

Abstract
TGF-β utilizes receptor-activated SMAD signaling to mediate growth suppression; however, non-SMAD signaling that modulates the TGF-β response in epithelial cells become apparent when the SMAD signaling is abrogated, a common occurrence in pancreatic cancers. Here, we examined whether TGF-β utilized NF-κB to downregulate PTEN, a gene that is rarely mutated in pancreatic cancers. SMAD4-null BxPc3 and CAPAN-1 pancreatic cancer cells were treated with TGF-β (10 ng/ml) and lysed, and cellular proteins were analyzed by Western blots using p-IκB, p65, and PTEN antibodies. PTEN promoter and NF-κB activities were assessed by PTEN-luc and p-NF-luc constructs, respectively. Dominant negative p-IκB-α-M (NF-κB superrepressor) was used to block activation of NF-κB. Cell motility was assessed by Boyden chamber migration assay. TGF-β induced IκB-α phosphorylation followed by NF-κB p65 subunit nuclear translocation and increased NF-κB activity. IκB-α-M blocked TGF-β-induced NF-κB activity, reversed downregulated PTEN promoter activity and PTEN expression, and prevented augmentation of cell motility induced by TGF-β. SMAD4 restoration, but not knockdown of SMAD2 and/or 3, reversed TGF-β-induced NF-κB activity. Thus TGF-β suppresses PTEN in pancreatic cancer cells through NF-κB activation and enhances cell motility and invasiveness in a SMAD4-independent manner that can be counteracted when TGF-β-SMAD signaling is restored. The TGF-β/NF-κB/PTEN cascade may be a critical pathway for pancreatic cancer cells to proliferate and metastasize.
Keywords: pancreatic cancer, TGF-β, NF-κB, PTEN, cell motility
the transforming growth factor-β (TGF-β) family of peptides regulates several biological processes including cell proliferation, differentiation, and apoptosis (25). In normal epithelial cells, including pancreatic cells and intestinal epithelium, TGF-β exerts growth-inhibitory effects, promotes terminal differentiation, and serves as a tumor suppressor. Neoplastic transformation results in loss of this normal growth-inhibitory response (12) in many cancer cells types, including pancreatic (2, 13), breast (1), and colorectal carcinoma cells (16). SMAD4 is a key intracellular mediator for TGF-β signaling. SMAD4 deficiency by biallelic deletion is one of the most common genetic lesions found in pancreatic cancer, affecting more than 50% of patients (17, 27).
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is localized to chromosome 10q23 (21, 31). It is a dual-specificity phosphatase and antagonizes phosphoinositide-3-kinase (PI3K)/ATP-dependent tyrosine kinases (Akt) signaling pathway (24) and thus plays a functional role in cell cycle arrest and apoptosis (6, 30). In fact, our previous findings indicate that TGF-β-induced PTEN downregulation is accompanied by Akt activation (4). Inactivation of PTEN by mutation is observed in many types of cancer, but its mutation is rarely found in pancreatic cancer (26, 28). PTEN expression has been shown to be regulated by TGF-β1 (4, 20, 21). PTEN mRNA levels are reduced in a model of TGF-β1-overexpressing transgenic mice that developed pancreatic fibrosis (8). Reduction of PTEN mRNA levels in the pancreatic cancer cell line PANC-1 following incubation with TGF-β1 has also been reported (8). In pancreatic cancer, TGF-β1 is commonly overexpressed, which may be secondary to loss of negative feedback from a disrupted TGF-β-SMAD signaling cascade or other mechanism (10). Although PTEN is not found mutated in pancreatic cancers, its reduction in expression gives pancreatic cells an additional growth advantage and may be an instrumental pathway for TGF-β-induced cell proliferation (4). However, the signaling pathway involved in TGF-β-induced PTEN suppression has not been fully characterized. We have previously demonstrated the SMAD-independent activation of PKC-α by TGF-β to deregulate PTEN, but other downstream effectors directly mediate TGF-β-induced PKC-α activation. Interestingly, PTEN expression is reported to be transcriptionally suppressed by tumor necrosis factor-α (TNF-α) through activation of NF-κB (19, 33).
NF-κB consists of the transactivation subunit RelA/p65 and the DNA-binding subunits p50 (NFκB1) and p52 (NFκB2), which are processed from the precursors p105 and p100, respectively (11). NF-κB is sequestered in the cytoplasm by the inhibitor IκB-α to prevent transcriptional activation in unstimulated conditions. Addition of inflammatory cytokines or peptide growth factors causes phosphorylation of I-κB-α, which in turn undergoes proteasome-dependent degradation while simultaneously releasing NF-κB, which subsequently translocates into the nucleus to transcriptionally affect the expression of target genes with key roles in the prevention of apoptosis, promotion of tumor growth, and activation of inflammatory responses (7).
Here, we hypothesized that TGF-β downregulates PTEN expression via the activation of NF-κB to increase cell motility, a marker for metastatic behavior. We demonstrate that TGF-β promotes cell survival and mobility by suppression of PTEN expression through activation of NF-κB.
MATERIALS AND METHODS
Materials and reagents.
Human recombinant TGF-β1 (10 ng/ml; Peprotech, Rocky Hill, NJ) was dissolved in PBS that contains BSA. The solution was used after dilution in cell culture medium for each assay. All other reagents were purchased from Sigma. Cell culture media and supplements came from Invitrogen (Carlsbad, CA) unless otherwise indicated.
Dominant negative and wild-type constructs.
