

Abstract
Nicotinamide phosphoribosyltransferase (Nampt) is an important coenzyme involved in cellular redox reactions. Inside the cell, Nampt (iNampt) functions as a rate-limiting enzyme in the NAD salvage pathway, and outside the cell (eNampt), it acts as a proinflammatory cytokine. High-circulating levels of Nampt are reported in different pathological conditions. This study was designed to examine the role of Nampt in the development of cardiac hypertrophy and ventricular remodeling. We studied the hypertrophic response in Nampt heterozygous (+/−) knockout and cardiac-specific overexpressing Nampt transgenic mice. Whereas Nampt+/− mice were protected against agonist (isoproterenol and angiotensin II)-induced hypertrophy, Nampt transgenic mice spontaneously developed cardiac hypertrophy at 6 mo of age. Experiments conducted to gain insight into the mechanism revealed that treatment of cardiomyocytes with recombinant (eNampt) or overexpression with Nampt-synthesizing adenovirus vector (Ad.Nampt) induced cardiomyocyte hypertrophy. The prohypertrophic effects of eNampt and Ad.Nampt were blocked by the addition of a Nampt-blocking antibody into cultures, thus suggesting that Nampt was in fact invoking hypertrophic response of cardiomyocytes by acting on the cell surface receptors. We also found increased Nampt levels in the supernatant of cardiomyocyte cultures subjected to stress by either serum starvation or H2O2 treatment. Exploration of signaling pathways in Nampt-induced cardiac hypertrophy and fibrosis revealed increased activation of mitogen-activated protein kinases, namely, JNK1, p38, and ERK. This was also associated with increased calcineurin levels and nuclear factor of activated T-cell localization into the nucleus. From these studies we conclude that cardiomyocytes are capable of secreting Nampt during stress, and exogenous Nampt is a positive regulator of cardiac hypertrophy and adverse ventricular remodeling.
Keywords: cytokines, hypertrophy, inflammation, Nampt, remodeling
in conditions of sustained increase of workload and/or activation of neurohumoral factors, the heart increases ventricular muscle mass because of hypertrophy of cardiomyocytes. This change may be initially a compensatory response to increased workload on the heart; however, sustained increase in workload and/or activation of neurohumoral factors leads to induction of pathological hypertrophy, which is associated with cardiomyocyte growth and consequent induction of fetal gene program, apoptosis, and interstitial fibrosis, a phenomenon called adverse ventricular remodeling, an important precursor of heart failure. A multitude of factors like obesity, hypertension, coronary artery diseases, diabetes, valvular insufficiency, and stenosis can act solely or together to cause adverse ventricular remodeling (28, 32). At the cellular level ventricular remodeling is induced by biomechanical stretch of cardiomyocytes and/or release of hormones like adipokines, chemokines, cytokines, and growth factors. These factors, whose source can be exogenous or endogenous, act topically on cardiomyocytes to regulate their growth and survival. Persistent stress can cause imbalance in their secretion, leading to aberrant growth of cardiomyocyte and induction of adverse ventricular remodeling that progresses to heart failure.
Nicotinamide phosphoribosyltransferase (Nampt) is a proinflammatory cytokine that has gained considerable attention in recent years with respect to induction of cardiovascular diseases (30). Other than being a cytokine, Nampt is also a key enzyme necessary for the nicotinamide adenine dinucleotide (NAD) synthesis, thus making it unique among the group of cytokines. Although NAD is synthesized de novo from tryptophan, the majority of it is synthesized from a salvage pathway that starts with nicotinamide or nicotinic acid. In mammals, Nampt catalyzes the rate-limiting step of converting nicotinamide to nicotinamide mononucleotide (NMN), which is then converted to NAD with the help of another enzyme, Nmnat (15). The role of Nampt as a NAD-synthesizing enzyme is cardinal in combating stress since severe stress can cause NAD depletion because of direct loss of NAD from cells or because of increased activity of NAD-consuming enzymes like poly(ADP-ribose) polymerase, sirtuins, and cyclic-ADP ribose synthases (13, 31). This is particularly important in the setting of cardiac hypertrophy because pathological hypertrophy is associated with reduced levels of NAD (31). Thus, inside the cell, Nampt (iNampt) helps to maintain intracellular NAD levels and hence participates in regulating activity of several enzymes implicated in mediating cell survival, stress resistance, and regulation of metabolism.
Like many other cytokines, Nampt is released into circulation by a wide variety of cell types including adipose tissue, liver, fetal membranes, and lymphocytes (10, 21, 34). Contrary to its role inside the cell, extracellular Nampt (eNampt) acts as a proinflammatory adipokine, cytokine, and growth factor (33, 41). It was first reported to be secreted from visceral fat (visfatin), and, subsequently, it was found to be identical to the pre-cell colony enhancing factor (PBEF), a cytokine acting on early B lineage precursor cells. These characteristics of Nampt suggested that it can act locally and distally through systemic circulation. Elevated levels of circulating Nampt were reported in different clinical conditions, such as obesity, type 2 diabetes, and chronic inflammatory diseases (12, 30). Upregulation of Nampt was also observed in response to hypoxia (35). In the context of cardiovascular diseases, a strong correlation exists between Nampt expression and atherosclerosis (19). Nampt was found to induce endothelial dysfunction and contribute to destabilization of atheromatous plaque. Patients with coronary artery disease were reported to have increased epicardial Nampt levels, compared with control subjects (37). So far, three studies have addressed the direct effect of Nampt in the heart. Lim at al. (18) reported that addition of Nampt at the time of myocardial reperfusion significantly reduced myocardial infract size. Similarly, cardiac-specific expression of Nampt reduced myocardial infract size in response to prolonged ischemia and ischemia-reperfusion injury (14). In contrast to these findings, inhibition of Nampt reduced neutrophil-mediated injury in myocardial infarction (25). Besides this, the role of Nampt in the development of cardiac hypertrophy and ventricular remodeling has never been investigated.
In this study, we show for the first time that cardiomyocytes can secrete Nampt under stress conditions, and exogenous Nampt causes cardiac hypertrophy and fibrosis, which can be blocked by use of a Nampt-specific antibody. Cardiac-specific overexpression of Nampt induced pathological hypertrophy and cardiac failure. The prohypertrophic effect of Nampt was found to be mediated through a calcineurin/nuclear factor of activated T-cell (NFAT) signaling pathway. On the basis of these results here we describe the pathological consequences of the sustained activation of a prosurvival molecule, Nampt.
MATERIALS AND METHODS
Primary cultures of cardiomyocytes, transfection/infection, and luciferase assay.
Neonatal rat cardiomyocytes were cultured and infected with adenoviral vectors as described earlier (38). Luciferase activity assay was performed using Luciferase activity assay kit from Promega according to the manufacturer's protocol. The COOH-terminal human influenza hemagglutinin epitope-tagged Nampt was generated from pCXN2 Nampt (mouse) plasmid by Vector Biolabs. For determination of cell size, ImageJ software was used to calculate cardiomyocyte surface area of at least 50 cells (actinin-positive cells) per experimental group. Cardiomyocyte size was expressed as fold change with respect to untreated control.
Induction of hypertrophy in mice.
