration of endothelial function during the diabetic process. The comparable endothelium-independent dilator responses in all groups (Fig. 2) reflected normal smooth muscle responsiveness.
NO and superoxide interaction in shear stress-induced dilation.
Emerging evidence indicates the role of superoxide in the pathogenesis of cardiovascular disorders by interacting with endothelial nitric oxide (35). As one of the primary enzymes responsible for the generation of superoxide in the vasculature, NAD(P)H oxidase was upregulated in vessels of db/db mice (Fig. 3, A and B). Because endothelium-independent dilator responses in vessels of both db/db and WT mice were comparable, we hypothesized that an impaired NO bioavailability is responsible for the attenuated SSID in db/db mice. Indeed, we demonstrated that NADPH oxidase-dependent production of superoxide in arteries of db/db mice continuously increased along with the progression of the disease (Fig. 3C). This elevated oxidative stress impairs NO bioavailability, leading to the attenuated SSID. Mechanisms responsible for the upregulated Nox-2 in vessels of db/db mice remain unclear. Obesity and insulin resistance-associated inflammatory factors, including leptin, have been suggested to induce oxidative stress (9, 29). Increased gene expression of Nox-1 has also been found in mesenteric arteries of db/db mice, as indicated by the fact that correcting insulin resistance reduces Nox-1 expression and improves acetylcholine-induced dilation (1). There is growing debate as to the specificity of apocynin as a specific inhibitor NADPH oxidase (14). For this reason, VAS2870, a specific NADPH oxidase inhibitor, was used. The specificity of VAS2870 to inhibit NADPH oxidase in mesenteric arteries of db/db mice was confirmed by its inhibitory effect on ANG II-induced superoxide production (Fig. 3D). But, the specificity of VAS2870 to the isoform(s) of NADPH oxidase needs to be further determined. To further characterize the role of increased Nox-2-derived superoxide production in the endothelial dysfunction of db/db mice, SSID was evaluated before and after administration of VAS2870 (Fig. 4). The results indicated that VAS2870 that did not affect vascular responses in control mice (Fig. 4, A and B), restored significantly the attenuated dilator responses in vessels of 3M db/db mice and also the restored portion of the responses was prevented by l-NAME (Fig. 4C), indicating that NADPH oxidase-derived superoxide impairs SSID through scavenging of NO. In agreement with our findings, other studies reported that suppression of oxidative stress improves glucose metabolism, insulin resistance, and vascular function in type II diabetes (18). Other vasodilator mechanisms seem less affected by the increased oxidative stress in diabetes. In db/db mice, endothelium-derived hyperpolarizing factor (EDHF)-induced dilation in mesenteric arteries is preserved (25). Promoting EDHF-mediated response may compensate for the impaired NO-dependent vasodilator responses in vessels of db/db mice (26, 38).
Role of nitrotyrosine in the deterioration of endothelial function.
Unlike the restoration of NO-mediated SSID with inhibition of NADPH oxidase in arteries of 3M db/db mice, VAS2870 failed to restore the responses in vessels of 9M db/db mice (Fig. 4D). Thus, some additional mechanism(s) may contribute to the failure of scavenging superoxide in the prevention of endothelial dysfunction in advanced diabetes. Because p-eNOS is essential for shear stress-stimulated activation of eNOS (4) and the insulin-related activation of eNOS employs a phosphorylation-dependent mechanism, which involves insulin receptor tyrosine kinase, PI3 kinase, Akt, and eNOS (21), we hypothesized the presence of impaired eNOS activity in diabetic mice. As expected, p-eNOS was attenuated in the early stage of diabetes, but then basically disappeared in the advanced stage (Fig. 5B), implying a deficiency of functional eNOS. Additionally, these arteries expressed iNOS that was undetectable in vessels of other groups (Fig. 5A). Our results were supported by other studies indicating that pathogenesis of diabetes involves expression of iNOS (6, 8) that serves as a source of NO to trigger the production of free radicals/peroxynitrite (13, 23). In type 1 diabetes, impairment of endothelium-dependent relaxation was also demonstrated to be related to an increased iNOS expression (12). Moreover, Western blot analysis showed