

Abstract
Spontaneous plaque rupture in mouse models of atherosclerosis is controversial, although numerous studies have discussed so-called “vulnerable plaque” phenotypes in mice. We compared the morphology and biomechanics of two acute and one chronic murine model of atherosclerosis to human coronaries of the thin-cap fibroatheroma (TCFA) phenotype. Our acute models were apolipoprotein E-deficient (ApoE−/−) and LDL receptor-deficient (LDLr−/−) mice, both fed a high-fat diet for 8 wk with simultaneous infusion of angiotensin II (ANG II), and our chronic mouse model was the apolipoprotein E-deficient strain fed a regular chow diet for 1 yr. We found that the mouse plaques from all three models exhibited significant morphological differences from human TCFA plaques, including the plaque burden, plaque thickness, eccentricity, and amount of the vessel wall covered by lesion as well as significant differences in the relative composition of plaques. These morphological differences suggested that the distribution of solid mechanical stresses in the walls may differ as well. Using a finite-element analysis computational solid mechanics model, we computed the relative distribution of stresses in the walls of murine and human plaques and found that although human TCFA plaques have the highest stresses in the thin fibrous cap, murine lesions do not have such stress distributions. Instead, local maxima of stresses were on the media and adventitia, away from the plaque. Our results suggest that if plaque rupture is possible in mice, it may be driven by a different mechanism than mechanics.
Keywords: atherosclerosis, plaque rupture, mouse model, vulnerable plaque, biomechanics
plaque rupture is a major cause of atherosclerosis-related thrombosis in the modern world that, despite concerted research efforts, still is not fully understood. Most knowledge of plaque rupture in humans comes from histological evaluation of autopsy specimens or from in vivo imaging of high-risk patients after plaque rupture has already occurred; neither technique has led to direct observations of plaque rupture in progress. Therefore, scientists are forced to study the etiology of plaque rupture either retrospectively in patients who already had a lesion rupture or prospectively in patients who may or may not provide evidence of rupture at a later date. Extensive morphological studies of histology specimens and mathematical modeling of biomechanics have led to the identification of the thin-cap fibroatheroma (TCFA) as the atherosclerotic plaque phenotype currently thought to be most “vulnerable” to rupture (33).
Despite the identification of this phenotype, it is still not possible to accurately predict rupture. Because such research to identify rupture is impractical in humans, scientists frequently study atherosclerosis-prone mice, which are known to rapidly and reliably form atherosclerotic plaques over a period of weeks to months instead of years to decades, as is the case in humans. Rodents are naturally resistant to atherosclerosis, but a wide variety of techniques for inducing atherosclerosis exist (38). As the mechanism behind each of these techniques is unique, the severity and specific characteristics of the atherosclerotic burden vary among models (38). Among the factors modulated by researchers are transgenic strain [with the apolipoprotein E-deficient (ApoE−/−) and low-density lipoprotein receptor (LDLR−/−) strains being the most popular], diet (especially lipid composition), age, sex, and pharmaceutical agents [e.g., angiotensin II (ANG II)]. ANG II induces hypertension, which is a common comorbidity of atherosclerosis in humans. However, much of the effect of ANG II on the vessel wall is proinflammatory (35). The anatomic location of the plaque is another variable, as the brachiocephalic artery has been reported to produce lesions with some similarities to human vulnerable plaques (23). Regardless of which permutation of conditions any particular research group uses, the overall goal is the same: to recapitulate features of human atherosclerosis in a mouse model to better understand the pathophysiology of the disease and, eventually, to develop treatments for its prevention and management.
In addition to the need for a robust animal model of atherosclerotic plaque formation, researchers desire a relevant animal model for the study of thrombotic end points, such as plaque rupture. However, spontaneous plaque rupture in mice is a controversial topic (2, 11, 15–17, 26). Several published reports (5, 25) of murine plaque rupture exist, but others (26) question whether plaque rupture is really occurring in mice, suggesting that reports may actually be sectioning artifacts, intraplaque hemorrhage, or a completely different form of lesion disruption not seen in human plaques. Bond and Jackson (3), who developed a putative mouse model of plaque rupture, contest these claims and suggest that a mouse model need not be morphologically identical to human plaques to be useful. Many publications discuss ruptured and vulnerable plaques in mice, although a unifying definition of the phenomenon does not exist. Schwartz and colleagues (26) have argued that, because the topic is so controversial, researchers should avoid using the terms “vulnerable” and “stable” to describe phenotypes of murine plaques until the community better understands plaque disruption in mice.
