

Abstract
Electrical communication and its role in blood flow regulation are built on an examination of charge movement in single, isolated vessels. How this process behaves in broader arterial networks remains unclear. This study examined the nature of electrical communication in arterial structures where vessel length and branching were varied. Analysis began with the deployment of an existing computational model expanded to form a variable range of vessel structures. Initial simulations revealed that focal endothelial stimulation generated electrical responses that conducted robustly along short unbranched vessels and to a lesser degree lengthened arteries or branching structures retaining a single branch point. These predictions matched functional observations from hamster mesenteric arteries and support the idea that an increased number of vascular cells attenuate conduction by augmenting electrical load. Expanding the virtual network to 31 branches revealed that electrical responses increasingly ascended from fifth- to first-order arteries when the number of stimulated distal vessels rose. This property enabled the vascular network to grade vasodilation and network perfusion as revealed through blood flow modeling. An elevation in endothelial-endothelial coupling resistance, akin to those in sepsis models, compromised this ascension of vasomotor/perfusion responses. A comparable change was not observed when the endothelium was focally disrupted to mimic disease states including atherosclerosis. In closing, this study highlights that vessel length and branching play a role in setting the conduction of electrical phenomenon along resistance arteries and within networks. It also emphasizes that modest changes in endothelial function can, under certain scenarios, impinge on network responsiveness and blood flow control.
Keywords: endothelial cells, gap junctions, computational modeling, smooth muscle cells, conducted response
the wall of small resistance arteries comprises a single layer of endothelium and one or more layers of smooth muscle. For these vascular cells to function as an integrated unit, they must electrically communicate with one another (17; 28; 33). The exchange of electrical information is facilitated by the expression of gap junctions among endothelial cells, among smooth muscle cells, and between the two cell layers (10; 22; 26). Functionally, electrical communication can be assayed by deploying a “conduction protocol” as popularized by Segal and Duling (28). This approach entails discretely stimulating a small portion of a resistance artery while monitoring voltage and/or vasomotor responses at sites distal to and upstream from the point of agent application.
Early investigations using the conduction protocol typically applied neurotransmitters, vasoactive metabolites, and ion channel modulators to cheek pouch, skeletal muscle, and mesenteric arteries (6; 17; 28; 33). Over time, a general pattern emerged, in which endothelial dependent responses spread more robustly along small arteries than those initiated in smooth muscle (8; 16). This pattern fostered the view that the endothelium was the primary pathway for conduction, a perspective later confirmed in functional experiments through the focal disruption of this cell layer (8). Biophysically, electrical phenomena spread effectively along the endothelium as these cells' parallel orientation and the robust gap junctional expression ensure that intercellular resistivity is low (4; 31). It should be noted that as electrical responses spread, a modicum of charge will be lost through the membrane and high resistance myoendothelial gap junctions, the latter of which mediates electromechanical coupling. In pursuit of this understanding, the vascular field has largely concentrated on the electrical behavior of isolated vessel segments (3; 5; 31). While this reductionist approach has been valuable, there is a paucity of information in regard to arterial networks and how vessel branching influences electrical communication. Such knowledge will help advance current observations and provide a clearer understanding of how electrical communication influences blood flow control.
In this investigation, we explored the nature of electrical communication in arterial networks and ascertain how charge distribution influences blood flow control in normal and diseased tissues. Using an existing model of electrical communication, we first observed that lengthening a virtual artery or introducing a branch point attenuated the conducted response. This reduction corresponded with experimental findings and was consistent with the idea that additional vascular cells augment electrical load. Using a symmetrical virtual network consisting of 15 branch points, we subsequently showed that electrical responses could increasingly ascend from fifth- to first-order arteries as the number of stimulated distal vessels increased. This property enabled our virtual network to grade vasomotor and perfusion responses as determined in an established blood flow model. Simulating the system-wide changes in gap junctional conductance that likely occurs during sepsis (21) revealed that this disease can in theory attenuate the ascension of electrical and hemodynamic responses. In summary, the current investigation emphasizes how network structure shapes electrical communication and consequently blood flow regulation. Additionally, it re-emphasizes the endothelium's essential role in this biological process and how a modest change in this layer's coupling not only attenuates electrical conduction but also compromises network perfusion.
MATERIALS AND METHODS
Animal and tissue preparations.
Animal handling procedures were approved by the Animal Care and Use Committee at the University of Calgary. Briefly, each male golden Syrian hamster (10- to 12 wk of age) was overdosed with pentobarbital sodium (65 mg/kg ip). The mesentery was removed and put into PBS containing (in mM) 138 NaCl, 3 KCl, 10 Na2HPO4, 2 NaH2PO4, 5 glucose, 0.1 CaCl2, and 0.1 MgSO4. Fifth-order arteries, with or without their fourth-order parent vessel, were dissected free from connective tissue and prepared for cannulation.
Vessel myography.
Mesenteric arteries were cannulated in a customized arteriograph, positioned on an inverted microscope, and pressurized to 15 mmHg, as described previously (12; 16). Tissues were equilibrated for 30 min in a physiological saline solution (37°C; 5% CO2; 21% O2) containing (in mM) 119 NaCl, 4.7 KCl, 1.7 KH2PO4, 1.2 MgSO4, 1.6 CaCl2, 5 glucose, and 20 NaHCO3. Next, intravascular pressure was slowly raised from 15 to 60 mmHg; phenylephrine (0.1 μM) was then added to the superfusate to ensure a consistent level of tone among preparations and treatments. Internal diameters were monitored using a 10× objective and manual calipers. Vessels that did not robustly respond to superfused KCl (55 mM) were excluded, a priori, from additional experimentation.
Experimental protocols.
For conduction along single isolated arteries, KCl (250 mM, 10 psi, 20-s pulse) was discretely applied via a micropipette to a small portion of a fifth-order mesenteric artery to initiate local changes in endothelial membrane potential (Vm). Arterial diameter was then monitored at 0, 450, 900, 1,350, and 1,800 μm distal to the site of stimulation. To induce endothelial disruption at a site 900 μm distal to the point of stimulation, fluorescein-conjugated dextran (70 kDa; 0.5% solution) was intraluminally perfused and excited with fluorescent light focused through a 40× objective as described previously (8).