Many signal transduction pathways that result in NF-κB activation culminate in serine phosphorylation of I-κB on residues 32 and 36. Phosphorylation of the COOH-terminal PEST sequence has been implicated in constitutive turnover of I-κB. The dominant-negative mutant of the NF-κB pathway, I-κB-α-M, was deposited to Addgene (Cambridge, MA) and was created by digestion of plasmids pCMX-IκBS32/36A and pCMX-IκBmutF with EcoNI and BstEII and ligation of the small S32/36A fragment to the large vector/IκBmutF fragment (32). Wild-type SMAD4 construct was kindly provided by Dr. Masayuki Funaba (Azabu University, Sagamihara, Kanagawa, Japan) and was used to restore SMAD4 expression in SMAD4-null pancreatic cancer cells. SMAD4 was cloned into pcDNA3 vector that was ligated with XbaI and EcoRI sites. Dominant negative (DN)-SMAD2 and DN-SMAD3 were provided by Dr. Rik Derynck (University of California, San Francisco, CA) (5). PTEN-luc was a gift from Dr. Eileen Adamson (Burnham Institute, La Jolla, CA) (34). The 5′ PTEN regulatory sequences were amplified via PCR from genomic DNA with use of a pair of appropriate primers (5′-KpnI-GCCGGGTTTCACGCGGC-3′ and 5′-HindIII-GTCTGGGAGCCTGTGG-3′) located respectively at the position −1 and −1,978 from the ATG. The amplified product was purified and cloned into the KpnI/HindIII-digested pGL3 basic reporter gene to make the PTEN-Luc construct. pNFκB-Luc (Clontech, Mountain View, CA) is designed to measure the reporter gene activity, providing a direct measurement of activation for the NF-κB pathway.
Cell cultures.
BxPC3 and CAPAN-1 cells were obtained from ATCC and were maintained respectively in RPMI and Iscove's DMEM media (GIBCO-BRL, Gaithersburg, MA) supplemented with 10% fetal calf serum (GIBCO-BRL) in an incubator at 37°C and 5% CO2. They are both derived from human ductal pancreatic adenocarcinoma (9, 23), and both of them are SMAD4 null(29). To analyze the effect of TGF-β1 on PTEN expression, pancreatic cancer cell lines were grown to 70–80% confluence in medium containing 10% FBS. Afterward, cells were washed twice in PBS, incubated for 30 min in serum-free medium, and treated for 24 and 48 h with 10 ng/ml TGF-β1 or medium alone without serum throughout the experiment.
RNA interference-induced PTEN gene silencing.
BxPc3 cells were transfected with PTEN siRNA or control nonspecific small interfering RNA (siRNA) by use of SignalSilence PTEN siRNA (Cell Signal Technology). Cells were seeded at a density of 2 × 105 cells per 35-mm dish in medium with 10% FBS and without antibiotics and allowed to adhere overnight before transfection. PTEN siRNA (500 nM) or scramble siRNA was added to 500 μl of Opti-MEM I reduced serum medium, whereas 10 μl of Lipofectamine 2000 (Invitrogen) was added to 500 μl Opti-MEM I reduced serum medium. Each was mixed gently and incubated for 5 min, after which diluted siRNA and diluted Lipofectamine 2000 were then combined, gently mixed, and allowed to incubate for 20 min at room temperature. The siRNA plus Lipofectamine 2000 complex were added directly to the culturing cells. After 4-h incubation, complete medium with 10% FBS was added and cells were cultured for another 24 h.
Subcellular fractionation.
Cells were lysed and separated into various compartments to determine p65 subunit nuclear translocation when NF-κB is activated. This was carried out by using a Cell Compartment Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. After treatment, cells were lysed in extraction buffer CE1 on ice for 10 min, which disrupts the plasma membrane without solubilizing the cells. The lysates were then centrifuged at 1,000 g for 10 min at 4°C. The supernatant were removed, and this fraction contained cytosolic proteins. The pellet was resuspended in extraction buffer CE2 that solubilized the plasma membrane as well as all organelle membranes but not the nuclear membrane when pipetting up and down with a 1-ml pipette tip. The lysates were then incubated at 4°C for 30 min on a shaker. Lysates were then centrifuged at 6,000 g for 10 min at 4°C. The supernatants, which contained the membrane proteins, were discarded. The pellets consisted of nuclei were solubilized by use of extraction buffer CE3 in which all soluble and most membrane-bound nuclear proteins are extracted. The lysates were incubated for 10 min at 4°C on a shaker. Thereafter, the lysates were centrifuged at 6,800 g for 10 min at 4°C with the supernatants containing the nuclear proteins.
Western blotting.
Cells were washed three times with ice-cold phosphate-buffered saline or Tris-buffered solution, then lysed by use of total lysis buffer (150 mM NaCl, 10 mM Tris · HCl, pH 7.8, 1 mM EDTA, 0.5% Triton X-100, 1 mM sodium orthovanadate) containing protease inhibitors (1 μg/ml leupeptin, and 100 μg/ml PMSF). Cells were then incubated at 4°C for 30 min with constant shaking. The cells were then scraped into microcentrifuge tubes, and the samples were centrifuged at 12,000 g for 15 min to remove insoluble material. The protein content in each sample was determined by Lowry assay. Equal loading was assessed by using an antibody against the housekeeping gene product GAPDH. For immunoprecipitation studies, lysates were incubated with the immunoprecipitating antibody for 1 h at 4°C, followed by another 1-h incubation with protein A-agarose at 4°C. Pellets were then resuspended in 2× loading buffer (50 mM Tris, pH 6.8, 2% SDS, 100 mM dithiothreitol, 0.2% bromphenol blue, 20% glycerol) and boiled for 5 min. Loading buffer supernatants from immunoprecipitation studies or cell lysates were then resuspended in 2× gel loading buffer, boiled for 5 min, and then separated by SDS-PAGE (9% polyacrylamide). Resolved proteins were transferred overnight at 4°C onto a polyvinylidene difluoride membrane (Millipore, Billerica, MA). Membranes were then blocked with a 5% solution of skim milk for 30 min at room temperature, followed by further incubation with specific monoclonal antibodies PKC-α, 1:1,000 (BD Transduction Laboratories, San Diego, CA), IκB-α, phospho-IκB-α (Ser32/36), and NF-κB p65 1:1,000 (Cell Signaling Technology, Danvers, MA), PTEN 1:500 (Santa Cruz Biotechnology, Santa Cruz, CA), and GAPDH 1:5,000 (Ambion, Austin, TX). After washing with phosphate-buffered saline (PBST) or Tris-buffered solution with 0.1% Tween (TBST), the secondary antibody was applied to the membrane. After being washed with either PBST or TBST, the membrane was treated with a chemiluminescent solution according to manufacturer's instructions and exposed to X-ray film. Densitometric analysis of the blots was performed by use of an AlphaImager digital imaging system (Alpha Innotech, San Leandro, CA).