Within this context, it is prudent to evaluate the morphological and mechanical stress differences between mouse and human plaques. In the existing literature, several morphological risk factors for human plaque rupture exist. The most well known of such parameters is the thickness of the fibrous cap overlying a necrotic core, but others, such as the size and shape of the necrotic core and the presence of microcalcifications embedded in the fibrous cap, are emerging as well (21). The functional consequence of these features is that a concentration of mechanical stress occurs on the fibrous cap or shoulders of the plaque, and, if the magnitude of the stress is sufficient, the lesion will rupture (6). Biological factors, such as inflammation, are closely associated with plaque rupture as well, but the final failure is, by definition, a physical, mechanical event. However, little is known about whether these geometric and mechanical risk factors translate to mice. Histological inspection of murine plaques reveals distinct morphological differences from human atherosclerotic lesions, the most obvious of which is size: a mouse aorta's diameter is only a fraction of the diameter of a human coronary artery.
In this study, we set out to quantify the morphological and mechanical phenotypes across a range of mouse models. To consider the extremes of atherosclerosis-prone mice, on one end of the spectrum, we investigated a chronic, more intrinsic model of atherogenesis, and, on the other end, we equipped two popular strains of mice with an aggressive set of provocative factors for acute atherosclerosis. Here, we provide a side-by-side quantification of the morphological and mechanical differences between mouse and human plaques. We hypothesized that the plaque extent and composition would be significantly different between mouse and human plaques. We further hypothesized that the relative distribution of locations of peak mechanical stress characteristically found in mice is different from that found in human atherosclerotic lesions.
METHODS
Mouse models.
We considered three models of advanced atherosclerosis in mice: two models that acutely form plaques as well as a chronic model. The first acute model included 15 male ApoE−/− mice aged 8 wk fed a high-fat (HF) diet (D12336, Research Diets) for 8 additional weeks with simultaneous subcutaneous infusion of ANG II via an osmotic minipump (0.75 mg·kg−1·day−1, Sigma Aldrich). The second acute model included 12 male LDLR−/− mice aged 8 wk fed a HF diet for 8 additional weeks with ANG II via a minipump (8, 34). The chronic model of spontaneous lesion formation included 12 male ApoE−/− mice fed a regular chow diet (LabDiet 5001 Rodent Diet, PMI) for 1 yr without ANG II. All animal research was approved by the Institutional Animal Care and Use Committee of Emory University.
Tissue harvest.
One day preceding harvest, each animal's systolic blood pressure was recorded via tail-cuff measurement (BP-2000, Visitech Systems). Animals were harvested using humane euthanasia techniques (slow inhalation of CO2). Upon expiration, animals were perfused with saline followed by 10% buffered formalin for 5 min at 80 mmHg (approximate diastolic blood pressure) by direct cardiac injection. To preserve the conformation of the lumen as closely as possible, mice were then perfused with Batson's no. 17 Vascular Casting Compound (Polysciences) at each mouse's diastolic blood pressure (assumed to be 40 mmHg below the mouse's measured systolic blood pressure) to preserve the in vivo conformation of the lumen. Pressure was maintained using an intravenous pressure infuser (Infu-Surg, Ethox) and measured with an inline manometer. Upon hardening of the vascular casting compound, the aortic tree between the heart and the descending thoracic aorta was excised, fixed 48 h in formalin, and then transferred to 70% ethanol.
Human specimens.
Human coronary artery plaques of the TCFA phenotype were selected from an existing tissue bank by an expert in cardiovascular pathology (R. Virmani, CVPath Institute). Specimens were obtained as part of a consultation service for the Office of the Chief Medical Examiner of the State of Maryland. Fresh hearts were perfused with 10% neutral buffered formalin at 100 mmHg for 15 min, and the coronary arteries were then excised and further fixed in formalin before being embedded.