For conduction along a branching arterial structure, a fourth-fifth order branching structure was placed in a widen arteriograph chamber. Next, each end of the branching structure was cannulated onto a glass pipette held by a manual micromanipulator and the preparation was continuously superfused with physiological saline solution. Tubing to the upstream (fourth-order) artery (∼1,200 μm, length) was attached to a pressurizing servomotor, whereas tubing to the downstream (fifth-order) branches (∼1,100 μm in length) were clamped. KCl was focally applied to one daughter branch using pressure ejection while vasomotor responses were evaluated at 0, 450, 900, 1,350, and 1,800 μm distal to the point of agent application as shown in Fig. 5. Note, the internal diameter of the daughter branches were comparable with one another (range, ±10 μm).
Fig. 5.
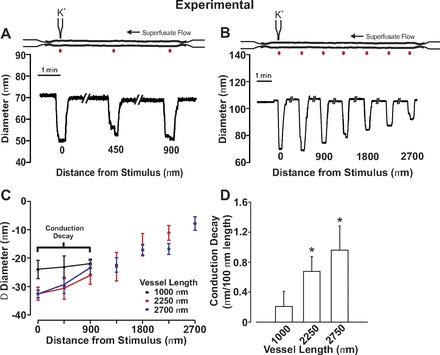
Electrical decay increases with the length of mesenteric arteries. For experimentation, endothelial cells in a small portion of a fifth-order mesenteric artery were focally stimulated with KCl while vasomotor responses were monitored along the artery. A and B: representative traces showing conducted constriction in arteries of different length. C and D: summary data were plotted as a change in vessel diameter (C) or as conduction decay (D). Resting, minimal, and maximal diameters were as follows: 1,000 μm in length (n = 7), 101 ± 7 μm, 24 ± 4 μm, 134 ± 7 μm; 2,250 μm in length (n = 6), 109 ± 7 μm, 27 ± 4 μm, 134 ± 8 μm; 2,700 μm in length (n = 6), 114 ± 8 μm, 32 ± 6 μm, 140 ± 9 μm. *Significant difference from 1,000 μm.
Electrical modeling.
A computational model originally developed by Diep et al. (4) was used to advance our understanding of electrical communication in arterial networks (Fig. 1A). The base parameters were similar to those of the original model. Briefly, each cell was characterized electrically as a capacitance linked in parallel with a nonlinear resistor, the latter representing ionic flow through endothelial and smooth muscle channels (Fig. 1B). In contrast, gap junction channels were represented by ohmic resistors. Cells were connected electrically to one another with gap junctions, represented as ohmic resistances. Membrane capacitance was set to 1 μF cm−2. Our initial virtual artery was structured to be 2.0 mm long, ∼75 μm in diameter, and composed of one layer of endothelium circumscribed by one layer of smooth muscle. The artery was subdivided into 40 arterial segments, each 50 μm long, and consisting of an outer layer of 30 smooth muscle cells (arranged as 10 consecutive bands, each consisting of 3 cells) and an inner layer of 48 endothelial cells (arranged side by side parallel to the artery's longitudinal axis). The physical dimensions of each vascular cell were as follows: endothelium, 50 μm length, 5 μm width, 1μm height; smooth muscle, 80 μm length, 5 μm width, 5 μm height (Fig. 1A). To assemble a single branch point, the smooth muscle cells at the end of each individual vessel were electrically coupled with one another. Next, endothelial cells from the parent vessel were connected equally to the daughters while half of the daughter endothelial cells were coupled to the parent and the adjacent daughter, respectively. The length and diameter of each arterial segment was set manually. Specific simulations were as follows.
Fig. 1.
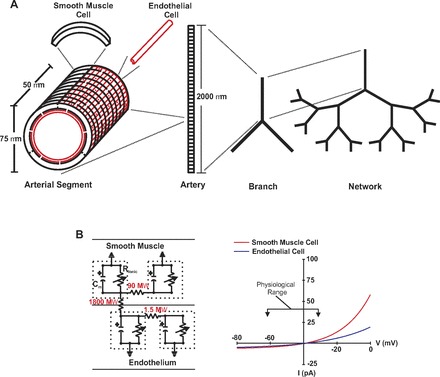
Physical and electrical representation of a virtual artery. A: virtual artery was comprised 1 endothelial and 1 smooth muscle layer. The standard arterial segment consisted of 48 endothelial cells and 30 smooth muscle cells. Cells were treated as discrete elements with defined dimension, coupling, and ionic conductance. Neighboring smooth muscle cells were connected to one another as well as neighboring endothelial cells. Every smooth muscle cell was randomly connected to 2 endothelial cells. Arterial segments were assembled to build an isolated vessel and these in turn were assembled to form a branch structure or network. B: in the equivalent circuit diagram (left), each cell was modeled as a capacitator coupled in parallel with a nonlinear resistor representing ionic conductance; gap junctions were represented by linear resistors. I-V properties of nonlinear resistor are displayed on the right panel and are referenced to resting Vm of −40 mV.
For Figs. 2 and 3, one arterial segment of the endothelium was voltage clamped 15 mV positive or negative to the resting Vm (−40 mV, 200 ms) while smooth muscle and endothelial voltage changes were monitored along virtual arteries under control conditions or following disruption of one arterial segment of the endothelium, 900 μm distal to the point of stimulation. Endothelial cell disruption was simulated by increasing endothelial-endothelial and endothelial-smooth muscle coupling resistances 1,000-fold. Boundary cells were sealed. The preceding stimulation paradigm was also deployed on virtual vessels that varied in length from 1,000 to 8,000 μm.
Fig. 2.
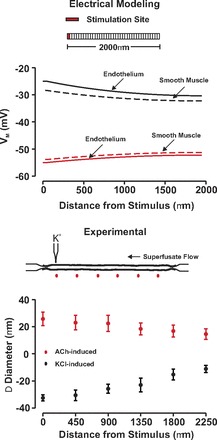
Endothelial-initiated responses conduct along resistance arteries independently of the stimuli polarity. Top: for simulations, endothelial cells within 1 arterial segment were voltage clamped 15 mV negative (red) or positive (black) to resting Vm (−40 mV, 200 ms) while steady-state endothelial (solid line) and smooth muscle (dotted line) electrical response was monitored along the vessel. Bottom: for experimentation, ACh (red, 10 mM) or KCl (black, 250 mM) was pressure ejected onto a small region of a fifth-order mesenteric artery. Vasomotor responses were monitored at sites 0, 450, 900, 1,350, 1,800, and 2,250 μm distal to the point of agent application. Resting, minimal, and maximal diameters were as follows ACh (n = 4): 108 ± 5 μm; 18 ± 2 μm; 140 ± 7 μm; KCl (n = 6): 108 ± 5 μm; 17 ± 1 μm; 138 ± 7 μm.