Angiotensin II or isoproterenol, dissolved in 150 mM NaCl and 1 mM acetic acid, were delivered chronically at a rate of 3.0 mg·kg body wt−1·day−1 for 14 days or 8.7 mg·kg body wt−1·day−1 for 7 days, respectively, by implanting osmotic mini pumps (model 2002, ALZET) in the peritoneal cavity of mice. Control mice underwent the same procedure, except that the respective pumps were filled only with vehicle (150 mM NaCl and 1 mM acetic acid).
Histology and immunohistochemistry.
Fibrosis was detected using Masson's trichrome stain kit from Sigma according to the manufacturer's protocol. FITC-conjugated anti-CD31antibody (Millipore) was used for determination of capillary density. Terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) staining was performed using dead-end colorimetric TUNEL system (Promega) as per manufacturer's instruction. Cardiomyocyte staining and atrial natriuretic factor (ANF) release were measured by staining with antibodies specific for α-actinin and ANF, respectively.
Real-time PCR analysis of mRNA levels.
β-Myosin heavy chain (MHC) and collagen-1a1 and smooth muscle actin mRNA levels were quantified by SYBR green real-time PCR as per the protocol previously described (8). Total RNA was isolated from hearts by using TRIzol reagent (Invitrogen). Residual genomic DNA was digested by incubating the RNA preparation with 0.5 units of RNase-free Dnase-1 per microgram of RNA in 1× reaction buffer for 15 min at room temperature, followed by heat inactivation at 90° for 5 min. The quality of Dnase-1-treated RNA was tested by formaldehyde agarose gel electrophoresis. Two micrograms of DNase-treated RNA was reverse transcribed using the Superscript III (Invitrogen). The resultant cDNA was diluted 10-fold before PCR amplification. A reverse transcriptase minus reaction served as a negative control.
[3H]leucine incorporation.
Immediately after the treatment of cardiomyocytes with phenylephrine, cells were incubated with [3H]leucine (1.0 mCi/ml and 163 Ci/mmol specific activity, Amersham Biosciences) in leucine-free MEM medium (Invitrogen) for 48 h. Cells were harvested, washed with PBS, and then incubated in 10% trichloro acetic acid to precipitate proteins. The resultant pellet was solubilized in 0.2 N NaOH, and the lysates were diluted with one-sixth volume of scintillation fluid and counted in a scintillation counter. Values were normalized with DNA content, which was measured by using Quant-iT picogreen dsDNA assay kit (Invitrogen).
Antibodies used.
Nampt antibody was purchased from Alexis, GAPDH and activating transcription factor-2 antibody were purchased from Santa Cruz, and all other antibodies were purchased from Cell Signaling. Ployclonal PBEF (Nampt) neutralizing antibody was generated in Garcia's laboratory (12).
Nampt ELISA.
Nampt ELISA was performed using Nampt dual ELISA kit from AdipoGen as per the manufacturer's instruction.
3-(4,5-Dimethylthiazol-2-yl)-diphenyltetrazolium bromide assay.
Cardiac fibroblasts (1 × 104) were plated in 96-well plate and grown until 70–80% confluent, synchronized in medium containing 0.1% FCS, and then stimulated with 100 ng/ml recombinant Nampt (eNampt) in the presence or absence of 1 mg/ml neutralizing antibody. Media was replenished every 12 h for 48 h. 3-(4,5-Dimethylthiazol-2-yl)-diphenyltetrazolium bromide (MTT) was added to each well under sterile conditions (with a final concentration of 0.5 mg/ml), and the plates were incubated for 4 h at 37°C. Untransformed MTT was removed by aspiration, and formazan crystals were dissolved in dimethyl sulfoxide (200 μl/well). Formazan was quantified spectroscopically at 560 nm. The experiments were performed in triplicate with different preparations of fibroblasts.
Imaging of cardiac fibroblasts.
Cardiac fibroblasts on 12-mm coverslips were treated with recombinant Nampt or infected with adenovirus vector (Ad.Nampt) for 72 h. Cells were washed with PBS and fixed with 3.7% formaldehyde in PBS for 15 min, followed by permeabilization with 0.1% Triton X-100 for 5 min. It is then blocked with 10% BSA in PBS, followed by primary antibody overnight at 4°C and thereafter with either Alexa Fluor 594 or FITC-conjugated secondary antibody for 1 h. Cells were washed and mounted in ProLong Gold antifade reagent with 4,6-diamidino-2-phenylindole. Cells were visualized using a Leica SP2 laser-scanning microscope.
Nampt knockout and transgenic mice.
Homozygous Nampt (−/−) mice do not survive. Hemizygous Nampt knockout (+/−) mice were generously provided by Garcia's laboratory (12). With transgenic (Tg) mice generation, all animal protocols were reviewed and approved by the University of Chicago Institutional Animal Care and Use Committee. Mice with cardiac-restricted expression of Nampt were generated by amplifying DNA from plasmid encoding Nampt using the following primers, 5′-AATGTCGACGCCCGAGATGAATGCTG-3′ and 5′-GATAAGCTTCTATTATCAAGCGTAATCTGGAACATCGTATGGGTAATGAGGTGCCACGTCCTGCTC-3′, which contain restriction sites for SalI and HindIII, respectively. The amplified PCR fragment was cloned directly into SalI and HindIII restriction sites of expression plasmid, containing the pBluescript II KS plasmid (Stratagene), containing α-MHC promoter and human growth hormone poly(A) signal (a gift from J. Robbins, Cincinnati Children's Hospital, Cincinnati, OH). The orientation of Tg construct was confirmed by sequencing. Constructs were linearized and the PBS backbone was digested out of the plasmid with the use of the enzyme NotI. The purified DNA construct was injected in the pronuclear stage zygotes of the CD1 mouse strain, according to the standard Tg procedure of the University of Chicago Tg facility. At 2 to 3 wk of age, tail DNA was analyzed to confirm which mice were Tg. The following primers were used for genotyping: forward, 5′-CTTCCAGCCCTCTCTTTCTC-3′; reverse, CTGTACACTTCTTTGGCCTC; forward, 5′-TTCACATTGCATGTGTGG-3′; and reverse, 5′-TAGCCTGCGTAGTGTTGGTG-3′.
Echocardiography of mice.
Chest hairs of mice were removed with a topical depilatory agent, and transthoracic echocardiography was performed under inhaled isoflurane (∼1%) for anesthesia, delivered via nose cone. Limb leads were attached for electrocardiogram gating, and the animals were imaged in the left lateral decubitus position with a VisualSonics Vevo 770 machine, using a 30-MHz high-frequency transducer. Body temperature was maintained using a heated imaging platform and warming lamps. Two-dimensional images were recorded in parasternal long- and short-axis projections, with guided M-mode recordings at the midventricular level in both views. Left ventricular (LV) cavity size and wall thickness were measured in at least three beats from each projection and averaged. LV wall thickness [interventricular septum (IVS) and posterior wall (PW) thickness] and internal dimensions at diastole and systole (LVIDd and LVIDs, respectively) were measured. LV fractional shortening ([LVIDd − LVIDs]/LVIDd) and relative wall thickness [(IVS thickness + PW thickness)/LVIDd] were calculated from the M-mode measurements.