significantly greater nitrotyrosine levels for immunoprecipitated eNOS and total vascular protein in 9M db/db vessels (Fig. 5, D and E). This suggests that additional increases in nitrosative stress may further worsen eNOS function, leading to an abolished NO-mediated SSID. Although nitrotyrosine was initially believed to be a specific marker of peroxynitrite generation, other pathways can also induce tyrosine nitration. Thus, nitrotyrosine is now generally considered to be a collective index of reactive nitrogen species, rather than a specific indicator of peroxynitrite formation (24). Oxidative/nitrosative damage plays critical roles in diabetic cardiovascular dysfunction, as evidenced by the fact that its pathophysiological alterations can be prevented by antioxidants, NOS inhibitors, as well as by peroxynitrite neutralization agents (10). Consistently, we, as well as others provided evidence for the role of nitrotyrosine in the endothelial dysfunction via uncoupling of eNOS to increase superoxide production (33, 36). On the basis of the proposed feed-forward mechanism (20), initial activation of Nox-2 in diabetic arteries promotes the activation of other oxidases to produce more reactive oxygen species. This mechanism, therefore, may also function in the diabetic process presented in our study. In addition, superoxide activates redox-sensitive transcription factors to initiate induction of genes that may participate in the modulation of endothelial function. In this context, we did find an association between the upregulation of Nox-2 and elevated expressions of phospho-p38 and phospho-JNK MAP kinase (data not shown); the causal relationship between them and the significance of which (so called “oxidative-stress kinase cascade” that appears to be responsible for a variety of complications observed in advanced stages of diabetes) are currently being studied in our laboratory.
Changes in the basal tone of vessels, as a function of iNOS-derived NO, were not observed in the present study in which basal tone was statistically identical in all groups. This phenomenon could be due to the coexistence of other altered constrictor mechanisms, such as changes in the prostanoid synthesis via upregulation of COX-2 derived constrictor agents (7, 39) or increases in endothelin production (7, 11, 27, 28), all of which could affect vascular tone in diabetes.
Although 9M mice cannot be defined as aged animals, the concern about whether a natural maturity compromises vascular function may challenge our conclusions. To this end, our results indicate that all data obtained on SSID (Fig. 1), superoxide production and NOXs expression (Fig. 3), changes in p-eNOS and iNOS expression (Fig. 5), as well as circulating glucose and insulin (Table 1) were comparable in vessels of 3M and 9M WT mice. This suggests that during this period, natural aging is unlikely to be responsible for the endothelial dysfunction, which is rather the consequence of the process of diabetes per se.
Perspective and Significance
We characterized complexities of endothelial dysfunction related to the changes in oxidative stress, NO bioavailability, and NOS signaling in different stages of diabetes. The endothelial dysfunction was reversible in the early stage of the disease when NADPH oxidase was inhibited. During the progression of the disease, specific upregulation of iNOS occurs, followed by increases in tyrosine nitration of eNOS, which, in turn, damages further eNOS activity, leading to an irreversible impairment of SSID. Results of our studies suggest that antioxidant therapy could be effective in the treatment of diabetes-induced cardiovascular dysfunction, especially in an early stage of diabetes; however, sophisticated therapeutic plans aimed to recruit eNOS function may be important in the treatment of more advanced diabetes. In this context, our next study will focus on specific mechanisms involving the contribution of iNOS to the deterioration of cardiovascular function, by chronic treatment of the late stage of db/db mice with iNOS inhibitors and/or peroxynitrite decomposition catalysts. We will then assess whether inhibition of iNOS, in addition to antioxidants is able to alleviate the endothelial dysfunction. These studies will highlight the functional significance of iNOS and protein nitration in diabetic progression and moreover, may provide new ideas for the development of effective therapeutic strategies for patients.