Fig. 3.
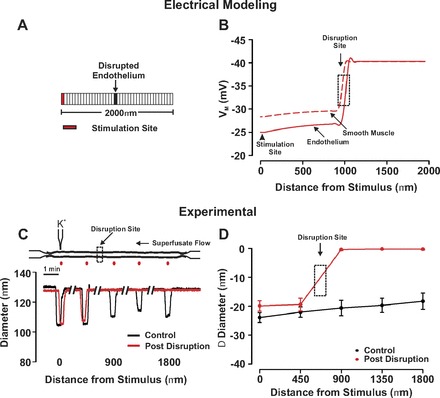
Endothelium is the principal pathway for conduction. A and B: for simulations, 1 arterial segment of endothelium was voltage clamped 15 mV positive to the resting Vm (−40 mV, 200 ms) while steady state Vm was monitored in a virtual artery when 1 arterial segment of endothelium was disrupted. C and D: for experimentation, KCl (250 mM) was pressure ejected onto a small region of a fifth-order mesenteric artery. Vasomotor responses were monitored at sites 0, 450, 900, 1,350, and 1,800 μm distal to the point of agent application under control conditions (black) and following focal endothelial disruption (red) at a site positioned ∼900 μm from the stimulus pipette. A representative trace and summary data are in C and D, respectively. Resting, minimal, and maximal diameters were as follows (n = 6): 89 ± 5 μm; 27 ± 4 μm; 129 ± 8 μm.
For Fig. 6, a simplified branching structure consisting of one parent (75 μm diameter, 2,000 μm length) and two daughters (60 μm diameter, 2,000 μm length) was constructed to examine the effect of branch points on electrical conduction. To accommodate the reduction in daughter vessel diameter, the number of endothelial cells per arterial segment was reduced to 38, which smooth muscle length was shortened to 63 μm. One arterial segment of endothelium at the distal end of one or both daughter branches were voltage clamped 15 mV positive to resting Vm (−40 mV) for 200 ms. Endothelial and smooth muscle Vm responses were monitored along the virtual artery.
Fig. 6.
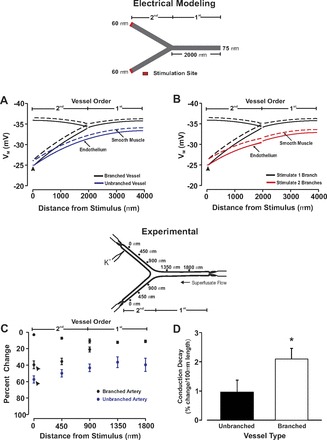
Electrical decay in a branching arterial structure. A and B: for simulations, an endothelial segment in 1 or both daughter arteries was voltage clamped 15 mV positive to resting Vm (−40 mV, 200 ms). Steady-state depolarization was plotted along the branching structure and compared with a similar length of unbranched virtual artery. C and D: for experimentation, 1 fifth-order mesenteric artery from a fourth- to fifth-order branch point was stimulated with KCl (250 mM, 20-s pulse) while vasomotor responses were monitored along the network. Summary data were plotted as a percent change of vasoconstrictor capacity (C) or as conduction decay (D). Resting, minimal, and maximal diameters for the branching arterial structure (n = 9) were as follows: parent vessel, 152 ± 10 μm, 56 ± 7 μm, 207 ± 10 μm; stimulated fifth-order artery, 105 ± 9 μm, 36 ± 8 μm, 160 ± 14; unstimulated fifth-order artery, 114 ± 10 μm, 33 ± 6 μm, 147 ± 9 μm. For the unbranched fifth-order vessels (n = 7), the resting, minimum, and maximal diameters were 121 ± 10 μm, 32 ± 6 μm, and 161 ± 15 μm, respectively. *Significant difference from the unbranched artery.
A symmetrical arterial network consisting of five orders was subsequently constructed to better resolve the influence of branch points on electrical communication. The physical dimensions of originating first-order artery was preset to values comparable with those of hamster skeletal muscle feed vessels (75 μm diameter, 2,000 μm length). The length and diameter of each subsequent branch was then generated based on the dendritic tree structure equation (23). Note, this equation generated physical dimensions that approximated values reported for the porcine coronary vasculature (13; 32). Values were as follows: second order (47 μm in diameter, 1,450 μm in length); third order (30 μm in diameter,1,050 μm in length); fourth order (19 μm in diameter, 750 μm in length); and fifth order (12 μm in diameter, 550 μm in length). As above, the number of endothelial cells per arterial segment along with smooth muscle length was adjusted to accommodate the change arterial diameters. One endothelial segment in every fourth-, second-, or all fifth-order arteries was voltage clamped 15 mV negative to resting Vm for 200 ms while voltage changes along the network were monitored. Voltage responses were also monitored under conditions in which: 1) endothelial-endothelial coupling resistance was increased to from 1.5 to 4.5–6.0 MΩ; and 2) 10 arterial segments of endothelium were disrupted at a site positioned downstream from the first-second order branch point. As above, endothelial disruption was induced by increasing endothelial-endothelial and endothelial-smooth muscle coupling resistances 1,000-fold.
Blood flow modeling.
A previously developed theoretical model (9; 24; 25) was used to calculate two-phase (red blood cell and plasma) steady-state flow in the arteriolar networks considered. This model implements conservation of blood and red blood cell volume flow at each node joining three vessels and includes known blood rheology, specifically, the Fahraeus and Fahraeus-Lindqvist effects in unbranched segments and phase separation at diverging bifurcations. Besides geometric information (segment diameters and network connectivity), the model requires specification of boundary conditions in the form of hematocrit at the inlet segment (upstream end of the first-order artery) and pressure changes between the inlet node and outlet nodes (downstream ends of the fifth-order arteries).