Boyden Transwell chamber migration assay.
After coating the Corning Costar insert chambers as well as Transwell 24-well plates (8-μm pores; Corning, Corning, NY) with 0.1% fibronectin (Sigma Chemical, St. Louis, MO) and blocking with 1% BSA in 1× PBS for 1 h at 37°C, BxPc3 and CAPAN-1 cells were seeded in triplicate at 50,000 cells per well in serum-free media containing 1% BSA with or without TGF-β (10 ng/ml) and/or siRNA treatment. Cells were then allowed to migrate for 3 h. After removal of medium from both the chamber and the Transwell and followed by three washes with 1× PBS, the chamber was gently wiped with a cotton swab. Migrated cells were fixed in 100% methanol for 1 h and then allowed to air dry overnight. Cell staining was performed with a modified Giemsa stain (Sigma Chemical) at 1:10 for 1 h. After careful rinsing of the Transwell and the chamber with water, images were captured by use of an Axiovert 2000 microscope with an AxioCAM HRC Camera (both Zeiss Microimaging). Images were taken from six microscopic fields at the center of each well, and the stained cells on the bottom of the chamber were counted.
Cell transfection and reporter gene assays.
Cells were transfected in six-well plates with Fugene 6 (Roche) according to the manufacturer's instructions. To measure luciferase activity for the reporter gene assays, NF-κB-Luc or PTEN promoter reporter vector (PTEN-Luc) was cotransfected with a pRL-TK vector, which expresses Renilla luciferase as an internal transfection control. Luciferase activity was measured 24 h after transfection by a dual luciferase reporter system (Promega). Relative luciferase activity was normalized to the Renilla luciferase activity.
Statistical analysis.
All data are expressed as the means for a series of n experiments ± SE. Data were analyzed by one-way ANOVA followed by Tukey‘s post hoc test or by Student’s t-tests for unpaired samples using GraphPad Prism 3.0 (San Diego, CA). P < 0.05 was considered statistically significant.
RESULTS
TGF-β induces IκB-α phosphorylation in human pancreatic cancer cell lines.
NF-κB is not a single gene but a family of closely related transcription factors that includes five genes NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), c-Rel, and RelB. These five genes give rise to seven proteins that share a Rel Homology Domain (RHD) in their sequence (18). Dimers contain either RelA or c-Rel, together with p50. The dimers are held inactive in the cytoplasm by their interaction with IκB proteins (14). Upon activation, the IKKβ subunit is the major unit responsible for IκB phosphorylation on serine residues. After phosphorylation, IκB proteins undergo ubiquitin-dependent degradation by the proteasome, and NF-κB is translocated to the nucleus, where it acts as nuclear transcription factor (15). In the present study, we examined the effect of TGF-β on activation of the NF-κB pathway. To study whether TGF-β can stimulate IκB-α phosphorylation in cancer cells, we treated SMAD4-null BxPC3 human pancreatic epithelial cancer cells with a single dose of human recombinant TGF-β1 (10 ng/ml) and observed IκB-α phosphorylation over 4 h by Western blots using an anti-p-IκB-α antibody. As shown in Fig. 1A, IκB-α was phosphorylated within the first 30 min of treatment. This was followed by a gradual reduction of IκB-α phosphorylation and total IκB-α over 4 h after treatment with TGF-β. This result suggests that TGF-β causes the phosphorylation of the inhibitor IκB-α and subsequent ubiquitination of IκB-α and therefore activation and subsequent nuclear translocation of NF-κB. To confirm this result, we tested whether TGF-β induced IκB-α phosphorylation in another pancreatic cancer cell, CAPAN-1. Similar to the BxPC3 cells, IκB-α phosphorylation occurred after TGF-β treatment, with gradual reduction of phosphorylation and total IκB-α over 4 h (Fig. 1B). These findings indicate that TGF-β induces phosphorylation of IκB-α in pancreatic cancer cells and hence activation of the NF-κB pathway.
Fig. 1.
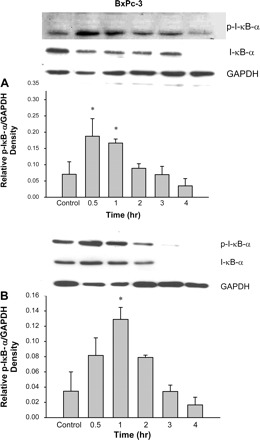
TGF-β increases IκB-α phosphorylation in pancreatic cancer cells. BxPc-3 (A) and Capan-1 (B) cells were treated with TGF-β (10 ng/ml) for the times shown. Cells were lysed and lysates were subjected to electrophoresis through 7.5% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were probed with monoclonal antibody specific for total and phosphorylated IκB-α. GAPDH was used as an internal standard. TGF-β significantly induces IκB-α serine phosphorylation by 30 min after treatment. The phosphorylation event came to basal level 2 h after treatment in both cell types. Total IκB-α was reduced over time as the protein was degraded once phosphorylated in both cell lines. *P < 0.05 vs. control. Data were means ± SE of 3–5 experiments.