Histology and microscopy.
Murine aortic specimens were embedded in methyl methacrylate (MMA) resin and then sectioned at 5 μm using an automated rotary microtome. Consecutive serial sections were obtained at the brachiocephalic artery immediately distal to the bifurcation from the arch and at three sites in the descending thoracic aorta, each separated by ∼1 cm. Tissues were deplasticized and stained to identify plaque morphology using a hybrid Masson's trichrome and Von Kossa's calcium staining technique (10). Human specimens were embedded in paraffin, sectioned at 5 μm, and then stained with Movat's pentachrome. To ensure that the difference in stains between humans and mice did not bias our evaluation of the morphology, we stained nine mouse sections (3 sections/phenotype) with Movat's pentachrome. Images of all stained slides were obtained via bright-field microscopy. Sections with major sectioning artifacts were discarded.
Image segmentation.
We classified the tissue of atherosclerotic blood vessels into four primary components (10): 1) cellular tissue (primarily media), 2) fibrous tissue, 3) lipid, and 4) calcification (Fig. 1). Images were imported into custom image-analysis software implemented in Matlab (R2011b) and segmented using k-means clustering based on staining color (27). Manual revision, when necessary, was performed using an implementation of the Live Wire edge-detection algorithm (7).
Fig. 1.
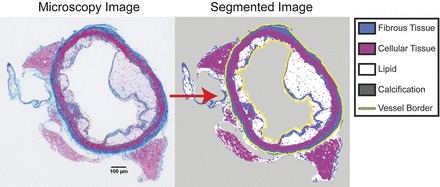
Semi-automated segmentation of atherosclerotic lesions: a sample cross-section of an atherosclerotic low-density lipoprotein receptor-deficient (LDLR−/−) mouse aorta stained with a combination of Masson's trichrome and Von Kossa's calcium stain. The microscopy image (left) was loaded into custom Matlab software and segmented (right) using a semi-automated algorithm. In brief, tissue types were automatically identified using a k-means clustering algorithm and then touched up (for example, to distinguish lipid from background) using an implementation of the Live Wire algorithm (7). The perimeter of the lumen and adventitia were also identified with Live Wire (yellow line).
Morphological analysis.
We measured four morphological parameters and one histological parameter in our microscopy images using Matlab, as shown in Fig. 2. In brief, we traced the internal elastic lamina (IEL), external elastic lamina (EEL), and lumen border in all images. A method for accurately measuring tissue thickness developed by Yezzi et al. (37) was used to compute the distance between any two contours, and the radius of each vessel was defined as the radius of the minimum bounding circle around the EEL. We calculated the following:
1. Lesion coverage of the wall (defined as the fractional portion of the circumference of the vessel where the thickness between the IEL and lumen was nonzero)
2. Plaque burden (percentage of the area inside the IEL containing lesion)
3. Normalized mean lesion thickness, a measure of how far plaques protrude into the lumen (defined as the distance between the IEL and the lumen only where nonzero, normalized to media thickness, defined as the distance between the IEL and EEL)
4. Normalized lesion eccentricity (the distance between the centroid of the lumen border contour and the EEL contour, normalized to vessel radius)
5. Relative composition of the lesion based on the segmented image (the lesion was defined as any tissue between the IEL and the lumen contour)
Statistical differences between models were computed by one-way ANOVA followed by Tukey's honestly significant difference test, with α = 0.05. Results are shown in Figs. 3 and 4, and statistics are shown in Table 1.
Fig. 2.