To apply the blood flow model to the arteriolar networks containing 31 vessels, a subdivision of each vessel into segments 50 μm (endothelial cell length) long was used. The luminal diameter di of each cylindrical segment was determined from the baseline diameter di0 using Vm, the segment's average smooth muscle cell Vm membrane potential: di = di0 [1 − ((40 + Vm)/30)] di = di0 for Vm = −40 mV and di varies linearly between di0/2 and 3 times (di0/2) as Vm varies between −25 and −55 mV. All flow simulations used an inlet hematocrit of 0.4, and inlet and outlet pressures of 75 and 20 mmHg, respectively.
Statistical analysis.
Data are expressed as means ± SE, and n indicates the number of mesenteric arteries. One artery was used per animal. Unpaired t-tests were used to compare arterial responses among independent groups. P values ≤0.05 were considered statistically significant.
Solutions and chemicals.
All buffers, chemicals, and reagents were purchased from Sigma-Aldrich (St. Louis, MO).
RESULTS
Our examination of electrical communication began with simulations highlighting the ability of endothelial-initiated responses, albeit hyperpolarization and depolarization, to longitudinally conduct. In this regard, one endothelial segment of a virtual artery (75 μm diameter, 2,000 μm length) was voltage clamped 15 mV negative or positive to resting Vm (−40 mV) while voltage responses were resolved along the structure. As shown in Fig. 2A, hyperpolarization and depolarization first spread with limited decay along the endothelial layer and then a near parallel response in the overlying smooth muscle. This virtual electrical behavior was consistent with observations from fifth-order mesenteric arteries where the local delivery of acetylcholine or KCl elicited conducted vasodilation and vasoconstriction, respectively (Fig. 2B). Subsequent simulations were designed to illustrate the importance of the endothelium as a primary pathway for longitudinal conduction. In this context, we monitored the spread of depolarization in a virtual vessel where the endothelium had been discretely disrupted. As findings in Fig. 3A note, endothelial-initiated electrical responses effectively conduct up to but not beyond the site of simulated disruption. To keep with these conceptual observations, biological experiments on fifth-order mesenteric arteries similarly revealed that a 20-s KCl pulse induced conducted constriction up to but not passed the site of endothelial disruption (Fig. 3, B and C). Control experiments confirmed that endothelial disruption did not prevent the overlying smooth muscle from focally constricting to a KCl pulse or the focal application of phenylephrine (data not shown).
Subsequent work was undertaken to ascertain the impact of vessel length and branching on the spread of electrical responses along arterial structures. We began with simulations in which the spread of endothelial-initiated depolarization was monitored along virtual arteries varying in length from 1,000 to 8,000 μm. As highlighted in Fig. 4, increasing vessel length through the addition of new vascular cells enhanced the electrical decay. This conceptual behavior coincided well with experimental observations where conducted constriction in fifth-order mesenteric arteries was shown to decay more extensively when vessels were ∼2,750 μm rather than 1,000 μm in length (Fig. 5). This is best denoted in the calculation of conduction decay (Fig. 5D), a parameter that varied approximately threefold between the shorter and longer vessels. Simulations were next progressed to a simplified branching structure comprising a parent vessel (75 μm diameter, 2,000 μm length) and two daughter vessels (60 μm diameter, 2,000 μm length). Voltage clamping one endothelial segment in a daughter vessel 15 mV positive to the resting Vm (−40 mV) elicited a depolarization that conducted across the branch point into the parent vessel (Fig. 6). Electrical decay was greater than observed for an unbranched virtual vessel of comparable length (4,000 μm length), an observation consistent with the branch point enhancing the electrical load (Fig. 6A). It is interesting that the attenuating effects of a branch point could be minimized by simultaneously depolarizing one endothelial segment in both daughter branches (Fig. 6B). To experimentally support the model predictions in Fig. 6A, a fourth-fifth order branching structure was isolated from the mesentery and KCl focally applied to one fifth-order branch. The constrictor response spread along the stimulated fifth-order vessel into the adjacent daughter and fourth-order parent arteries (Fig. 6C). Vasomotor decay was greater in this branching structure than an unbranched fifth-order mesentery of equal length (Fig. 6D). Vasomotor decay was calculated on a percentage based from the formulation: (KCl minus induced diameter minus resting diameter)/(minimal diameter minus resting diameter).
Fig. 4.
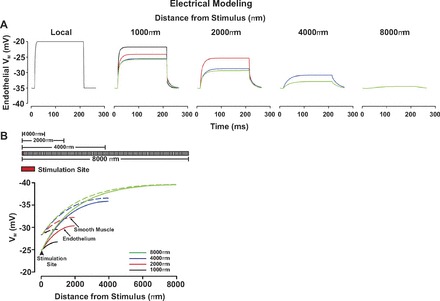
Electrical decay increases with the length of the virtual artery. A and B: for simulations, endothelial cells in 1 arterial segment were voltage clamped 15 mV positive to the resting Vm (−40 mV, 200 ms) in vessels 1,000, 2,000, 4,000, and 8,000 μm in length. A: time course of the endothelial electrical response to a depolarizing step. B, top: representation of virtual arteries varying in lengths; B, bottom: steady-state depolarization plotted along vessels varying in length.
With the preceding work showing that branch points attenuate the longitudinal spread of charge, we began to consider the nature of electrical communication in broader arterial networks and its impact on blood flow control. In this regard, we constructed a symmetrical virtual network comprising 31 vessels and 15 branch points. The physical dimensions of first-order artery were predetermined, with the length and diameter of each subsequent artery (second- to fifth-order) set in accordance with dendritic tree structure equation (23). Simulations consisted of voltage clamping one endothelial segment in a variable number fifth-order vessels and resolving the response over the entire structure. Note that a hyperpolarizing stimulus was used in place of depolarization since it is more functional relevant to the endothelium. Findings in Fig. 7, A and B, demonstrate that hyperpolarizing (−40 to −55 mV) one endothelial segment in every fourth distal artery elicited an electrical response that decayed dramatically as it spread into the first-order artery. For reference, note that only those vessel responses within the dotted box are plotted. Increasing the number of stimulated distal vessels, to every second or all, attenuated the decay and augmented the ability of hyperpolarizing current to ascend in the vascular tree (Fig. 7, C-F). To ascertain the impact of distal arterial hyperpolarization on tissue perfusion, an existing blood flow model (9) was applied to our symmetrical arterial network. The relationship between vasomotor response and Vm was as previously discussed in the materials and methods. Figure 8, A and B, highlights that when the number of stimulated distal arteries was constrained to every fourth vessel, vasodilation was principally limited to the stimulated fifth-order artery, its upstream fourth-order parent, and the adjacent fifth-order unstimulated daughter vessel. Network blood flow rose modestly from rest, and this augmentation was directed primarily to regions near the stimulated distal artery (Fig. 8, C and D). Calculations of percent flow change indicate that the stimulated vessel and its immediate upstream parent as well as the adjacent unstimulated daughter vessel received the greatest increase in flow. This enhancement of flow substantially reduced in vessels that are more proximal (i.e., third- to first-order vessels). It is interesting that in aspects of the network removed from the stimulation site, the phenomenon of blood flow “steal” was observed. As the number of stimulated daughter vessels increased from 4 to 16, vasodilation was enhanced and responses spread increasingly upward through the arterial network. This enhanced vasomotor effect fostered a stepwise increase in the magnitude of tissue blood flow, a finding consistent with the physiological behavior of ascending vasodilation (11; 29).