TGF-β induces nuclear translocation of RelA/p65 of NF-κB.
The phosphorylation of IκB-α releases NF-κB, allowing its subsequent translocation to the nucleus. To verify whether nuclear translocation occurs, we inspected the translocation of the RelA/p65 subunit of NF-κB in both BxPc3 and CAPAN-1 pancreatic cell lines after treatment with TGF-β. As shown in Fig. 2, RelA/p65 translocation occurred, shifting from the cytoplasm into the nucleus by 2 h after TGF-β treatment. These data indicate that TGF-β induces the nuclear translocation of RelA/p65 in pancreatic cancer cells subsequent to IκB-α phosphorylation, which is evidence for the activation of NF-κB.
Fig. 2.
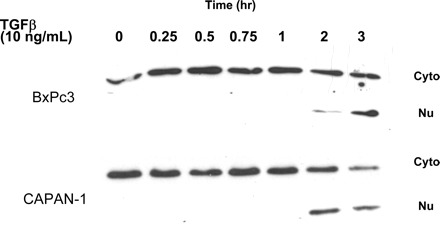
TGF-β induces nuclear translocation of NF-κB p65 in pancreatic cancer cells. BxPc-3 and Capan-1 cells were treated with TGF-β (10 ng/ml) for the times shown. Cells were then lysed and separated into cytosolic and nuclear fraction as described in materials and methods. Cell lysates were subjected to electrophoresis through 7.5% SDS-polyacrylamide gels and transferred to PVDF membranes. Membranes were probed with an antibody specific for the NF-κB p65 subunit. GAPDH was used as an internal standard. TGF-β significantly induces NF-κB p65 subunit to translocate from the cytosolic to the nuclear fraction by 2 h after treatment, indicating that NF-κB is activated.
TGF-β induces NF-κB reporter gene activity.
To confirm that NF-κB activity is induced by TGF-β, we assessed NF-κB reporter gene activity utilizing the dominant negative mutant IκB-α-M, which prevents serine phosphorylation of IκB-α such that NF-κB will always be sequestered by IκB-α and remains in the cytoplasm. BxPc3 pancreatic cancer cells were treated with TGF-β for 24 h and then the reporter gene activity of pNF-κB-luc was measured. TGF-β increased the pNF-κB-luc reporter gene activity in the absence of the superrepressor (IκB-α-M) (Fig. 3). Transfection of the cells with IκB-α-M significantly reduced TGF-β-induced NF-κB binding activity to basal levels for NF-κB activity. This result confirms that TGF-β induces pNF-κB-luc reporter gene activity in the pancreatic cancer cells.
Fig. 3.
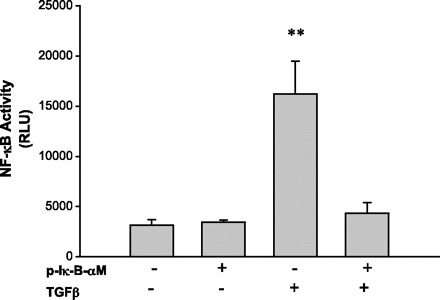
Transfection of a dominant negative mutant construct of IκB-α (IκB-αM, a superrepressor of NF-κB) reverses TGF-β-induced pNF-κB-luc reporter gene activity. To further confirm whether TGF-β could activate the NF-κB pathway, cells were transfected with NF-luc with (+) or without (−) cotransfection with IκB-αM 24 h before TGF-β treatment. Twenty-four hours after TGF-β treatment, cells were lysed and subjected to luciferase assay. TGF-β induces pNF-κB-luc reporter gene activity, suggesting activation of NF-κB. However, IκB-αM transfection inhibited TGF-β-induced activation of NF-κB.
Restoration of SMAD4 reversed TGF-β-induced NF-κB promoter activity and induction of NF-κB by TGF-β is independent of SMAD2 or SMAD3.
Most pancreatic cancers have SMAD4 deleted, which is an important intracellular mediator for the TGF-β signaling pathway as a tumor suppressor. To investigate whether the presence of SMAD4 affected TGF-β-mediated induction of NF-κB activity, we restored SMAD4 by transfecting the wild-type plasmid in pancreatic cancer cells (BxPc3) and observed NF-κB binding activity after TGF-β treatment. As shown in Fig. 4, inset, SMAD4 protein was present in BxPC3 cells 24–48 h after transient transfection of the SMAD4 plasmid. Interestingly, when SMAD4 was restored in BxPc3 cells, TGF-β-induced NF-κB binding activity was abolished (Fig. 4). We also studied the effects of TGF-β on the expression levels of the native suppressor of NF-κB, I-κB-α, in the presence or absence of SMAD4. In SMAD4-null BxPC3 cells, a single dose of TGF-β suppressed total I-κB-α expression starting 3 h after treatment, and the expression level was completely suppressed by 6 through 24 h, consistent with its proteasome-induced destruction. Forty-eight hours after the single dose of TGF-β, however, the expression level of I-κB-α returned to basal levels (Fig. 5). On the other hand, with SMAD4 present, the expression levels of I-κB-α were induced 1 h after TGF-β treatment and remained elevated through 24 h (Fig. 5). Thus restoration of TGF-β-SMAD signaling countered the NF-κB activation that occurs when SMAD4 is absent. Because TGF-β-SMAD signaling utilizes SMADs 2 and 3 in addition to SMAD4 for growth suppression in cancer cells, we explored whether SMAD2 or SMAD3 had any effects on NF-κB activation. Pancreatic cancer cells were transfected with dominant negative (DN)-SMAD2 and/or DN-SMAD3 in the SMAD4-null BxPC3 cells, followed by treatment with TGF-β. As shown in Fig. 4, the NF-κB binding activity induced by TGF-β treatment after either DN-SMAD2 or DN-SMAD3 transfection was similar to the control cells. However, in the absence of both SMAD2 and SMAD3, NF-κB activity was elevated. These data suggest that restoration of SMAD4 in SMAD4-null pancreatic cancer cells reversed the NF-κB activation that is observed when SMAD4 is absent by preventing the destruction of I-κB-α after TGF-β treatment. The loss of SMAD4 is permissive for TGF-β activation of NF-κB, and this is amplified by the loss of other SMADs.