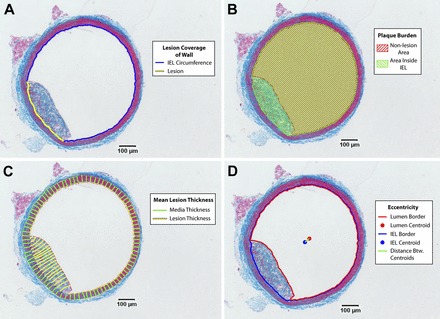
Measurement of morphological parameters from histology. A: lesion coverage of the wall was defined as the percentage of the vessel circumference with nonzero thickness between the internal elastic lamina (IEL) and the lumen border. B: plaque burden was defined as the percentage of the area inside the IEL occupied by the lesion. C: mean lesion thickness was defined as the mean thickness of the lesion normalized to the mean thickness of the media (37). D: eccentricity was defined as the distance between the centroid of the lumen border and the centroid of the IEL border, normalized to vessel radius.
Fig. 3.
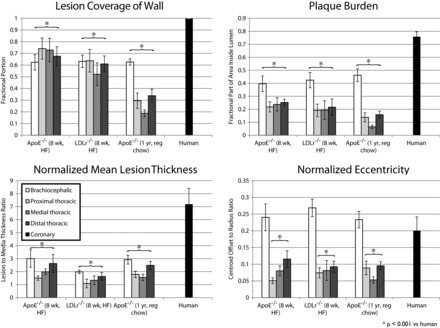
Morphological measures of murine and human plaques. Geometric features of atherosclerotic plaques were measured from histology specimens of lesions from humans and mice. Error bars represent SE. Tukey's honestly significant difference test results are noted for comparisons of brachiocephalic aorta versus the human coronary artery and all descending aortae pooled together versus the human coronary artery. Top left: lesion coverage of the wall. Top right: plaque burden. Bottom left: normalized mean lesion thickness. Bottom right: normalized eccentricity. ApoE−/− mice, apolipoprotein E-deficient mice; HF diet, high-fat diet; reg, regular.
Fig. 4.
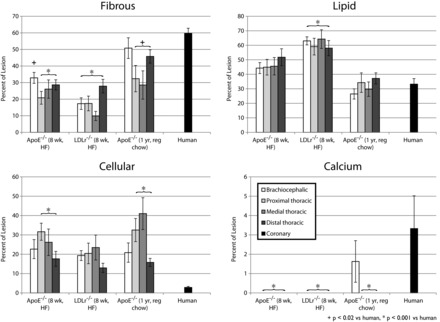
Relative composition of atherosclerotic plaques in mice and humans. Based upon segmented atherosclerotic plaques (Fig. 1), we calculated the relative composition of lesions [fibrous (top left), lipid (top right), cellular (bottom left), and calcified (bottom right)] for humans and mice. Error bars represent SE. Tukey's honestly significant difference test results are noted for comparisons of brachiocephalic aorta vs. human coronary and all descending aortae pooled together vs. human coronary.
Table 1.
Statistical results from Tukey's honestly significant difference post hoc test for morphological metrics
| Metric | Young ApoE−/− Mice Versus Young LDLR−/− Mice | Young ApoE−/− Mice Versus Old ApoE−/− Mice | Young LDLR−/− Mice Versus Old ApoE−/− Mice | Young ApoE−/− Mice Versus Humans | Young LDLR−/− Mice Versus Humans | Old ApoE−/− Mice Versus Humans |
|---|---|---|---|---|---|---|
| Percentage of wall covered with lesion | 0.29219 | <0.00001 | 0.00010 | 0.00198 | 0.00001 | <0.00001 |
| Plaque burden | 0.97702 | 0.14574 | 0.27482 | <0.00001 | <0.00001 | <0.00001 |
| Lesion thickness | 0.19357 | 0.99999 | 0.14242 | <0.00001 | <0.00001 | <0.00001 |
| Eccentricity | 0.95805 | 0.98027 | 0.77581 | 0.09288 | 0.17335 | 0.04070 |
| Percent fibrous | 0.11130 | 0.00146 | <0.00001 | <0.00001 | <0.00001 | 0.00445 |
| Percent lipid | 0.00036 | 0.00138 | <0.00001 | 0.08164 | <0.00001 | 0.99999 |
| Percent cellular | 0.30812 | 0.99999 | 0.25562 | 0.00044 | 0.01725 | 0.00029 |
| Percent calcium | 1.00000 | 0.84934 | 0.83289 | <0.00001 | <0.00001 | 0.00001 |
P values are shown for comparisons among the following groups: young apolipoprotein E-deficient (ApoE−/−) mice, young low-density lipoprotein receptor-deficient (LDLR−/−) mice, old ApoE−/− mice, and humans.