Statistical analysis.
Statistical differences among groups were determined with either Student's t-test (for 2 groups) or one-way analysis of variance. P values < 0.05 were considered significant.
RESULTS
Cardiac-specific expression of Nampt promotes cardiac hypertrophy and dysfunction: To determine the effect of Nampt in an intact heart, we generated cardiac-specific Nampt overexpressing (4-fold over control) Tg mice (Fig. 1A). Nampt-Tg mice produced nearly 25% cardiac hypertrophy (Fig. 1B), which was associated with accumulation of fibrosis in the interstitial space (Fig. 1, C and D). Nampt-Tg mice showed significantly reduced LV fractional shortening and increased wall thickness (Fig. 1, E and F). Nampt-Tg mice also showed significantly increased expression of β-MHC, collagen-1, and smooth muscle actin mRNAs in their hearts compared with control hearts (Fig. 1G). Furthermore, Nampt-Tg mice showed reduced capillary density compared with control mice as revealed by decreased CD31 staining (Fig. 1, H and I). These data thus suggest that Nampt-Tg mice spontaneously develop pathological cardiac hypertrophy.
Fig. 1.
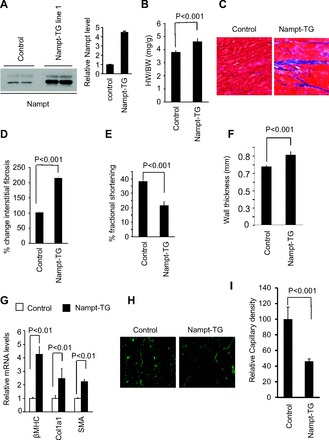
Extracellular nicotinamide phosphoribosyltransferase (eNampt) is prohypertrophic in vivo. A: cardiac Nampt levels in the nontransgenic (N-Tg) control and in the Nampt-Tg mice. B: heart weight (HW)-to-body weight (BW) ratio of control and Nampt-Tg mice. C: sections of hearts treated with Masson's trichrome to detect fibrosis (blue). D: quantification of cardiac fibrosis in control and Nampt-Tg mice. E and F: echocardiographic measurements of left ventricular fractional shortening and wall thickness of control and Nampt-Tg hearts. G: β-myosin heavy chain (β-MHC), collagen-1 (Col1a1), and α-smooth muscle actin (α-SMA) mRNA levels in the heart samples of control and Nampt-Tg mice. H: representative heart sections of control and Nampt-Tg mice showing CD31 staining. I: quantification of relative capillary density in 2 groups of hearts. Means ± SD; n = 5–7.
Nampt heterozygous knockout (+/−) mice are protected from agonist-induced cardiac hypertrophy.
To further examine the role of Nampt in the induction of hypertrophy, we used heterozygous Nampt knockout (Nampt+/−) mice which express half the amount of Nampt compared with their wild-type littermates (Fig. 2A). We subjected heterozygous Nampt+/− mice along with their wild-type controls to agonist-induced hypertrophy. At basal level Nampt+/− mice displayed similar heart weight body weight ratio, fractional shortening and ventricular wall thickness compared with wild-type control mice. Basal level of fibrosis was absent in both groups of mice. Isoproterenol infusion in control mice resulted in consistently elevated heart weight, body weight ratio and produced nearly 50% hypertrophy in control mice (Fig. 2B). This was associated with increased accumulation of fibrosis, increased ventricle wall thickness and reduced cardiac function (Fig. 2, C–E). These pathological changes were not seen in Nampt+/− mice infused with isoproterenol. The expression level of other hypertrophy markers such as mRNA levels of β-MHC, collagen-1, and smooth muscle α-actin were also elevated in isoproterenol-induced control mice, but not in Nampt+/− mice (Fig. 2F). Similarly, Nampt+/− mice were protected from developing angiotensin II-mediated cardiac hypertrophy (data not shown). These results thus confirmed that Nampt plays a crucial role in the induction of cardiac hypertrophic response.
Fig. 2.
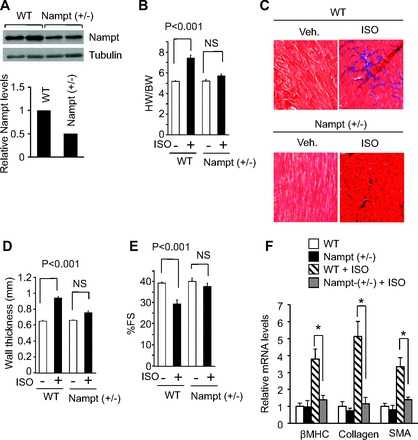
Nampt+/− hearts are protected from agonist-induced hypertrophy. A: cardiac Nampt levels in the wild-type (WT) control and Nampt+/− mice. B: HW-to-BW ratio of control and Nampt+/− mice infused with either vehicle (Veh) or isoproterenol (Iso) for 7 days. NS, not significant. C: Masson's trichrome-stained heart sections from WT and Nampt+/− mice infused with Iso. D and E: echocardiographic measurements of left ventricular wall thickness and fractional shortening of control and Nampt+/− mice. F: β-MHC Col1a1 and α-SMA mRNA levels in the heart samples of Iso infused or untreated WT and Nampt+/− mice. Means ± SD; n = 6. *Statistically significant.
Nampt is released from cardiomyocytes under stress.
Although first thought to be an adipokine, Nampt has been found to be secreted by a variety of cell types including liver, fetal membrane, and lymphocytes. Since Nampt was found to be proinflammatory and hence could be prohypertrophic, we first sought to investigate whether cardiomyocytes are a source of exogenous Nampt (eNampt). Primary cultures of cardiomyocytes were infected with 10 or 100 multiplicity of infection of adenovirus vector expressing Nampt for 48 h. Subsequently, supernatant was collected without disturbing layers of cells in the bottom, and it was analyzed for the presence of Nampt by ELISA. The results showed that Nampt was present in supernatants and its levels increased with expression level of the protein inside the cell (Fig. 3, A and B). Western blot analysis of the supernatant confirmed these results (Fig. 3C). To determine whether endogenous Nampt (iNampt) is also released from cells, cardiomyocytes were serum starved for 24 or 48 h and the supernatant was again analyzed for the presence of Nampt in the media as well as in cells. As shown, supernatants from cardiomyocytes subjected to 24-h serum starvation had a significantly higher concentration of Nampt compared with serum-fed cardiomyocytes, and it remained elevated up to 48 h of starvation (Fig. 3D). These cells also showed increased intracellular levels of Nampt (Fig. 3E). Since serum starvation is a stress condition, we investigated whether Nampt is released during severe stress of cardiomyocytes. For this purpose, we treated cardiomyocytes with a sublethal dose of H2O2 and checked the supernatant and cells lysates for the presence of Nampt at 1, 2, and 4 h following H2O2 treatment (Fig. 3, F and G). A linear increase of eNampt levels was observed in the supernatants, suggesting that eNampt is released from cardiomyocytes following oxidative stress. The cells from which supernatant was obtained were also stained with annexin V and analyzed by fluorescent-activated cell-sorting analysis, and the results showed an absence of cell death in all these experiments.