Fig. 4.
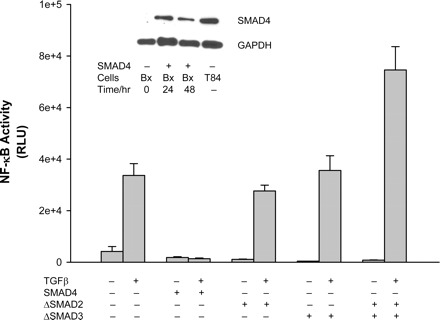
Effects of SMADs on TGF-β-induced NF-κB activity. To verify the effects of SMAD proteins on TGF-β-induced NF-κB activity, wild-type SMAD4 construct or dominant negative mutants of SMAD2 or 3 were cotransfected to the cells with NF-luc construct for 24 h. The cells were then treated with TGF-β for another 24 h. Cells were then lysed and subjected to luciferase assay. SMAD4 protein expression was present 24 and 48 h after transfection of the wild-type SMAD4 construct in BxPc-3 cells, (Bx; inset). T84 cells were used as a positive control for SMAD4 protein expression. GAPDH was used as an internal standard. SMAD4 restoration completely abolished TGF-β-induced NF-κB activity. Interestingly, inhibition of SMAD2 or SMAD3 by their corresponding dominant negative mutant constructs did not affect TGF-β-induced NF-κB activity. When both SMAD2 and SMAD3 were inhibited, the TGF-β-induced NF-κB activity was increased. RLU, relative light units.
Fig. 5.
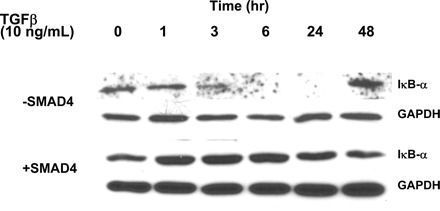
TGF-β induces downregulation of I-κB-α protein in SMAD4-null (−SMAD4) but not SMAD4-restored (+SMAD4) pancreatic cancer cells. Parental BxPc-3 cells were transfected with either an empty plasmid or wild-type SMAD4 construct. Twenty-four hours after transfection, cells were treated with TGF-β for 48 h. Cell lysates were subjected to electrophoresis through 7.5% SDS-polyacrylamide gels and transferred to PVDF membranes. Membranes were probed with an antibody specific for the I-κB-α. GAPDH was used as an internal standard. In the parental cells, TGF-β significantly induced removal of I-κB-α starting at 3 h and lasted for 24 h after treatment. Protein levels of I-κB-α exceeded the basal level 48 h after treatment. On the other hand, TGF-β induces upregulation of I-κB-α protein 1 h after treatment in the SMAD4-restored cells.
TGF-β downregulates PTEN promoter activity and protein expression via activation of NF-κB.
We have previously shown that PTEN expression is modulated by TGF-β1 in pancreatic cancer cells (3, 4), a finding verified by others (8). The mechanisms by which TGF-β uses to regulate PTEN expression are not fully understood but appear to involve SMAD-independent signaling. We examined whether TGF-β-induced PTEN expression can be suppressed through activation of NF-κB. We again utilized the p-IκB-αM superrepressor that sequesters NF-κB in the cytoplasm, and PTEN promoter activity and expression were examined by luciferase assay and Western blotting, respectively, after TGF-β treatment. As shown in Fig. 6, TGF-β induced downregulation of PTEN promoter activity. Transfection of p-IκB-αM reversed the TGF-β-induced PTEN downregulation. Similarly, TGF-β suppressed PTEN protein, as we have previously published (3); this occurs 48 h after treatment (Fig. 7A). Transfection of p-IκB-αM reversed TGF-β-induced PTEN protein suppression in both BxPc3 (Fig. 7A) and CAPAN-1 (Fig. 7B) cells. This result indicates that PTEN expression is downregulated by TGF-β at the transcriptional level owing to activation of NF-κB.
Fig. 6.
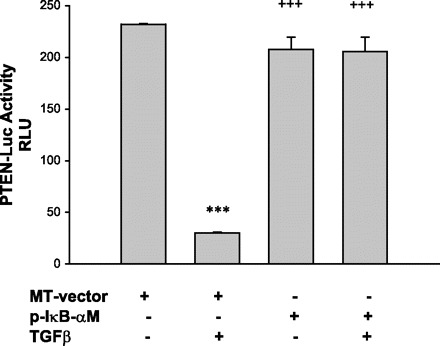
Inhibition of NF-κB reverses TGF-β-induced downregulation of PTEN promoter activity in BxPc-3 cells. Cells were transfected with PTEN-luc construct together with an empty vector (MT-vector) or the dominant negative mutant IκB-αM for 24 h. They were then treated with TGF-β for 24 h and subjected to luciferase assay. TGF-β downregulates PTEN promoter activity but dominant negative mutant IκB-αM completely reversed downregulation of PTEN promoter activity. ***P < 0.001 vs. Control; +++P < 0.001 vs. TGF-β. Data were means ± SE of 6 samples.