Fig. 7.
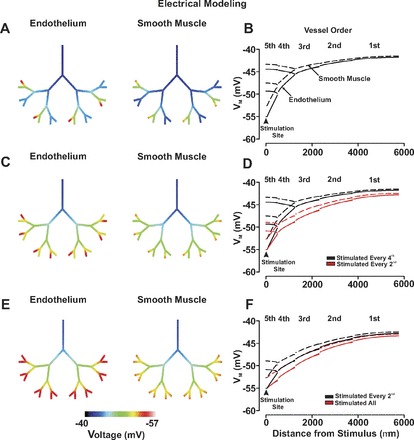
The ascension of electrical responses in a virtual arterial network. For simulations, an endothelial segment of every fourth (A and B), every second (C and D), or all (E and F) fifth-order arteries were voltage clamped 15 mV negative to the resting Vm (−40 mV) for 200 ms. Endothelial (solid line) and smooth muscle (dotted line) voltage responses were displayed in steady state with the use of color maps (A, C, and E) or 2-dimensional plots (B, D, and F). Arrow heads denote sites of stimulation.
Fig. 8.
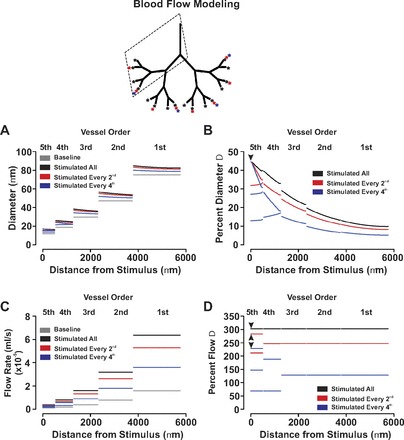
The ascension of vasomotor and blood flow responses in a virtual arterial network. For simulations, vasomotor and blood flow responses were calculated for the virtual network under conditions in which every fourth-, every second-, or all fifth-order arteries were voltage clamped 15 mV negative to the resting Vm (−40 mV, 200 ms). Inset: 1) the stimulation pattern of every fourth- (blue star), every second- (red star), and all fifth-order vessels (black stars); and 2) which part of the arterial network was sampled (dotted box). Data are presented as absolute measurements (A and C) or as a percent change from baseline (B and D). Arrow heads denote site of stimulation.
With the use of the symmetrical network model, this study subsequently expanded its theoretical examination to explore how disease states might impact electrical communication and blood flow control. Our initial work focused on the rise in endothelial coupling resistance that accompanies the production of lipopolysaccharides, potent activators of inflammatory cells and participants in the development of septic shock (21). Figure 9 highlights that as endothelial coupling resistance increased from 1.5 MΩ (control) to 4.5 MΩ and 6.0 MΩ, the ability of distal hyperpolarizing stimuli to rise through the arterial network was sizably reduced. The reduction in electrical communication diminished the ascension of vasodilation as expressed in absolute and relative terms (Fig. 10, A and B). This attenuation in mechanical responsiveness translated into a generalized 30% reduction in network perfusion (Fig. 10, C and D). Additional simulations explored the influence of focal endothelial disruption on longitudinal charge movement and arterial network function. In this regard, we fashioned an endothelial disruption site consisting of 10 sequential segments and positioned it downstream from the first-second order branch point. Figure 11, A and B, revealed that when all fifth-order arteries were simultaneously stimulated, the endothelial disruption site modestly altered the ascent of hyperpolarization. More specifically, hyperpolarization was augmented in arteries positioned below the disruption site while those above displayed an attenuated response. Although these electrical alterations drove predictable changes in vasodilation (Fig. 11C), their impact on the magnitude and distribution of tissue blood flow was limited (Fig. 11D). Other distal stimulation patterns or changing the site of focal endothelial disruption site produced a qualitatively similar outcome (data not shown).
Fig. 9.
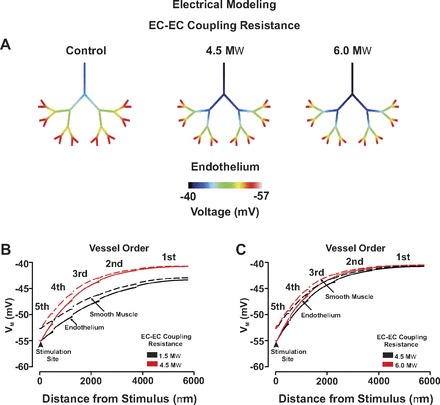
Endothelial coupling resistance influences the ascension of electrical responses in a virtual arterial network. For simulations, an endothelial segment in all fifth-order arteries was voltage clamped 15 mV negative to resting Vm (−40 mV, 200 ms) under control conditions and in which endothelial-endothelial (EC-EC) coupling resistance was increased from 1.5 to 4.5–6.0 MΩ. Endothelial (solid line) and smooth muscle (dotted line) voltage responses were displayed in steady state with the use of color maps (A) or 2-dimensional plots (B and C). Arrow heads denote sites of stimulation.
Fig. 10.