Fig. 3.
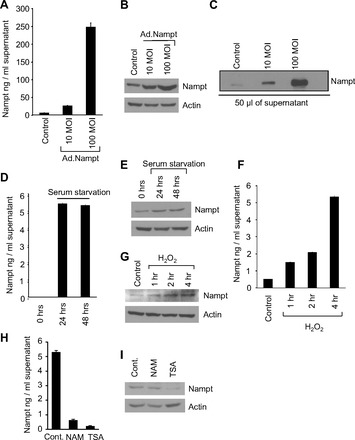
Nampt is released from cardiomyocytes under stress. A and B: primary cultures of cardiomyocytes were infected with 10 or 100 multiplicity of infection (MOI) of adenovirus expressing Nampt (Ad.Nampt) for 24 h. Cont, control. Supernatant was analyzed for the presence of Nampt by ELISA, and cells were analyzed for the Nampt expression levels by immunoblotting. C: equal amount of supernatants from different group of cells were analyzed for the presence of Nampt by immunoblotting with anti-Nampt antibody. D and E: primary cultures of cardiomyocytes were serum starved for 24 or 48 h, the supernatants were analyzed for the presence of Nampt by ELISA, and cells were analyzed for the Nampt expression levels by immunoblotting. F and G: primary cultures of cardiomyocytes, serum starved for 6 h, were treated with nonlethal dose of H2O2 (50 μM) for 1, 2, and 4 h. Supernatants were analyzed for the presence of Nampt by ELISA, and cells were analyzed for the Nampt expression levels by immunoblotting. H and I: primary cultures of cardiomyocytes were serum starved for 24 h and then treated with 25 mM nicotinamide (NAM) or 500 nM trichostatin A (TSA). After 24 h of treatment, supernatants were analyzed for the presence of Nampt by ELISA, and cells were analyzed for the Nampt expression levels by immunoblotting. Means ± SD; n = 4–6.
Nampt does not contain a classic secretory domain, suggesting that its secretion from cells must be regulated by a noncanonical mechanism. To understand the mechanism behind its release from cells, we treated cells with histone deacetylase inhibitors, trichostatin A, or nicotinamide. We found that both the treatments reduced the intracellular levels of Nampt and completely blocked Nampt release from cardiac myocytes after stress, suggesting that an acetylation-dependent mechanism is likely to participate in the release of Nampt from cardiomyocytes (Fig. 3, H and I).
eNampt is a prohypertrophic molecule.
Several studies have shown that agents that are cytoprotective are also prohypertrophic upon persistent activation. Therefore, to test whether eNampt can induce cardiomyocyte hypertrophy, we treated cardiomyocytes with recombinant eNampt (100 ng/ml). The hypertrophic response of cells was measured by increased protein synthesis as determined by increased [3H]leucine incorporation into total cellular proteins and by release of ANF from nuclei, a hallmark of cardiac hypertrophy. We found significantly higher [3H]leucine incorporation in eNampt-treated cardiomyocytes compared with controls (Fig. 4A). Phenylephrine was used as a positive control. An addition of a specific Nampt-blocking antibody to cultures prevented eNampt-mediated increased leucine incorporation into proteins, thus suggesting that eNampt is a prohypertrophic molecule. To confirm these results, we examined the effect of eNampt on the promoter activity of the hypertrophy signal-sensitive gene NFAT. The results indicated that eNampt treatment significantly induced the activation of NFAT promoter, thus again demonstrating the prohypertrophic potential of eNampt (Fig. 4B). This was further confirmed by the ANF release from cardiomyocyte nuclei (Fig. 4C) and increased cardiomyocyte size (Fig. 4D). We next examined the prohypertrophic effect of intracellular Nampt in cardiomyocytes. To this end cardiomyocytes were infected with adenovirus expressing Nampt and prohypertrophic response was measured by increased protein synthesis, NFAT activation, and ANF release (Fig. 4, A–D). Similar to eNampt treatment, infection with the Nampt-synthesizing adenovirus vector induced hypertrophic response in cardiomyocytes, and this was blocked by an addition of the Nampt-blocking antibody. Since very high levels of Nampt were detected in supernatants from adenovirus-infected cardiomyocytes, these results suggested that Nampt-adenovirus infection caused release of Nampt into the supernatant that, in turn, induced hypertrophic response of cardiomyocytes.
Fig. 4.
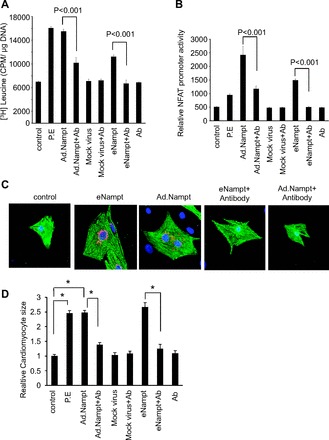
eNampt is prohypertrophic in vitro. A: primary cultures of cardiomyocytes labeled with [3H]leucine were treated with 100 ng/ml recombinant eNampt or infected with 10 MOI of Ad.Nampt (or mock virus) in the presence or absence of Nampt blocking antibody (Ab). Forty-eight hours after treatment, cells were harvested and incorporation of [3H]leucine into total cellular proteins was measured. cpm, Count per minute. B: cardiomyocytes were infected with a nuclear factor of activated T-cell (NFAT) responsive promoter-luciferase reporter adenovirus vector. Twelve hours after infection, cells were treated with recombinant eNampt in the presence or absence of Nampt-blocking antibody. Ad.Nampt virus infection was performed 12 h before NFAT adenovirus infection. C: cardiomyocytes were treated with recombinant Nampt or infected with Ad.Nampt in the presence or absence of Nampt-blocking antibody. Cardiomyocytes were identified by α-actinin staining (green). The release of atrial natriuretic factor (ANF) from nuclei was determined by staining cells with an anti-ANF antibody (red). 4,6-Diamidino-2-phenylindole stain was used to mark the position of nuclei. In all, these experiments cardiomyocytes treated with 20 μM phenylephrine (PE), and mock virus, and antibody alone served as controls. D: size of cardiomyocytes identified by α-actinin staining was quantified using ImageJ software. Means ± SD; n = 5. *P < 0.001.
Nampt activates hypertrophy via activation of calcineurin-NFAT and MAPK signaling pathways.
Since increased transcription of fetal genes is associated with nuclear translocation of NFAT, we analyzed the nuclear fraction of heart lysates for the presence of NFAT by immunoblotting. Increased abundance of NFAT was observed in Nampt-Tg mice (Fig. 5A). This was associated with increased calcineurin levels in the whole cell lysate of Nampt-Tg hearts. In the heart, increased calcineurin levels were shown to activate MAPK signaling (23). We therefore examined the three main branches of MAPK signaling cascade: ERK, JNK, and p38. The results showed significant activation of JNK1, ERK, and p38. Furthermore, activating transcription factor 2, the downstream target of JNK1 and p38, was also found to be activated (Fig. 5B). Since increased activation of Akt is also reported during development of cardiac hypertrophy, we examined the Akt activation and its downstream target GSK3-β. No notable difference in Akt and GSK3-β activation was detected between wild-type and Nampt-Tg hearts (Fig. 5B). In addition to the induction of cardiac hypertrophy, increased activation of MAPK signaling is associated with cardiomyocyte apoptosis. To gain insight into this, we tested the expression of apoptotic markers in wild-type and Nampt-Tg hearts. As expected, increased abundance of FAS, Bax, Bim, and TNF-related apoptosis-inducing ligand was found in Nampt-Tg hearts (Fig. 5C). This was further confirmed by the increased level of TUNEL-positive cells in Nampt-Tg mice, compared with wild-type controls (Fig. 5D). These data suggested that Nampt induces cardiac hypertrophy by augmenting the activity of MAPK signaling cascade, which also induced cardiomyocyte apoptosis.