Fig. 7.
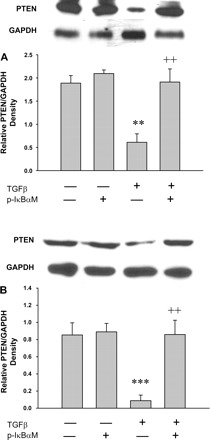
IκB-αM prevents TGF-β-induced downregulation of PTEN expression in BxPc-3 (A) and CAPAN-1 (B) cells. Cells were transfected with IκB-αM. The cells were then treated with TGF-β for 48 h. Cell lysates were subjected to electrophoresis through 7.5% SDS-polyacrylamide gels and transferred to PVDF membranes. Membranes were probed with an antibody specific for the PTEN. GAPDH was used as an internal standard. TGF-β significantly reduces expression of PTEN. Transfection with I-κB-αM reversed downregulated PTEN. **P < 0.01, ***P < 0.001 vs. Control; ++P < 0.01 vs. TGF-β. Data were means ± SE of 3–4 samples.
Inhibition of the NF-κB pathway reduces TGF-β-induced cell motility in human pancreatic cancer cells.
We have previously reported that TGF-β induces cell motility when it downregulates PTEN expression in pancreatic cancer (3). To assess whether TGF-β-induced cell motility is dependent on NF-κB activation, we utilized a Transwell Boyden chamber migration assay using pancreatic cancer cells transfected with p-IκB-αM. As shown in Fig. 8, TGF-β significantly increased cell motility in the absence of the p-IκB-αM, and this effect was reversed when p-IκB-αM was transfected. This finding indicates that TGF-β-induced cell motility is dependent on NF-κB activation and subsequent downregulation of PTEN.
Fig. 8.
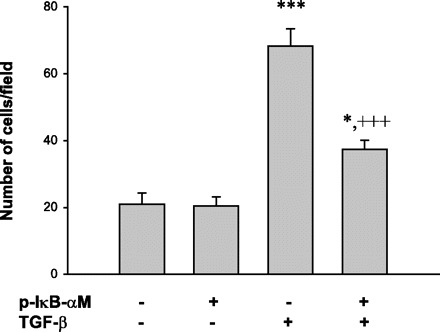
IκB-αM prevents TGF-β-induced cell motility in pancreatic cancer cells. Cells were transfected with the mutant IκB-αM for 24 h. The cells were then seeded on the Boyden chambers, incubated with TGF-β for 4 h, then fixed in methanol. The fixed cells on the bottom of the membrane in each chamber were stained with Giemsa stain and observed under the microscope. Cells in 5 fields were recorded, and the number of cells that cross the membrane were counted and averaged. TGF-β induced cell motility, and transfection with the mutant IκB-αM reversed the number of cells that cross the membrane after TGF-β treatment. *P < 0.05, ***P < 0.001 vs. Control; +++P < 0.001 vs. TGF-β. Data were means ± SE of 3 samples.
Direct inhibition of PTEN increases cell motility in human pancreatic cancer cells.
Although our findings clearly indicate that downregulation of PTEN by TGF-β via activation of NF-κB causes increased pancreatic cancer cell motility, and regulation of cell motility in fibroblasts by PTEN was postulated in study by others (22), a direct link of regulation of cell motility by PTEN in pancreatic cancer cells has not been shown. To assess whether PTEN is key to regulation of pancreatic cancer cell motility, we used PTEN siRNA to inhibit PTEN (see Fig. 9A), followed by Transwell Boyden chamber migration assay using the pancreatic cancer cells. As shown in Fig. 9B, knockdown of PTEN significantly enhanced pancreatic cancer cell motility. This finding indicates that PTEN is an important and direct mediator of pancreatic cancer cell motility.
Fig. 9.
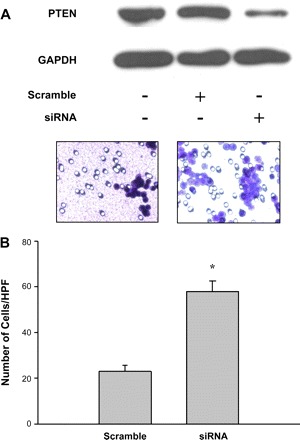
Inhibition of PTEN by small interfering RNA (siRNA)-enhanced cell motility in pancreatic cancer cells. Cells were transfected with either scramble siRNA or PTEN siRNA (500 nM) for 24 h. The cells were then seeded on the Boyden chambers and incubated in medium for 4 h, then fixed in methanol. The fixed cells on the bottom of the membrane in each chamber were stained with Giemsa stain and observed under the microscope. Cells in 5 fields were recorded, and the number of cells that cross the membrane were counted and averaged. Cells transfected with the PTEN siRNA showed a reduction of PTEN expression (A). Motility of cells transfected with PTEN siRNA was significantly increased compared with those that were transfected with scramble siRNA (B). *P < 0.05 vs. scramble siRNA transfection. Data were means ± SE of 3 samples. HPF, high-powered fields.