Computational solid mechanics.
We computed the relative distribution of stresses in each segmented vessel using an established lesion-specific computational mechanical modeling technique in Ansys 13.0 APDL software (10). We used a two-dimensional plane strain large deformation finite-element model. The cross-sectional shape of each lesion was imported into Ansys and finely meshed with tens of thousands of elements. Based on image segmentation, linear elastic isotropic material properties were calculated for each element using a constrained rule-of-mixtures approach (1, 14). We then simulated a pressure of 40 mmHg (representing the incremental pressure of the pulse from diastole to systole) normal to the lumen surface and calculated the relative distribution of Von Mises stress (Fig. 5). Modeling assumptions are explained in further detail in the appendix.
Fig. 5.
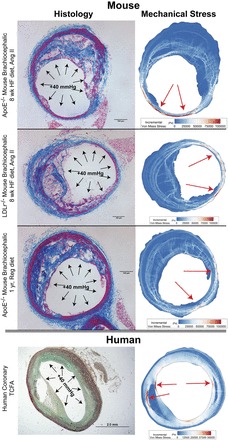
Computer model of stress distributions in plaques. We harvested atherosclerotic plaques from three different mouse models (top, middle top, and middle bottom) and obtained thin-cap fibroatheroma (TCFA) specimens from human coronary arteries (bottom). Using finite-element analysis techniques, we computed the distribution of solid mechanical stresses in each vessel cross-section. Local maxima of stress (red arrows) indicate that in mice, highest stresses are on the media and adventitia, particularly on plaque-free walls, whereas in humans, local maxima tend to be on the thin fibrous cap or shoulders of plaques.
Postprocessing analysis.
We examined the relative distribution of stresses by mapping sites of highest stress by plotting each Von Mises stress distribution in Tecplot 360 software. We displayed stresses greater than a threshold value, defined as the vessel's mean stress + 4 SD. The locations of stresses greater than this threshold were tallied based on their location (Fig. 6): fibrous cap over lipid core, shoulder of plaque, elsewhere on or in plaque other than fibrous cap over lipid core, or on media/adventitia. Media was defined as a circular ring staining positively for cellular tissue (red in Masson's trichrome), and adventitia was defined as all contiguous tissue outside the EEL. Contiguous tissue between the IEL and the lumen was considered plaque, with fibrous cap over lipid core specifically defined as fibrous tissue (blue in Masson's Trichrome) forming a thin strip separating the lumen from lipid. Plaque shoulder was defined as the zone where fibrous or cellular tissue of a plaque meets the media at the periphery of the plaque. For each section, we quantified the location of stress peaks, recording a binary tally for the presence of a local maximum of stress in each region of each section. Some sections had multiple local maxima but were only counted once per location per section: for example, a plaque with one local maximum on the shoulder of the plaque and two distinct peaks in the media would receive one tally for the shoulder and one for the media. The threshold of 4 SD was determined empirically as the highest integer multiple of the SD that identified at least one local maximum per section. To ensure that our choice of stains (Masson's trichrome and Von Kossa's calcium in mice and Movat's pentachrome in humans) did not affect our stress analysis, we performed identical analysis on consecutive serial section pairs stained with both protocols and confirmed that the tallies of local maxima of stress were exactly identical (Fig. 7).
Fig. 6.
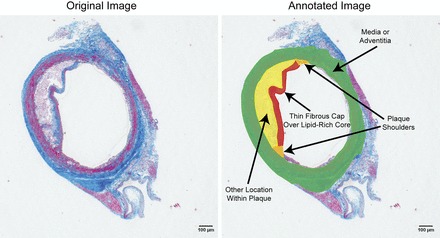
Annotation of locations: We identified the location of local maxima of Von Mises stresses (Fig. 5) according to several anatomic criteria, denoted here. Peak stresses were either identified as being located on a thin fibrous cap over a lipid-rich core or plaque shoulders, the two locations where plaque rupture are known to occur in human lesions, or on the media or adventitia or elsewhere in the plaque.