Fig. 5.
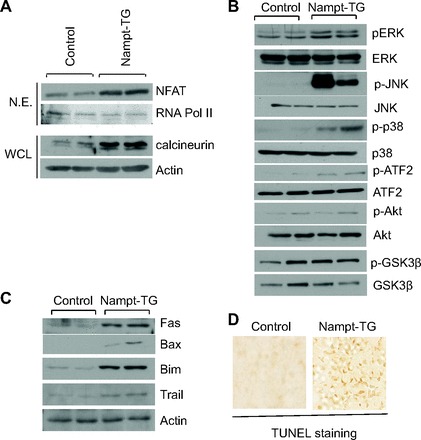
Nampt induces cardiac hypertrophy via activation of MAPK pathway. A: nuclear extract (NE) and whole cell lysates (WCL) of control and Nampt-Tg mice hearts were analyzed by immunoblotting for the expression of NFAT and calcineurin, respectively. Results of 2 hearts in each group are shown. Poll II, polymerase II. B: heart extracts prepared from N-Tg control and Nampt-Tg mice were analyzed by Western blot analysis with antibodies as indicated. p-ATF2, phospho-activating transcription factor 2. C: heart extracts prepared from N-Tg control and Nampt-Tg mice were analyzed by Western blot analysis with apoptotic marker antibodies. D: heart sections from N-Tg control and Nampt-Tg mice subjected to terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) staining.
We then asked whether similar signaling mechanism operates in cardiac hypertrophy-induced by eNampt in vitro (Fig. 6A). The results showed that same as in vivo, cardiomyocytes treated with recombinant Nampt (eNampt) or infected with adenovirus vectors expressing Nampt (iNampt) showed increased NFAT accumulation in the nucleus and increased level of calcineurin in whole cell lysate (Fig. 6A). To gain further evidence for the involvement of calcineurin-Nampt and MAPK signaling pathways in the induction of hypertrophy, we examined the effect of Nampt in the presence of FK506, a calcineurin inhibitor. Cells were pretreated with FK506 and then stimulated with recombinant Nampt or transduced with Nampt adenovirus vector. Analysis of cell lysates revealed that both recombinant Nampt and Ad.Nampt activated the MAPK signaling pathway, whereas treatment with FK506 blocked its hyperactivation (Fig. 6B). The hypertrophic response of cardiomyocytes was examined by [3H]leucine incorporation into total cellular protein. Treatment with FK506 blocked both iNampt and eNampt induced [3H]leucine incorporation (Fig. 6C) and activation of NFAT promoter (Fig. 6D). Furthermore, FK506-treated cells showed reduced cardiomyocyte cell size when treated with recombinant Nampt and Ad.Nampt (Fig. 6E). These data thus confirmed that Nampt-induced cardiomyocyte hypertrophy is indeed mediated through calcineurin-NFAT and MAPK signaling pathways.
Fig. 6.
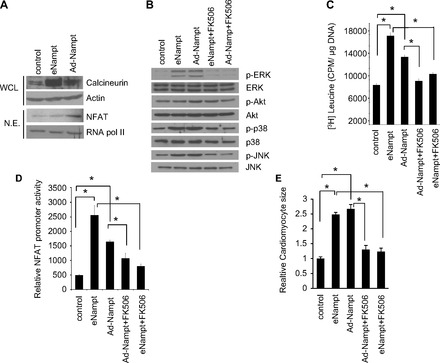
Calcineurin inhibitor blocks Nampt-induced hypertrophy. A: NE or WCL from cardiomyocytes treated with recombinant Nampt or Ad.Nampt were analyzed by Western blot analysis with antibodies as indicated. B: lysates from cardiomyocytes treated with recombinant Nampt (eNampt) or Ad.Nampt in the presence or absence of FK506 were analyzed by Western blot analysis with antibodies as indicated. C: primary cultures of cardiomyocytes labeled with [3H]leucine were treated with 100 ng/ml recombinant Nampt (eNampt) or infected with 10 MOI of Ad.Nampt in the presence or absence of 100 ng/ml FK506. Forty-eight hours after treatment, cells were harvested and incorporation of [3H]leucine into total cellular proteins was measured. D: cardiomyocytes were infected with a NFAT responsive promoter-luciferase reporter adenovirus vector. Twelve hours after infection, cells were treated with recombinant eNampt in the presence or absence of 100 ng/ml FK506. Ad.Nampt virus infection was performed 12 h before NFAT adenovirus infection. E: cardiomyocytes were identified by α-actinin staining, and cardiomyocyte cell size was quantified. Means ± SD; n = 4. *P < 0.001.
Nampt regulates cardiac fibroblast proliferation and differentiation.
An important component of myocardial remodeling is activation of cardiac fibroblast proliferation and their transformation into myofibroblasts. For the reason that cardiac-specific Nampt-Tg mice showed increased fibrosis, we next investigated the effect of Nampt on cardiac fibroblast proliferation, differentiation, and apoptosis. Proliferation of cardiac fibroblast in the presence of recombinant Nampt and adenoviral-transduced Nampt was assessed by MTT assay. Both recombinant and Ad.Nampt increased cardiac fibroblast proliferation, which was inhibited by Nampt-blocking antibody (Fig. 7A). We next determined the effect of Nampt on transformation of cardiac fibroblast to myofibroblasts. α-Smooth muscle actin and collagen 1 are critical determinants of myofibroblast differentiation. We observed marked increase of these markers in fibroblast treated or infected with Nampt, suggesting that Nampt promotes myofibroblast differentiation (Fig. 7B). These observations were confirmed by Western blot analysis (Fig. 7C). We next determined whether Nampt has any role in protecting cardiac fibroblasts from apoptosis induced by oxidative stress. To this end, we treated cardiac fibroblasts with a cell death-inducing dose of H2O2 in the presence or absence of Nampt and measured the apoptosis by Annexin V staining followed by fluorescent-activated cell-sorting analysis. Apoptosis was found to be reduced by 50% in eNampt-treated cardiac fibroblasts, suggesting that Nampt blocks myofibroblast apoptosis (Fig. 7, D and E). These findings corroborate with our observation that Nampt-Tg mice showed extensive fibrosis in the heart. Together, these data suggest that chronic overexpression of Nampt is deleterious to cardiomyocytes but favors the growth and differentiation of cardiac fibroblasts.
Fig. 7.