DISCUSSION
The treatment of pancreatic cancer has not progressed significantly over recent years, and understanding its pathological mechanisms will be key to improving survival for patients. Utilizing human pancreatic cancer cells in which we manipulated both SMAD and NF-κB signaling pathways to study the effects of TGF-β, we demonstrate that TGF-β-induced activation of NF-κB is a key mediator in downregulating the expression of the tumor suppressor PTEN and subsequent motility capability of the pancreatic cancer cells. This pathway is SMAD4 independent, although intact TGF-β-SMAD signaling, when present, counteracts SMAD4-independent NF-κB activation. Overall, our findings support the following in the pathogenesis of pancreatic cancers: 1) inactivation of TGF-β-SMAD suppressive signaling, 2) loss of SMAD4 being permissive for TGF-β activation of NF-κB, 3) suppression of the tumor suppressor PTEN by TGF-β, and 4) subsequent enhanced pancreatic cell motility that could facilitate tumor spread and metastasis. We suggest that fully deciphering the TGF-β/NF-κB/PTEN cascade could provide pharmacological targets that could be used to combat pancreatic cancer.
We first demonstrated that the TGF-β-induced NF-κB activity occurs in the absence of SMAD4 and observed an increase in NF-κB activity when both SMAD2 and SMAD3 were inhibited by their corresponding dominant negative constructs. This observation suggests that, even in the absence of SMAD4, the combined activity of SMAD2 and SMAD3 after TGF-β has some influence on counteracting TGF-β-induced NF-κB activation, a finding that is greatly amplified in the presence of SMAD4. We have previously observed SMAD2 and SMAD3 nuclear translocation in the absence of SMAD4 in cancer cells (4) with evidence of TGF-β-SMAD-mediated transcriptional activity (25). Thus TGF-β-SMAD suppressive signaling is minimal but not zero when SMAD4 is absent, but the influence of this residual pathway is greatly diminished compared with activation of other signaling pathways when SMAD4 is lost. We have previously demonstrated that TGF-β-induced PTEN suppression is dependent on the mobilization of intracellular calcium and the specific activation of PKC-α (3). Although we have not tested this here, we suggest that TGF-β induces PKC-α and subsequently NF-κB. This suggestion is based on the fact that both PKC-α and NF-κB are activated by TGF-β when SMAD4 is absent, and both contribute to TGF-β-induced PTEN suppression. It is not known whether other mediators constitute this pathway in pancreatic cancer. Although it is clear from our data that TGF-β induces NF-κB activation and subsequent suppression of PTEN, other mediators have been described that could influence NF-κB and its regulation of PTEN. For instance, MAPK4 can activate NF-κB and subsequently suppress PTEN expression (35). Another study implicates TNF-α and NF-κB-inducing kinase or interaction between PI3K/Akt and NF-κB for activation and PTEN suppression (19). Another interesting finding by Vasudevan et al. (33) indicated that downregulation of PTEN may be due to sequestration of transcriptional coactivators by NF-κB so that PTEN transcription is not able to proceed. All of these findings suggest a complexity of signaling after TGF-β ligand binds to its receptor with ultimate suppression of PTEN.
In pancreatic cancer cells in which SMAD signaling is abrogated, TGF-β causes cell proliferation (4) and increases cell motility. We demonstrated that NF-κB activity is a mediator of TGF-β-induced cell motility as well as a mediator for reducing PTEN expression. Thus targeting NF-κB activity might be a strategy to reverse these effects. Blocking NF-κB in pancreatic cancer may elevate PTEN expression levels and reduce the potential of cancer metastasis. This will require further study in the clinical arena among pancreatic cancer patients.
GRANTS
This research was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (DK-067287 to J. M. Carethers and DK-073090 to J. Y. C. Chow), the UCSD Digestive Diseases Research Development Center (DK-080506), and the Veterans Affairs Research Service (Merit Review Award to J. M. Carethers).
DISCLOSURES
No conflicts of interest are declared by the author(s).
REFERENCES
- 1. Arteaga CL, Carty-Dugger T, Moses HL, Hurd SD, Pietenpol JA. Transforming growth factor beta 1 can induce estrogen-independent tumorigenicity of human breast cancer cells in athymic mice. Cell Growth Differ 4: 193–201, 1993 [PubMed] [Google Scholar]
- 2. Beauchamp RD, Lyons RM, Yang EY, Coffey RJ, Jr, Moses HL. Expression of and response to growth regulatory peptides by two human pancreatic carcinoma cell lines. Pancreas 5: 369–380, 1990 [DOI] [PubMed] [Google Scholar]
- 3. Chow JY, Dong H, Quach KT, Van Nguyen PN, Chen K, Carethers JM. TGF-β mediates PTEN suppression and cell motility through calcium-dependent PKC-α activation in pancreatic cancer cells. Am J Physiol Gastrointest Liver Physiol 294: G899–G905, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chow JY, Quach KT, Cabrera BL, Cabral JA, Beck SE, Carethers JM. RAS/ERK modulates TGFbeta-regulated PTEN expression in human pancreatic adenocarcinoma cells. Carcinogenesis 28: 2321–2327, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choy L, Skillington J, Derynck R. Roles of autocrine TGF-beta receptor and Smad signaling in adipocyte differentiation. J Cell Biol 149: 667–682, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell 100: 387–390, 2000 [DOI] [PubMed] [Google Scholar]