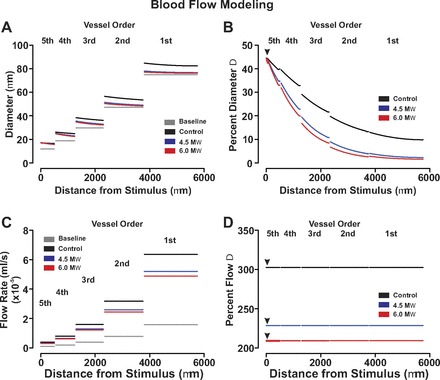
Endothelial coupling resistance influences the ascension of vasomotor and blood flow responses in a virtual arterial network. For simulations, vasomotor and blood flow responses were calculated for the virtual network under conditions in which all fifth-order arteries were voltage clamped 15 mV negative to the resting Vm (−40 mV, 200 ms). Endothelial-endothelial coupling resistance was set to 1.5, 4.5, or 6.0 MΩ. Data are presented as absolute measurements (A and C) or as a percent change from baseline (B and D). Arrow heads denote site of stimulation.
Fig. 11.
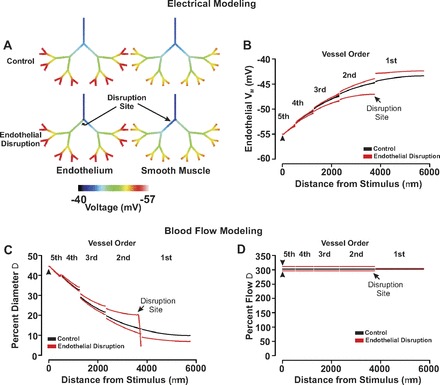
The effect of focal endothelial disruption on the ascension of responses in a virtual arterial network. A and B: for simulations, an endothelial segment in all of fifth-order arteries was voltage clamped 15 mV negative to resting Vm (−40 mV, 200 ms) under control conditions and following endothelial ablation. Endothelial (solid line) and smooth muscle (dotted line) voltage responses were displayed in steady state with the use of color maps (A) or 2-dimensional plots (B). C and D: for simulations, vasomotor and blood flow responses were calculated for the virtual network under conditions in which all fifth-order arteries were voltage clamped 15 mV negative to the resting Vm (−40 mV, 200 ms). The endothelial layer was intact or focally disrupted at a site distal to the first- and second-order branch. Flow data are presented in absolute terms (C) or as a percent change from baseline (D). Arrow heads denote site of stimulation.
DISCUSSION
In this study, we systematically explored how vessel structures, varying from single arteries to integrated networks shape electrical communication and blood flow control. Using an existing model of electrical communication, we first showed that vessel lengthening or branch point introduction diminished the ability of an electrical response to conduct along the arterial wall. This attenuation arose from the augmentation of electrical load and was consistent with experimental findings from hamster mesenteric arteries. Subsequently, we showed in a symmetrical network model consisting of 15 branch points that electrical responses could ascend into proximal arteries in accordance with the number of distal vessels that were initially stimulated. The property of electrical ascension enabled the virtual network to grade vasomotor and perfusion responses, as established in a blood flow model. Mimicking the broad based changes in vascular coupling that likely arise during sepsis (21) revealed that this disease state could limit the ascension of electrical responses and consequently the hemodynamic performance of the arterial network. In closing, this study showed that the physical structure of arteries and networks play a key role in shaping electrical communication and blood flow control. It secondarily highlights the endothelium's importance as a communication pathway and that significant reductions in organ perfusion are likely to occur when particular diseases limit this layer's coupling resistivity.
August Krogh (14) was the first to observe in the resistance vasculature that focal stimuli elicit vasomotor responses that radiated outward from the site of perturbation. This unique observation received sporadic attention for six decades until Segal and Duling (28) revisited the finding, systematically noting that focal neurotransmitter delivery also elicited robust “conducted responses.” It was subsequently surmised that the conducted response was a manifestation of electrical communication, a view later confirmed with arterial Vm measurements (7; 27; 33). Mechanistic investigations have, over time, defined the requisite events underlying this biological response. To begin, the initiating stimulus must elicit a focal change in endothelial Vm so that charge can effectively spread along this low resistance cellular pathway (4; 8; 34). Next, as the electrical response conducts, charge must be able to cross myoendothelial gap junctions to drive electromechanical behavior in the overlying smooth muscle, a phenomenon in agreement with previous work (7; 34). We highlighted these foundational concepts in Figs. 2 and 3 with the use of computational and biological approaches. In particular, we showed that when a small number of endothelial cells are focally depolarized or hyperpolarized, robust conducted response will ensue, except when the endothelium has been discretely disrupted, a behavior consistent with past studies (4).
Electrical conduction along resistance arteries is a passive phenomenon as vascular ion channels do not support action potential propagation. Although charge regeneration mechanisms are absent, resting ionic conductances do influence conduction decay by modifying membrane resistivity (12). Gap junction resistivity also impacts the robustness of the conducted response as does the overall shape of the vascular structure (4; 16; 30; 31). The latter aspect has received little attention since investigative efforts have largely focused on the mechanistic behavior of isolated vessels. It was in this context that we started to examine electrical communication in more diverse arterial structures. Work began with computational simulations where one arterial segment of endothelium was depolarized while the spread of electrical phenomenon was monitored in virtual vessels varying in length. As the number of vascular cells increased in a lengthened virtual artery, conduction decay per unit length rose (Fig. 4). This enhancement coincided well with biological observations, which showed that the decay of conducted constriction was more extensive in mesenteric arteries that were ∼2,750 μm than ∼1,000 μm in length (Fig. 5). Enhanced decay can be partly explained by the augmented electrical load of the arterial structure: the additional vascular cells along with their resting conductances provide further membrane pathways for charge loss. Branch points are another means of adding vascular cells to an arterial structure. Like vessel lengthening, the inclusion of a branch point to our virtual model diminished the ability of distally initiated events to conduct upward into the parent vessel (Fig. 6). This branch-induced attenuation was also observable in mesenteric branch structures where KCl-induced conduction was monitored from daughter to parent vessel and then compared with a similar length of unbranched artery (Fig. 6). It is interesting that further modeling revealed that the attenuating influence of a branch point could be ameliorated by stimulating both daughter vessels (Fig. 6). This tandem stimulation protocol facilitates upward conduction by introducing additional charge to the arterial structure and by eliminating the electrochemical gradient in the previously unstimulated daughter vessel.