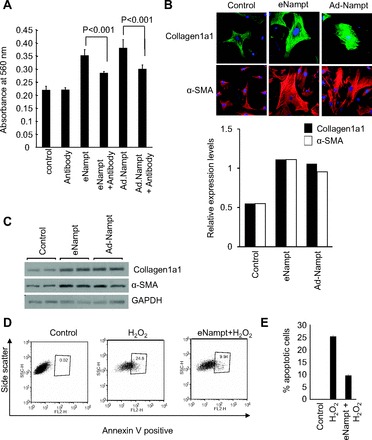
Nampt regulates cardiac fibroblast proliferation, differentiation, and apoptosis. A: primary cultures of cardiac fibroblasts were treated with 100 ng/ml Nampt or infected with Ad.Nampt, and cell proliferation was measured by 3-(4,5-dimethylthiazol-2-yl)-diphenyltetrazolium bromide (MTT) assay after 48 h of plating. Means ± SD; n = 5. B: primary cultures of cardiac fibroblasts were treated with 100 ng/ml recombinant eNampt or infected with 10 MOI of Ad.Nampt. Cells were immunostained for collagen 1 (green) and α-SMA (red). C: primary cultures of cardiac fibroblasts were treated with 100 ng/ml recombinant eNampt or infected with 10 MOI of Ad.Nampt. Cell lysates were analyzed by Western blot analysis with antibodies as indicated. D: primary cultures of cardiac fibroblasts were treated with cell death-inducing dose (500 μM) of H2O2 in the presence or absence of recombinant Nampt for 1 h. Extent of apoptosis was measured by estimating the percentage of annexin V-positive cells by fluorescent-activated cell-sorting analysis. E: results of 4 independent experiments. Means ± SD; n = 4.
DISCUSSION
This study was designed to investigate the consequence of continuous expression of Nampt in the heart. By using both in vitro and in vivo models, we show that Nampt induces cardiac hypertrophic response. Nampt-Tg mice spontaneously developed pathological cardiac hypertrophy by 6 mo of age. Induction of hypertrophy and failure are attributed to the exogenous effect of Nampt on cardiomyocytes, as its effects were blocked by use of a specific anti-Nampt antibody. Experiments carried out to identify the mechanisms of eNampt-mediated hypertrophy revealed increased activation of calcineurin-NFAT signaling pathway. Nampt-Tg mice showed an exacerbated level of apoptosis and MAPK signaling. These studies unveil the vulnerability of Nampt as a prosurvival molecule and warrants caution in approaches aimed to increase its activity in the heart.
Nampt as an adipokine and growth factor.
A great number of studies have provided substantial evidence that adipokines contribute to the list of major risk factors for cardiovascular diseases. Several adipokines like adiponectin, leptin, restin, apelin, amentin, and chemerin are all linked to cardiovascular diseases (20). A relatively new member in this group is visfatin or Nampt. Recent studies show that Nampt acts like a proinflammatory molecule in many disease conditions. Elevated levels of Nampt are observed in serum of patients with inflammatory bowel disease, Crohn's disease, psoriasis, lung injury, sepsis, and ammonites and patients with type 2 diabetes or gestational diabetes (2, 22, 26, 39).
From a cardiovascular disease perspective, visfatin/PBEF/Nampt have been proposed as a marker of atherosclerosis, a clinical condition characterized by low-grade chronic inflammation. Increased expression of Nampt in the periaortic and pericoronary fat of patients with coronary atherosclerosis suggests its potential paracrine role in the development of atherosclerotic lesions (37). Furthermore, Nampt levels were upregulated in the site of plaque rupture in patients with acute myocardial infarction as well as in plaques formed in the carotid artery of patients with symptoms of stroke (3). Elevated levels of Nampt were also found in the abdominal and epicardial fat tissues of coronary heart disease patients, suggesting a positive correlation between Nampt and cardiovascular diseases (2).
In a mouse model of ischemia-reperfusion study, an administration of intravenous Nampt at the time of myocardial reperfusion reduced the myocardial infract size (18). Furthermore, treatment of ventricular myocytes with Nampt at the time of reoxygenation following ischemia also reduced myocyte death (18). Contrary to this, inhibition of Nampt reduced neutrophil-mediated injury during myocardial infarction in vivo in a mouse model of ischemia-reperfusion injury (25). Our data presented in this study also contradicts the beneficial effects of Nampt on the heart, since we found that prolonged activation of Nampt promotes development of adverse cardiac remodeling.
One of the most important findings we report here is the elevated levels of Nampt in the supernatant of cardiomyocytes subjected to serum starvation and oxidative stress, indicating that Nampt is released when cardiomyocytes are subjected to severe stress, and sustained activation of Nampt induces cardiomyocyte hypertrophy and fibrosis. Our in vitro evidence suggests that extracellular Nampt is responsible for inducing hypertrophic response in cardiomyocytes since antibody against Nampt can block Ad.Nampt-induced cardiac hypertrophy. eNampt may also exacerbate hypertrophic response by activating macrophages. In macrophages, Nampt induces release of extracellular matrix metalloproteinase (MMP) inducer and MMP9 (9). Accordingly, activation of MMPs is associated with adverse cardiac remodeling in different models of cardiac injury and deletion of MMP9 attenuated myocardial contractile dysfunction in heart failure (7, 16, 17, 27). Overall, based on the data presented here and published before, it seems that the immediate role of Nampt could be to protect cardiomyocytes from stress, whereas chronic activation of Nampt is detrimental to the heart.
Nampt is also known to function as a growth factor causing cellular growth, proliferation, and differentiation. PBEF, the original name of Nampt, is derived because of its ability to induce growth and proliferation of pre-B cells (34). Nampt also promoted vascular smooth muscle cell maturation (40). Another well-documented characteristic of Nampt is its insulin mimetic effect (10). Nampt injected intravenously or expressed endogenously significantly lowered plasma glucose level in mice (10). Though activation of insulin/IGF1 signaling is also known to play a critical role in the physiological growth of the heart, overactivation of this pathway leads to development of pathological hypertrophy (6). In this study, exogenous Nampt-treated or Ad.Nampt-infected cardiac fibroblasts showed increased differentiation to myofibroblasts, thus attributing a profibrotic trait for sustained activation of Nampt. Since growth factors are also associated with protection of cells from stress under physiological conditions, the immediate effect of Nampt would be protecting cardiomyocytes from oxidative stress and apoptosis. However, persistent stress leading to chronic activation of Nampt is expected to cause an undesirable consequence to the heart. The aforementioned reasoning accounts for the paradox observed for Nampt and for many other growth factors in reference to cardioprotection and hypertrophy and is an extemporaneous remark to the riddle of the obesity paradox, where even though obesity increases one's susceptibility to develop cardiac diseases, in the event of heart attack obese patients survive better than their leaner counterparts (1).
Role of Nampt and cell survival signaling in cardiac hypertrophy.