Fig. 7.
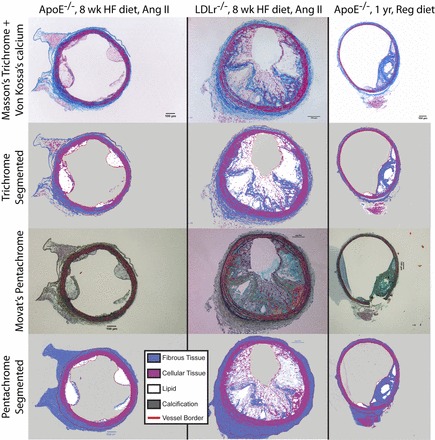
Validation of staining protocols. We stained consecutive serial sections of mouse tissue with Masson's trichrome and Von Kossa's calcium protocols (top) and with Movat's pentachrome protocol (bottom). Representative images paired with their segmentation demonstrated qualitative agreement. The location of local maxima of stress (not shown) did not differ between specimens. Only tissue inside the lines marked “vessel border” was included in our study, so periadventitial segmentation differences will not affect results. A representative problem with the Movat's pentachrome stain in our tissues is visible in the LDLR−/− and older ApoE−/− specimens: the vascular casting compound absorbed dye, partially obscuring nearby tissue.
Analysis of plaque cap thickness.
In only the subset of lesions exhibiting a local maximum of stress located on a fibrous cap over lipid-rich core, we computed the thickness of the fibrous cap at the site of peak stress relative to the mean thickness of the media around the entire vessel (37). Differences between groups were assessed using an unpaired, two-tailed t-test with α = 0.05 and Bonferroni correction for multiple comparisons.
Analysis of calcification.
To assess of the presence of microcalcifications, we scanned the brachiocephalic arteries and descending aortas of a subset of our mice (3 LDLR−/− mice fed HF diet for 8 wk with ANG II and 3 ApoE−/− mice fed regular diet for 1 yr) using micro-computed tomography (micro-CT; μCT 50, Scanco Medical, Brüttisellen, Switzerland). Tissues were scanned in 70% ethanol. A positive control scan of hydroxyapatite powder (Fisher Scientific) was analyzed with scanning electron microscopy (DS-130F, Topcon Positioning Systems, Livermore, CA) to determine its particle size. This powder was then scanned with micro-CT to confirm the equipment's ability to resolve microcalcifications. Additionally, adjacent sections of the ascending aorta were stained with alizarin red to further confirm the presence of calcium.
RESULTS
We computed morphological metrics and relative stress distributions in plaques without significant sectioning artifacts from mice without premature mortality. In total, we evaluated plaques from 10 young ApoE−/− mice, 11 young LDLR−/− mice, and 11 old ApoE−/− mice. Mortality was highest for the group of young ApoE−/− mice, with 5 mice expiring within 8 wk. This animal model has previously been reported to experience aortic aneurysms and dissection at approximately this rate (8, 24). One LDLR−/− mouse expired prematurely, and one older ApoE−/− mouse developed dermatitis and was euthanized. We evaluated brachiocephalic, proximal descending thoracic aorta, mid-descending thoracic aorta, and distal descending thoracic aorta plaques totaling 34 sections in young ApoE−/− mice, 42 sections in young LDLR−/− mice, and 49 sections in old ApoE−/− mice. In humans, we evaluated 11 coronary plaque sections.
Morphological analysis.
In all four morphological metrics we examined, atherosclerotic plaques from each of the three mouse models were significantly smaller than human lesions. Murine lesions covered less of the wall, the plaque burden was lower, the normalized mean lesion thickness was lower, and the normalized eccentricity of plaques was lower. Results are shown in Fig. 3, and statistics are shown in Table 1. In all mouse models, the brachiocephalic artery had greater plaque burden, normalized mean lesion thickness, and normalized eccentricity from any of the descending aorta plaques we considered, and the difference sites in the descending aorta were mostly not different from one another. In the chronic model of plaque formation, brachiocephalic artery lesions covered a greater fraction of the wall than the descending aorta plaques, but this trend was not observed in either of the acute models.