The preceding work with single branch points inspired interest in more elaborate arterial networks and whether responses elicited in small distal arteries could effectively ascend into larger proximal vessels. To pursue this idea, we assembled a symmetrical network consisting of 31 arteries and 15 branch points. The physical dimensions of the first-order artery were predetermined, whereas the length and diameter of each subsequent artery were set according to the dendritic tree structure. When the number of stimulated fifth-order arteries was restricted to every fourth, conducted hyperpolarizing responses decayed extensively as they ascended (Fig. 7). This enhanced decay occurs because the majority of daughter branches are unstimulated, a scenario which creates an electrochemical gradient for downstream charge flow. Increasing the number of stimulated distal arteries to every second or all effectively enhanced the conduction of charge into all proximal arteries (Fig. 7). This ascension of hyperpolarization through the vascular tree should in theory induce greater relaxation, lower arterial resistance, and increase network blood flow. To better draw this relationship, an existing blood flow model was applied to our symmetrical arterial network. In simulations where every fourth distal artery was hyperpolarized, robust dilatory responses were principally observed in the stimulated fifth-order artery, its upstream fourth-order parent, and the adjacent fifth-order unstimulated daughter vessel (Fig. 8). This stimulation pattern increased network blood flow approximately twofold, with the rise primarily directed to regions near the stimulated distal artery (Fig. 8). Increasing the number of stimulated distal arteries from 4 to 16 promoted the upward spread of vasodilation and fostered a stepwise increase in the magnitude of tissue blood flow. The preceding findings are consistent with the general concept of ascending vasodilation, a physiological behavior that was first described in skeletal muscle to explain the progressive increase in blood flow that accompanies tissue activity (11; 29).
Disease states: impact on electrical communication and blood flow control.
With the preceding computational approaches replicating known vascular behaviors, this study subsequently examined how disease states might impact electrical communication and blood flow control. Our initial exploration focused on sepsis, a chronic condition that facilitates organ failure by reducing tissue perfusion (19). Past studies have suggested that this diminishment might arise, in part, from the inability of hyperpolarization to ascend through the arterial tree due to an increase in endothelial-to-endothelial coupling resistance (1; 2; 21). The latter rise in coupling resistance can be induced by lipopolysaccharides, potent activators of inflammatory cells (21). To corroborate this novel perspective, the symmetrical network was deployed, all distal arteries stimulated, and the spread of hyperpolarization monitored under conditions in which endothelial-endothelial coupling resistance was elevated from ∼1.5 to 6.0 MΩFig. 9). Simulations highlighted that when coupling resistance was elevated across an arterial network, the ascension of hyperpolarization into proximal vessels was compromised. This increase in longitudinal resistivity limited ascending dilation and reduced network perfusion by ∼33%, as calculated from blood flow modeling (Fig. 10). These findings indicate that dramatic reductions in tissue perfusion can be driven by modest changes in coupling resistivity if they are induced on a network-wide level.
In contrast with sepsis which has system-wide effects, others disease states induce focal changes in endothelial function. For example, in regions downstream from branch points where turbulent blood flow occurs, endothelial disruption is prominent and such sites are predisposed to the formation of atherosclerotic plaques (15; 18; 20). With this in mind, simulations were performed in which the upward spread of hyperpolarization was mapped in the absence and presence of an endothelial disruption site placed downstream from the first-second order branch point. Consistent with the computational findings in Fig. 3, focal endothelial disruption near a branch point altered the pattern of electrical conduction (Fig. 11). More specifically, focal endothelial disruption enhanced hyperpolarization in branches downstream from the ablation site where, above, electrical responses were attenuated. Such finding was expected as the disruption site seals and separates the network into two unequal parts. Consequently, if comparable charge is introduced into each half, the smaller will hyperpolarize to a greater degree. Although the disruption of electrical communication translated into predictable changes in the conducted vasomotor response, the effects on network perfusion were limited. These observations suggest that focal disease-induced changes in endothelial function are by themselves unlikely to dramatically compromise tissue blood supply unless accompanied by a substantive physical plaque.
In presenting these observations, we acknowledge two important limitations. First, we recognize that vessels may possess more than one layer of smooth muscle. These additional layers will understandingly enhance conduction decay by increasing electrical load, akin to the discussion in this study of branch points and vessel length. Second, we recognize that vasomotor responses are used as an indirect index of arterial Vm changes. This practice is valid as long as the deployed stimulus initiates electromechanical coupling and elicits submaximal responses that display vasomotor decay along the length of the vessel (4; 8).
Summary.
This study revealed that arterial structure is an important determinant in shaping electrical communication in single vessels and networks. Specifically, we noted that vessel lengthening and the introduction of branch points diminished conducted responses due to augmented electrical load. The attenuating effect of branch points does not, however, prevent electrical responses from ascending in a vascular tree and eliciting profound changes in tissue perfusion. Disease states that induce network-wide rather than focal changes in endothelial coupling impair this ascension process along with blood flow responsiveness.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: V.C. and A.A.G. conception and design of research; V.C. and A.A.G. performed experiments; V.C. and A.A.G. analyzed data; V.C. and A.A.G. interpreted results of experiments; V.C. and A.A.G. prepared figures; V.C. and A.A.G. drafted manuscript; V.C. and A.A.G. edited and revised manuscript; V.C. and A.A.G. approved final version of manuscript.