Sustained elevation of intracellular calcium levels can activate calcineurin, a serine/threonine-specific phosphatase that dephosphorylates NFAT family of transcription factors, thereby facilitating their translocation to the nucleus to transcribe a plethora of genes involved in cardiac hypertrophic response (23). In the current study, we observed increased protein levels of calcineurin in Nampt-Tg hearts as well as in Ad.Nampt-infected cardiomyocytes. Correspondingly, an augmented level of NFAT was observed in the nuclear extract of Nampt-Tg hearts as well as in the Ad.Nampt-infected cardiomyocytes. Several studies have reported increased calcineurin activity in hypertrophied hearts from aortic banded rats and mice (23). Other forms of hypertrophy induced by salt sensitivity, mineralocorticoids, and neuroendocrine-mediated hypertrophy were all found to be associated with increased calcineurin activity (23). Failing human hearts also show increased protein levels or activity of calcineurin. These studies were supported by the Tg studies where cardiac-specific overexpression of calcineurin was found to produce profound hypertrophy and heart failure (24). But the same mice were protected from ischemia-reperfusion injury and apoptosis, which, in part, helps to explain the paradox observed in our study with the cardioprotective effects of Nampt previously reported by others (4). Several other signaling pathways widely implicated in the development of cardiac hypertrophy are no exception to this phenomenon. Mice having cardiac-specific deletion of GP130 showed normal cardiac function but developed dilated cardiomyopathy in response to pressure stimulus (42). In contrast to this, continuous activation of GP130 by overexpressing IL-6 and IL-6R induced cardiac hypertrophy in mice (11). Parallel observations were reported in cell signaling pathways regulated by Akt. Mice lacking Akt1 were resistant to develop swimming-induced physiological hypertrophy but developed an exacerbated form of hypertrophy in response to transverse aortic constriction (5). Again, cardiac-specific overexpression of Akt in mice caused spontaneous development of physiological hypertrophy, which later progressed to pathological hypertrophy and failure (36). These observations suggest that that chronic activation of prosurvival pathways can lead to maladaptive changes in the heart, resulting in cardiac hypertrophy and failure.
Role of intracellular Nampt.
Inside the cell, Nampt catalyzes a rate limiting step in the NAD synthetic salvage pathway, converting nicotinamide and phosphoribosyl pyrophosphate to nicotinamide mononucleotide (15). Previous studies have reported that cardiac-specific Nampt-Tg mice have significantly elevated levels of NAD and ATP in the heart (14). We too have previously reported that exogenous supplementation of NAD can block agonist-induced cardiac hypertrophy (31). In that study we found that both Nampt and consequently NAD levels were reduced in agonist-induced hypertrophic models. When these mice were treated with exogenous NAD, it helped to maintain endogenous NAD as well as Nampt levels and to protect the hearts from developing hypertrophy. However, current study revealed that chronic activation of Nampt in the heart resulted in cardiac hypertrophy and failure. In this context, it is of importance to notice that in the presence of a blocking antibody, transduction of Nampt by adenovirus failed to induce hypertrophy in cardiomyocytes, drawing attention to seemingly contrasting roles played by exogenous and endogenous Nampt.
In summary, Nampt is a proinflammatory cytokine as well as growth factor that has the unique ability to synthesize NAD. Our data presented here show that prolonged release of Nampt in the heart can cause cardiac hypertrophy and failure. Strategies aimed at limiting the temporal action of Nampt could be useful in the management of hypertrophy and progression to heart failure.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants RO1-HL-117041, HL-83423 and HL-111455 (to M. P. Gupta), and N. R. Sundaresan was supported by an American Heart Association postdoctoral fellowship.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
V.B.P., N.R.S., G.H.K., and L.M.-V. and S.S. performed experiments; V.B.P. interpreted results of experiments; J.G.G. and M.G. approved final version of manuscript; V.B.P. and M.G. designed study and drafted manuscript; M.G. edited and revised manuscript.
ACKNOWLEDGMENTS
The α-MHC promoter used to make cardiac-specific Nampt-Tg mice was provided by J. Robbins (Univ. of Cincinnati).
REFERENCES
- 1. Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev 88: 389–419, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cheng KH, Chu CS, Lee KT, Lin TH, Hsieh CC, Chiu CC, Voon WC, Sheu SH, Lai WT. Adipocytokines and proinflammatory mediators from abdominal and epicardial adipose tissue in patients with coronary artery disease. Int J Obes (Lond) 32: 268–274, 2008 [DOI] [PubMed] [Google Scholar]
- 3. Dahl TB, Yndestad A, Skjelland M, Oie E, Dahl A, Michelsen A, Damas JK, Tunheim SH, Ueland T, Smith C, Bendz B, Tonstad S, Gullestad L, Froland SS, Krohg-Sorensen K, Russell D, Aukrust P, Halvorsen B. Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis: possible role in inflammation and plaque destabilization. Circulation 115: 972–980, 2007 [DOI] [PubMed] [Google Scholar]
- 4. De Windt LJ, Lim HW, Taigen T, Wencker D, Condorelli G, Dorn GW, 2nd, Kitsis RN, Molkentin JD. Calcineurin-mediated hypertrophy protects cardiomyocytes from apoptosis in vitro and in vivo: an apoptosis-independent model of dilated heart failure. Circ Res 86: 255–263, 2000 [DOI] [PubMed] [Google Scholar]
- 5. DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, Muslin AJ. Akt1 is required for physiological cardiac growth. Circulation 113: 2097–2104, 2006 [DOI] [PubMed] [Google Scholar]
- 6. Delaughter MC, Taffet GE, Fiorotto ML, Entman ML, Schwartz RJ. Local insulin-like growth factor I expression induces physiologic, then pathologic, cardiac hypertrophy in transgenic mice. FASEB J 13: 1923–1929, 1999 [DOI] [PubMed] [Google Scholar]
- 7. Deschamps AM, Spinale FG. Pathways of matrix metalloproteinase induction in heart failure: bioactive molecules and transcriptional regulation. Cardiovasc Res 69: 666–676, 2006 [DOI] [PubMed] [Google Scholar]
- 8. Dong J, Sulik KK, Chen SY. Nrf2-mediated transcriptional induction of antioxidant response in mouse embryos exposed to ethanol in vivo: implications for the prevention of fetal alcohol spectrum disorders. Antioxid Redox Signal 10: 2023–2033, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fan Y, Meng S, Wang Y, Cao J, Wang C. Visfatin/PBEF/Nampt induces EMMPRIN and MMP-9 production in macrophages via the NAMPT-MAPK (p38, ERK1/2)-NF-kappaB signaling pathway. Int J Mol Med 27: 607–615, 2011 [DOI] [PubMed] [Google Scholar]
- 10. Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, Watanabe E, Takagi T, Akiyoshi M, Ohtsubo T, Kihara S, Yamashita S, Makishima M, Funahashi T, Yamanaka S, Hiramatsu R, Matsuzawa Y, Shimomura I. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science 307: 426–430, 2005 [DOI] [PubMed] [Google Scholar]