GRANTS
This study was supported by the American Heart Association Grant 0735540T and National Institutes of Health Grant HL-070653 and HL-43023.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
REFERENCES
- 1. Ali MI, Ketsawatsomkron P, Belin de Chantemele EJ, Mintz JD, Muta K, Salet C, Black SM, Tremblay ML, Fulton DJ, Marrero MB, Stepp DW. Deletion of protein tyrosine phosphatase 1b improves peripheral insulin resistance and vascular function in obese, leptin-resistant mice via reduced oxidant tone. Circ Res 105: 1013–1022, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bagi Z, Erdei N, Toth A, Li W, Hintze TH, Koller A, Kaley G. Type 2 diabetic mice have increased arteriolar tone and blood pressure: enhanced release of COX-2-derived constrictor prostaglandins. Arterioscler Thromb Vasc Biol 25: 1610–1616, 2005 [DOI] [PubMed] [Google Scholar]
- 3. Bagi Z, Koller A, Kaley G. Superoxide-NO interaction decreases flow- and agonist-induced dilations of coronary arterioles in Type 2 diabetes mellitus. Am J Physiol Heart Circ Physiol 285: H1404–H1410, 2003 [DOI] [PubMed] [Google Scholar]
- 4. Busse R, Fleming I. Regulation of NO synthesis in endothelial cells. Kidney Blood Press Res 21: 264–266, 1998 [DOI] [PubMed] [Google Scholar]
- 5. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87: 840–844, 2000 [DOI] [PubMed] [Google Scholar]
- 6. Calles-Escandon J, Cipolla M. Diabetes and endothelial dysfunction: a clinical perspective. Endocr Rev 22: 36–52, 2001 [DOI] [PubMed] [Google Scholar]
- 7. Cosentino F, Eto M, De Paolis P, van der LB, Bachschmid M, Ullrich V, Kouroedov A, Delli GC, Joch H, Volpe M, Luscher TF. High glucose causes upregulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells: role of protein kinase C and reactive oxygen species. Circulation 107: 1017–1023, 2003 [DOI] [PubMed] [Google Scholar]
- 8. Cosentino F, Hishikawa K, Katusic ZS, Luscher TF. High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation 96: 25–28, 1997 [DOI] [PubMed] [Google Scholar]
- 9. Dong F, Zhang X, Ren J. Leptin regulates cardiomyocyte contractile function through endothelin-1 receptor-NADPH oxidase pathway. Hypertension 47: 222–229, 2006 [DOI] [PubMed] [Google Scholar]
- 10. Esberg LB, Ren J. Role of nitric oxide, tetrahydrobiopterin and peroxynitrite in glucose toxicity-associated contractile dysfunction in ventricular myocytes. Diabetologia 46: 1419–1427, 2003 [DOI] [PubMed] [Google Scholar]
- 11. Gogg S, Smith U, Jansson PA. Increased MAPK activation and impaired insulin signaling in subcutaneous microvascular endothelial cells in type 2 diabetes: the role of endothelin-1. Diabetes 58: 2238–2245, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gunnett CA, Heistad DD, Faraci FM. Gene-targeted mice reveal a critical role for inducible nitric oxide synthase in vascular dysfunction during diabetes. Stroke 34: 2970–2974, 2003 [DOI] [PubMed] [Google Scholar]
- 13. Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 105: 1656–1662, 2002 [DOI] [PubMed] [Google Scholar]
- 14. Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51: 211–217, 2008 [DOI] [PubMed] [Google Scholar]
- 15. Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation 113: 1888–1904, 2006 [DOI] [PubMed] [Google Scholar]
- 16. Koller A, Huang A. Shear stress-induced dilation is attenuated in skeletal muscle arterioles of hypertensive rats. Hypertension 25: 758–763, 1995 [DOI] [PubMed] [Google Scholar]
- 17. Koller A, Kaley G. Shear stress-induced dilation of arterioles. Am J Physiol Heart Circ Physiol 274: H382–H383, 1998 [DOI] [PubMed] [Google Scholar]
- 18. Lamb RE, Goldstein BJ. Modulating an oxidative-inflammatory cascade: potential new treatment strategy for improving glucose metabolism, insulin resistance, and vascular function. Int J Clin Pract 62: 1087–1095, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Molnar J, Yu S, Mzhavia N, Pau C, Chereshnev I, Dansky HM. Diabetes induces endothelial dysfunction but does not increase neointimal formation in high-fat diet fed C57BL/6J mice. Circ Res 96: 1178–1184, 2005 [DOI] [PubMed] [Google Scholar]
- 20. Mueller CF, Laude K, McNally JS, Harrison DG. ATVB in focus: redox mechanisms in blood vessels. Arterioscler Thromb Vasc Biol 25: 274–278, 2005 [DOI] [PubMed] [Google Scholar]