The relative composition of human plaques was different from murine plaques as well. Human plaques were, on average, significantly more fibrotic and calcified and contained less cellular tissue than mouse plaques. The foam cell/necrotic core content was significantly higher in young mice than in humans, but our chronic ApoE−/− mouse model was not significantly different in lipid content than human coronary plaques. Differences are shown in Fig. 4, and statistics are shown in Table 1.
Mechanical analysis.
In mice, only 8 of 125 (6.4%) sections had high-stress regions on a thin fibrous cap over a lipid core, whereas all 125 of 125 (100%) plaques examined had regions of peak stresses located in the media and adventitia. All lesions with local maxima of stress on the fibrous cap were located in the descending aorta, not the brachiocephalic artery; 4 of 125 (3.2%) lesions had stress peaks on the plaque shoulder, most of which were in the brachiocephalic artery, and 19 of 125 (15.2%) lesions had stress peaks inside the plaque. In contrast, in humans, 10 of 11 (90.9%) plaques had regions of peak stress on the thin fibrous cap over a lipid core, and the 1 section without a peak stress region on the fibrous cap exhibited a concentration of stress on the plaque shoulder [1 of 11 (9.1%) lesions]; 4 of 11 (36.4%) regions of stress peaks were on the media or adventitia, and 2 of 11 (18.2%) regions had a local maximum of stress inside the plaque itself. The results are shown in Fig. 8 and Table 2.
Fig. 8.
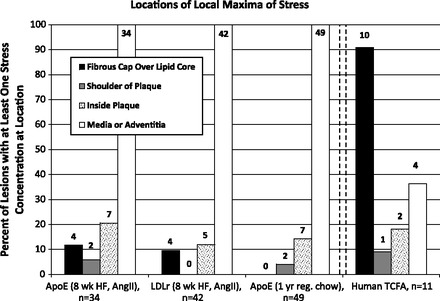
Locations of local maxima of stress. After computational solid mechanical modeling of Von Mises stress distributions in atherosclerotic plaques, we quantified the percentage of vessels exhibiting local maxima of stress in specific locations. A local maximum was defined as stresses > 4 SD above the vessel's mean stress. Very few mouse vessels exhibited peak stresses in a fibrous cap over lipid-rich core, whereas nearly all human TCFA did. In contrast, every single mouse vessel had peak stress in the adventitia or media, whereas less than half of human plaques did. Mice were fed HF or regular chow diets, and some mice received ANG II. Above each bar is the raw number of vessels with a local maximum at the specified location.
Table 2.
Tally of stress concentration locations
| Total Sections | Fibrous Cap Over Lipid Core | Shoulder of Plaque | Inside Plaque | Media or Adventitia | |
|---|---|---|---|---|---|
| ApoE−/− mice with 8-wk HF diet and ANG II | 34 | 4 | 2 | 7 | 34 |
| Brachiocephalic | 9 | 0 | 2 | 3 | 9 |
| Proximal descending | 9 | 1 | 0 | 2 | 9 |
| Medial descending | 10 | 2 | 0 | 1 | 10 |
| Distal descending | 6 | 1 | 0 | 1 | 6 |
| LDLR−/− mice with 8-wk HF diet and ANG II | 42 | 4 | 0 | 5 | 42 |
| Brachiocephalic | 11 | 0 | 0 | 2 | 11 |
| Proximal descending | 10 | 0 | 0 | 3 | 10 |
| Medial descending | 10 | 0 | 0 | 0 | 10 |
| Distal descending | 11 | 4 | 0 | 0 | 11 |
| ApoE−/− mice with 1-yr regular chow diet | 49 | 0 | 2 | 7 | 49 |
| Brachiocephalic | 8 | 0 | 1 | 3 | 8 |
| Proximal descending | 11 | 0 | 0 | 1 | 11 |
| Medial descending | 10 | 0 | 0 | 0 | 10 |
| Distal descending | 20 | 0 | 1 | 3 | 20 |
| Total mice | 125 | 8 | 4 | 19 | 125 |
| Brachiocephalic | 28 | 0 | 3 | 8 | 28 |
| Proximal descending | 30 | 1 | 0 | 6 | 30 |
| Medial descending | 30 | 2 | 0 | 1 | 30 |
| Distal descending | 37 | 5 | 1 | 4 | 37 |
| Human TCFA | 11 | 10 | 1 | 2 | 4 |
HF diet, high-fat diet; TCFA, thin-cap fibroatheroma.