- 11. Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci USA 92: 4862–4866, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hong SB, Huang Y, Moreno-Vinasco L, Sammani S, Moitra J, Barnard JW, Ma SF, Mirzapoiazova T, Evenoski C, Reeves RR, Chiang ET, Lang GD, Husain AN, Dudek SM, Jacobson JR, Ye SQ, Lussier YA, Garcia JG. Essential role of pre-B-cell colony enhancing factor in ventilator-induced lung injury. Am J Respir Crit Care Med 178: 605–617, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Houtkooper RH, Cantó C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 31: 194–223, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res 105: 481–491, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Imai S. The NAD world: a new systemic regulatory network for metabolism and aging—Sirt1, systemic NAD biosynthesis, and their importance. Cell Biochem Biophys 53: 65–74, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Janicki JS, Brower GL, Gardner JD, Forman MF, Stewart JA, Jr, Murray DB, Chancey AL. Cardiac mast cell regulation of matrix metalloproteinase-related ventricular remodeling in chronic pressure or volume overload. Cardiovasc Res 69: 657–665, 2006 [DOI] [PubMed] [Google Scholar]
- 17. Jugdutt BI. Matrix metalloproteinases as markers of adverse remodeling after myocardial infarction. J Card Fail 12: 73–76, 2006 [DOI] [PubMed] [Google Scholar]
- 18. Lim SY, Davidson SM, Paramanathan AJ, Smith CC, Yellon DM, Hausenloy DJ. The novel adipocytokine visfatin exerts direct cardioprotective effects. J Cell Mol Med 12: 1395–1403, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu SW, Qiao SB, Yuan JS, Liu DQ. Association of plasma visfatin levels with inflammation, atherosclerosis and acute coronary syndromes (ACS) in humans. Clin Endocrinol (Oxf) 71: 202–207, 2009 [DOI] [PubMed] [Google Scholar]
- 20. Matsuzawa Y. White adipose tissue and cardiovascular disease. Best Pract Res Clin Endocrinol Metab 19: 637–647, 2005 [DOI] [PubMed] [Google Scholar]
- 21. Mazaki-Tovi S, Romero R, Kusanovic JP, Erez O, Gotsch F, Mittal P, Than NG, Nhan-Chang CL, Hamill N, Vaisbuch E, Chaiworapongsa T, Edwin SS, Nien JK, Gomez R, Espinoza J, Kendal-Wright C, Hassan SS, Bryant-Greenwood G. Visfatin/Pre-B cell colony-enhancing factor in amniotic fluid in normal pregnancy, spontaneous labor at term, preterm labor and prelabor rupture of membranes: an association with subclinical intrauterine infection in preterm parturition. J Perinat Med 36: 485–496, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mazaki-Tovi S, Romero R, Kusanovic JP, Vaisbuch E, Erez O, Than NG, Chaiworapongsa T, Nhan-Chang CL, Pacora P, Gotsch F, Yeo L, Kim SK, Edwin SS, Hassan SS, Mittal P. Visfatin in human pregnancy: maternal gestational diabetes vis-a-vis neonatal birthweight. J Perinat Med 37: 218–231, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res 63: 467–475, 2004 [DOI] [PubMed] [Google Scholar]
- 24. Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93: 215–228, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Montecucco F, Bauer I, Braunersreuther V, Bruzzone S, Akhmedov A, Luscher TF, Speer T, Poggi A, Mannino E, Pelli G, Galan K, Bertolotto M, Lenglet S, Garuti A, Montessuit C, Lerch R, Pellieux C, Vuilleumier N, Dallegri F, Mage J, Sebastian C, Mostoslavsky R, Gayet-Ageron A, Patrone F, Mach F, Nencioni A. Inhibition of nicotinamide phosphoribosyltransferase reduces neutrophil-mediated injury in myocardial infarction. Antioxid Redox Signal 00: 1–12, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moschen AR, Gerner RR, Tilg H. Pre-B cell colony enhancing factor/NAMPT/visfatin in inflammation and obesity-related disorders. Curr Pharm Des 16: 1913–1920, 2010 [DOI] [PubMed] [Google Scholar]
- 27. Moshal KS, Rodriguez WE, Sen U, Tyagi SC. Targeted deletion of MMP-9 attenuates myocardial contractile dysfunction in heart failure. Physiol Res 57: 379–384, 2008 [DOI] [PubMed] [Google Scholar]
- 28. Nguyen NT, Nguyen XM, Wooldridge JB, Slone JA, Lane JS. Association of obesity with risk of coronary heart disease: findings from the National Health and Nutrition Examination Survey, 1999–2006. Surg Obes Relat Dis 6: 465–469, 2010 [DOI] [PubMed] [Google Scholar]
- 30. Peiro C, Romacho T, Carraro R, Sanchez-Ferrer CF. Visfatin/PBEF/Nampt: a new cardiovascular target? Front Pharmacol 1: 1–7, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem 285: 3133–3144, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Regitz-Zagrosek V, Seeland U, Geibel-Zehender A, Gohlke-Barwolf C, Kruck I, Schaefer C. Cardiovascular diseases in pregnancy. Dtsch Arztebl Int 108: 267–273, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Romacho T, Azcutia V, Vazquez-Bella M, Matesanz N, Cercas E, Nevado J, Carraro R, Rodriguez-Manas L, Sanchez-Ferrer CF, Peiro C. Extracellular PBEF/NAMPT/visfatin activates pro-inflammatory signalling in human vascular smooth muscle cells through nicotinamide phosphoribosyltransferase activity. Diabetologia 52: 2455–2463, 2009 [DOI] [PubMed] [Google Scholar]
- 34. Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol 14: 1431–1437, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Segawa K, Fukuhara A, Hosogai N, Morita K, Okuno Y, Tanaka M, Nakagawa Y, Kihara S, Funahashi T, Komuro R, Matsuda M, Shimomura I. Visfatin in adipocytes is upregulated by hypoxia through HIF1alpha-dependent mechanism. Biochem Biophys Res Commun 349: 875–882, 2006 [DOI] [PubMed] [Google Scholar]
- 36. Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 115: 2108–2118, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spiroglou SG, Kostopoulos CG, Varakis JN, Papadaki HH. Adipokines in periaortic and epicardial adipose tissue: differential expression and relation to atherosclerosis. J Atheroscler Thromb 17: 115–130, 2010 [DOI] [PubMed] [Google Scholar]
- 38. Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol 28: 6384–6401, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tilg H, Moschen AR. Role of adiponectin and PBEF/visfatin as regulators of inflammation: involvement in obesity-associated diseases. Clin Sci (Lond) 114: 275–288, 2008 [DOI] [PubMed] [Google Scholar]
- 40. van der Veer E, Nong Z, O'Neil C, Urquhart B, Freeman D, Pickering JG. Pre-B-cell colony-enhancing factor regulates NAD+-dependent protein deacetylase activity and promotes vascular smooth muscle cell maturation. Circ Res 97: 25–34, 2005 [DOI] [PubMed] [Google Scholar]
- 41. Wang P, Xu TY, Guan YF, Su DF, Fan GR, Miao CY. Perivascular adipose tissue-derived visfatin is a vascular smooth muscle cell growth factor: role of nicotinamide mononucleotide. Cardiovasc Res 81: 370–380, 2009 [DOI] [PubMed] [Google Scholar]
- 42. Yoshida K, Taga T, Saito M, Suematsu S, Kumanogoh A, Tanaka T, Fujiwara H, Hirata M, Yamagami T, Nakahata T, Hirabayashi T, Yoneda Y, Tanaka K, Wang WZ, Mori C, Shiota K, Yoshida N, Kishimoto T. Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders. Proc Natl Acad Sci USA 93: 407–411, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]