REFERENCES
- 1. Bolon ML, Kidder GM, Simon AM, Tyml K. Lipopolysaccharide reduces electrical coupling in microvascular endothelial cells by targeting connexin40 in a tyrosine-, ERK1/2-, PKA-, and PKC-dependent manner. J Cell Physiol 211: 159–166, 2007 [DOI] [PubMed] [Google Scholar]
- 2. Bolon ML, Peng T, Kidder GM, Tyml K. Lipopolysaccharide plus hypoxia and reoxygenation synergistically reduce electrical coupling between microvascular endothelial cells by dephosphorylating connexin40. J Cell Physiol 217: 350–359, 2008 [DOI] [PubMed] [Google Scholar]
- 3. Coleman HA, Tare M, Parkington HC. EDHF is not K+ but may be due to spread of current from the endothelium in guinea pig arterioles. Am J Physiol Heart Circ Physiol 280: H2478–H2483, 2001 [DOI] [PubMed] [Google Scholar]
- 4. Diep HK, Vigmond EJ, Segal SS, Welsh DG. Defining electrical communication in skeletal muscle resistance arteries: a computational approach. J Physiol 568: 267–281, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Domeier TL, Segal SS. Electromechanical and pharmacomechanical signalling pathways for conducted vasodilatation along endothelium of hamster feed arteries. J Physiol 579: 175–186, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dora KA, Martin PE, Chaytor AT, Evans WH, Garland CJ, Griffith TM. Role of heterocellular Gap junctional communication in endothelium-dependent smooth muscle hyperpolarization: inhibition by a connexin-mimetic peptide. Biochem Biophys Res Commun 254: 27–31, 1999 [DOI] [PubMed] [Google Scholar]
- 7. Emerson GG, Segal SS. Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res 87: 474–479, 2000 [DOI] [PubMed] [Google Scholar]
- 8. Emerson GG, Segal SS. Endothelial cell pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circ Res 86: 94–100, 2000 [DOI] [PubMed] [Google Scholar]
- 9. Goldman D, Popel AS. A computational study of the effect of capillary network anastomoses and tortuosity on oxygen transport. J Theor Biol 206: 181–194, 2000 [DOI] [PubMed] [Google Scholar]
- 10. Hill CE, Rummery N, Hickey H, Sandow SL. Heterogeneity in the distribution of vascular gap junctions and connexins: implications for function. Clin Exp Pharmacol Physiol 29: 620–625, 2002 [DOI] [PubMed] [Google Scholar]
- 11. Hilton SM. A peripheral arterial conducting mechanism underlying dilatation of the femoral artery and concerned in functional vasodilatation in skeletal muscle. J Physiol 149: 93–111, 1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jantzi MC, Brett SE, Jackson WF, Corteling R, Vigmond EJ, Welsh DG. Inward rectifying potassium channels facilitate cell-to-cell communication in hamster retractor muscle feed arteries. Am J Physiol Heart Circ Physiol 291: H1319–H1328, 2006 [DOI] [PubMed] [Google Scholar]
- 13. Kassab GS, Rider CA, Tang NJ, Fung YC. Morphometry of pig coronary arterial trees. Am J Physiol Heart Circ Physiol 265: H350–H365, 1993 [DOI] [PubMed] [Google Scholar]
- 14. Krogh A. Studies on the capillariometer mechanism: I. The reaction to stimuli and the innervation of the blood vessels in the tongue of the frog. J Physiol 53: 399–419, 1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ku DN, Giddens DP, Zarins CK, Glagov S. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis 5: 293–302, 1985 [DOI] [PubMed] [Google Scholar]
- 16. Kurjiaka DT, Bender SB, Nye DD, Wiehler WB, Welsh DG. Hypertension attenuates cell-to-cell communication in hamster retractor muscle feed arteries. Am J Physiol Heart Circ Physiol 288: H861–H870, 2005 [DOI] [PubMed] [Google Scholar]
- 17. Kurjiaka DT, Segal SS. Conducted vasodilation elevates flow in arteriole networks of hamster striated muscle. Am J Physiol Heart Circ Physiol 269: H1723–H1728, 1995 [DOI] [PubMed] [Google Scholar]
- 18. Kwak BR, Mulhaupt F, Veillard N, Gros DB, Mach F. Altered pattern of vascular connexin expression in atherosclerotic plaques. Arterioscler Thromb Vasc Biol 22: 225–230, 2002 [DOI] [PubMed] [Google Scholar]
- 19. Lam C, Tyml K, Martin C, Sibbald W. Microvascular perfusion is impaired in a rat model of normotensive sepsis. J Clin Invest 94: 2077–2083, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Le Brocq M, Leslie SJ, Milliken P, Megson IL. Endothelial dysfunction: from molecular mechanisms to measurement, clinical implications, and therapeutic opportunities. Antioxid Redox Signal 10: 1631–1674, 2008 [DOI] [PubMed] [Google Scholar]
- 21. Lidington D, Ouellette Y, Tyml K. Endotoxin increases intercellular resistance in microvascular endothelial cells by a tyrosine kinase pathway. J Cell Physiol 185: 117–125, 2000 [DOI] [PubMed] [Google Scholar]
- 22. Little TL, Beyer EC, Duling BR. Connexin 43 and connexin 40 gap junctional proteins are present in arteriolar smooth muscle and endothelium in vivo. Am J Physiol Heart Circ Physiol 268: H729–H739, 1995 [DOI] [PubMed] [Google Scholar]
- 23. Moore JA, Appenteng K. The morphology and electrical geometry of rat jaw-elevator motoneurones. J Physiol 440: 325–343, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pries AR, Secomb TW, Gaehtgens P, Gross JF. Blood flow in microvascular networks. Experiments and simulation. Circ Res 67: 826–834, 1990 [DOI] [PubMed] [Google Scholar]
- 25. Pries AR, Secomb TW, Gessner T, Sperandio MB, Gross JF, Gaehtgens P. Resistance to blood flow in microvessels in vivo. Circ Res 75: 904–915, 1994 [DOI] [PubMed] [Google Scholar]
- 26. Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ Res 86: 341–346, 2000 [DOI] [PubMed] [Google Scholar]
- 27. Segal SS, Beny JL. Intracellular recording and dye transfer in arterioles during blood flow control. Am J Physiol Heart Circ Physiol 263: H1–H7, 1992 [DOI] [PubMed] [Google Scholar]
- 28. Segal SS, Duling BR. Flow control among microvessels coordinated by intercellular conduction. Science 234: 868–870, 1986 [DOI] [PubMed] [Google Scholar]
- 29. Segal SS, Jacobs TL. Role for endothelial cell conduction in ascending vasodilatation and exercise hyperaemia in hamster skeletal muscle. J Physiol 536: 937–946, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Segal SS, Neild TO. Conducted depolarization in arteriole networks of the guinea-pig small intestine: effect of branching of signal dissipation. J Physiol 496: 229–244, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tran CH, Vigmond EJ, Plane F, Welsh DG. Mechanistic basis of differential conduction in skeletal muscle arteries. J Physiol 587: 1301–1318, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vanbavel E, Spaan JA. Branching patterns in the porcine coronary arterial tree. Estimation of flow heterogeneity. Circ Res 71: 1200–1212, 1992 [DOI] [PubMed] [Google Scholar]
- 33. Welsh DG, Segal SS. Endothelial and smooth muscle cell conduction in arterioles controlling blood flow. Am J Physiol Heart Circ Physiol 274: H178–H186, 1998 [DOI] [PubMed] [Google Scholar]
- 34. Yamamoto Y, Klemm MF, Edwards FR, Suzuki H. Intercellular electrical communication among smooth muscle and endothelial cells in guinea-pig mesenteric arterioles. J Physiol 535: 181–195, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]