- 21. Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev 28: 463–491, 2007 [DOI] [PubMed] [Google Scholar]
- 22. Nie H, Wu JL, Zhang M, Xu J, Zou MH. Endothelial nitric oxide synthase-dependent tyrosine nitration of prostacyclin synthase in diabetes in vivo. Diabetes 55: 3133–3141, 2006 [DOI] [PubMed] [Google Scholar]
- 23. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404: 787–790, 2000 [DOI] [PubMed] [Google Scholar]
- 24. Pacher P, Obrosova IG, Mabley JG, Szabo C. Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr Med Chem 12: 267–275, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pannirselvam M, Ding H, Anderson TJ, Triggle CR. Pharmacological characteristics of endothelium-derived hyperpolarizing factor-mediated relaxation of small mesenteric arteries from db/db mice. Eur J Pharmacol 551: 98–107, 2006 [DOI] [PubMed] [Google Scholar]
- 26. Park Y, Capobianco S, Gao X, Falck JR, Dellsperger KC, Zhang C. Role of EDHF in type 2 diabetes-induced endothelial dysfunction. Am J Physiol Heart Circ Physiol 295: H1982–H1988, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Potenza MA, Marasciulo FL, Chieppa DM, Brigiani GS, Formoso G, Quon MJ, Montagnani M. Insulin resistance in spontaneously hypertensive rats is associated with endothelial dysfunction characterized by imbalance between NO and ET-1 production. Am J Physiol Heart Circ Physiol 289: H813–H822, 2005 [DOI] [PubMed] [Google Scholar]
- 28. Potenza MA, Marasciulo FL, Tarquinio M, Quon MJ, Montagnani M. Treatment of spontaneously hypertensive rats with rosiglitazone and/or enalapril restores balance between vasodilator and vasoconstrictor actions of insulin with simultaneous improvement in hypertension and insulin resistance. Diabetes 55: 3594–3603, 2006 [DOI] [PubMed] [Google Scholar]
- 29. Singer G, Granger DN. Inflammatory responses underlying the microvascular dysfunction associated with obesity and insulin resistance. Microcirculation 14: 375–387, 2007 [DOI] [PubMed] [Google Scholar]
- 30. Su W, Guo Z, Randall DC, Cassis L, Brown DR, Gong MC. Hypertension and disrupted blood pressure circadian rhythm in type 2 diabetic db/db mice. Am J Physiol Heart Circ Physiol 295: H1634–H1641, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sun D, Huang A, Yan EH, Wu Z, Yan C, Kaminski PM, Oury TD, Wolin MS, Kaley G. Reduced release of nitric oxide to shear stress in mesenteric arteries of aged rats. Am J Physiol Heart Circ Physiol 286: H2249–H2256, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun D, Huang A, Zhao G, Bernstein R, Forfia P, Xu X, Koller A, Kaley G, Hintze TH. Reduced NO-dependent arteriolar dilation during the development of cardiomyopathy. Am J Physiol Heart Circ Physiol 278: H461–H468, 2000 [DOI] [PubMed] [Google Scholar]
- 33. Teng RJ, Wu TJ, Bisig CG, Eis A, Pritchard KA, Konduri GG. Nitrotyrosine impairs angiogenesis and uncouples eNOS activity of pulmonary artery endothelial cells isolated from developing sheep lungs. Pediatr Res 69: 112–117, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wind S, Beuerlein K, Armitage ME, Taye A, Kumar AH, Janowitz D, Neff C, Shah AM, Wingler K, Schmidt HH. Oxidative stress and endothelial dysfunction in aortas of aged spontaneously hypertensive rats by NOX1/2 is reversed by NADPH oxidase inhibition. Hypertension 56: 490–497, 2010 [DOI] [PubMed] [Google Scholar]
- 35. Wolin MS. Reactive oxygen species and the control of vascular function. Am J Physiol Heart Circ Physiol 296: H539–H549, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol 297: H1829–H1836, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zeng G, Nystrom FH, Ravichandran LV, Cong LN, Kirby M, Mostowski H, Quon MJ. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation 101: 1539–1545, 2000 [DOI] [PubMed] [Google Scholar]
- 38. Zhang LN, Vincelette J, Chen D, Gless RD, Anandan SK, Rubanyi GM, Webb HK, MacIntyre DE, Wang YX. Inhibition of soluble epoxide hydrolase attenuates endothelial dysfunction in animal models of diabetes, obesity and hypertension. Eur J Pharmacol 654: 68–74, 2011 [DOI] [PubMed] [Google Scholar]
- 39. Zou MH, Shi C, Cohen RA. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H(2) receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes 51: 198–203, 2002 [DOI] [PubMed] [Google Scholar]