- 7. Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch 446: 475–482, 2005 [DOI] [PubMed] [Google Scholar]
- 8. Ebert MPA, Fei G, Schandl L, Mawrin C, Dietzmann K, Herrera P, Friess H, Gress TM, Malfertheiner P. Reduced PTEN expression in the pancreas overexpressing transforming growth factor-beta 1. Br J Cancer 86: 257–262, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fogh J, Wright WC, Loveless JD. Absence of HeLa cell contamination in 169 cell lines derived from human tumors. J Natl Cancer Inst 58: 209–214, 1977 [DOI] [PubMed] [Google Scholar]
- 10. Friess H, Yamanaka Y, Buchler M, Ebert M, Beger HG, Gold LI, Korc M. Enhanced expression of transforming growth-factor-beta isoforms in pancreatic-cancer correlates with decreased survival. Gastroenterology 105: 1846–1856, 1993 [DOI] [PubMed] [Google Scholar]
- 11. Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell 109, Suppl: S81–S96, 2002 [DOI] [PubMed] [Google Scholar]
- 12. Gold LI. The role for transforming growth factor-beta (TGF-beta) in human cancer. Crit Rev Oncog 10: 303–360, 1999 [PubMed] [Google Scholar]
- 13. Grau AM, Zhang L, Wang W, Ruan S, Evans DB, Abbruzzese JL, Zhang W, Chiao PJ. Induction of p21waf1 expression and growth inhibition by transforming growth factor beta involve the tumor suppressor gene DPC4 in human pancreatic adenocarcinoma cells. Cancer Res 57: 3929–3934, 1997 [PubMed] [Google Scholar]
- 14. Greten FR, Karin M. The IKK/NF-kappaB activation pathway-a target for prevention and treatment of cancer. Cancer Lett 206: 193–199, 2004 [DOI] [PubMed] [Google Scholar]
- 15. Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev 18: 2195–2224, 2004 [DOI] [PubMed] [Google Scholar]
- 16. Hoosein NM, McKnight MK, Levine AE, Mulder KM, Childress KE, Brattain DE, Brattain MG. Differential sensitivity of subclasses of human colon carcinoma cell lines to the growth inhibitory effects of transforming growth factor-beta 1. Exp Cell Res 181: 442–453, 1989 [DOI] [PubMed] [Google Scholar]
- 17. Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin 57: 43–66, 2007 [DOI] [PubMed] [Google Scholar]
- 18. Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol 18: 621–663, 2000 [DOI] [PubMed] [Google Scholar]
- 19. Kim S, Domon-Dell C, Kang J, Chung DH, Freund JN, Evers BM. Down-regulation of the tumor suppressor PTEN by the tumor necrosis factor-alpha/nuclear factor-kappaB (NF-kappaB)-inducing kinase/NF-kappaB pathway is linked to a default IkappaB-alpha autoregulatory loop. J Biol Chem 279: 4285–4291, 2004 [DOI] [PubMed] [Google Scholar]
- 20. Lai CF, Cheng SL. Signal transductions induced by bone morphogenetic protein-2 and transforming growth factor-beta in normal human osteoblastic cells. J Biol Chem 277: 15514–15522, 2002 [DOI] [PubMed] [Google Scholar]
- 21. Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor β. Cancer Res 57: 2124–2129, 1997 [PubMed] [Google Scholar]
- 22. Liliental J, Moon SY, Lesche R, Mamillapalli R, Li D, Zheng Y, Sun H, Wu H. Genetic deletion of the Pten tumor suppressor gene promotes cell motility by activation of Rac1 and Cdc42 GTPases. Curr Biol 10: 401–404, 2000 [DOI] [PubMed] [Google Scholar]
- 23. Loor R, Nowak NJ, Manzo ML, Douglass HO, Chu TM. Use of pancreas-specific antigen in immunodiagnosis of pancreatic cancer. Clin Lab Med 2: 567–578, 1982 [PubMed] [Google Scholar]
- 24. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273: 13375–13378, 1998 [DOI] [PubMed] [Google Scholar]
- 25. Massague J. TGF-beta signal transduction. Annu Rev Biochem 67: 753–791, 1998 [DOI] [PubMed] [Google Scholar]
- 26. Okami K, Wu L, Riggins G, Cairns P, Goggins M, Evron E, Halachmi N, Ahrendt SA, Reed AL, Hilgers W, Kern SE, Koch WM, Sidransky D, Jen J. Analysis of PTEN/MMAC1 alterations in aerodigestive tract tumors. Cancer Res 58: 509–511, 1998 [PubMed] [Google Scholar]
- 27. Riggins GJ, Kinzler KW, Vogelstein B, Thiagalingam S. Frequency of Smad gene mutations in human cancers. Cancer Res 57: 2578–2580, 1997 [PubMed] [Google Scholar]
- 28. Sakurada A, Suzuki A, Sato M, Yamakawa H, Orikasa K, Uyeno S, Ono T, Ohuchi N, Fujimura S, Horii A. Infrequent genetic alterations of the PTEN/MMAC1 gene in Japanese patients with primary cancers of the breast, lung, pancreas, kidney, and ovary. Jpn J Cancer Res 88: 1025–1028, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seymour AB, Hruban RH, Redston M, Caldas C, Powell SM, Kinzler KW, Yeo CJ, Kern SE. Allelotype of pancreatic adenocarcinoma. Cancer Res 54: 2761–2764, 1994 [PubMed] [Google Scholar]
- 30. Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res 264: 29–41, 2001 [DOI] [PubMed] [Google Scholar]
- 31. Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 15: 356–362, 1997 [DOI] [PubMed] [Google Scholar]
- 32. Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 274: 787–789, 1996 [DOI] [PubMed] [Google Scholar]
- 33. Vasudevan KM, Gurumurthy S, Rangnekar VM. Suppression of PTEN expression by NF-kappa B prevents apoptosis. Mol Cell Biol 24: 1007–1021, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Virolle T, Adamson ED, Baron V, Birle D, Mercola D, Mustelin T, de Belle I. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat Cell Biol 3: 1124–1128, 2001 [DOI] [PubMed] [Google Scholar]
- 35. Xia D, Srinivas H, Ahn YH, Sethi G, Sheng X, Yung WK, Xia Q, Chiao PJ, Kim H, Brown PH, Wistuba II, Aggarwal BB, Kurie JM. Mitogen-activated protein kinase kinase-4 promotes cell survival by decreasing PTEN expression through an NF kappa B-dependent pathway. J Biol Chem 282: 3507–3519, 2007 [DOI] [PubMed] [Google Scholar]