Fibrous caps.
The relative thickness of murine lesions experiencing a stress peak on the fibrous cap was higher or indistinguishable from that in humans. In young ApoE−/− mice, the mean relative thickness (ratio of fibrous cap thickness at the site of peak stress to mean media thickness) was 0.842 ± 0.105 (n = 4 mice with local maximum stress on fibrous cap, mean ± SE cap thickness: 29.8 ± 4.0 μm, media thickness: 35.3 ± 1.0 μm); in young LDLR−/− mice, the mean ratio was 0.322 ± 0.056 (n = 4, mean ± SE cap thickness: 11.8 ± 1.9 μm, media thickness: 37.2 ± 1.4 μm); and in human coronary arteries, the mean ratio was similar at 0.349 ± 0.025 (n = 11, mean ± SE cap thickness: 43.8 ± 2.4 μm, media thickness: 156.3 ± 7.3 μm). The difference in relative thicknesses between humans and young ApoE−/− mice was significant (P = 0.019), but between humans and young LDLR−/− mice the difference in relative thicknesses was not (P = 0.867).
Micro- and macrocalcifications.
We histologically observed the presence of macrocalcifications in the brachiocephalic artery in our chronic model of plaque formation, the ApoE−/− mouse aged 1 yr on a regular chow diet (Fig. 9). We did not observe similar macrocalcifications in either of our 8-wk mouse models. Putative microcalcifications, in contrast, were observed in all three mouse models with both Von Kossa and alizarin red stains (Fig. 10). In our histological examination of the ascending aortas in our mice, we observed punctate positive staining for calcium on the aortic valve leaflets.
Fig. 9.
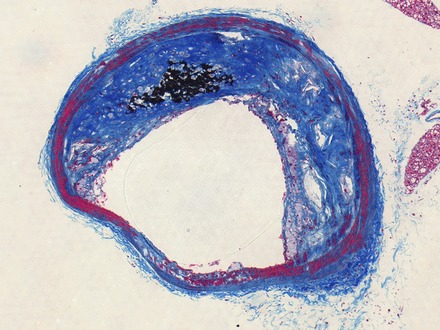
Macrocalcification in the brachiocephalic artery of ApoE−/− mouse. Von Kossa's calcium stain was performed in conjunction with a modified protocol for Masson's Trichrome, so black stain is positive for calcification.
Fig. 10.
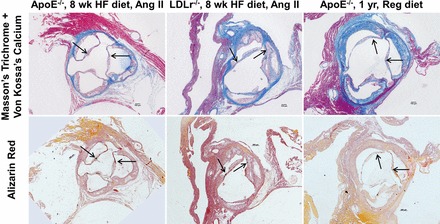
Microcalcifications in the ascending aorta of mouse models. Microcalcifications are visible in ApoE−/− (8-wk HF diet with ANG II; left), LDLR−/− (8-wk HF diet with ANG II; middle), and ApoE−/− (1 yr with regular chow diet; right) mouse models, as confirmed by Von Kossa's calcium stain (top) and alizarin red stain (bottom).
In micro-CT analysis of our older ApoE−/− mice (chronic model), regions of radiodensity similar to the hydroxyapatite powder (positive control) were present, including isolated voxels of positive signal that would appear to be “microcalcifications.” Whether these were located within the thin fibrous cap, as proposed by Vengrenyuk et al. (31), could not be discerned due to the low contrast between tissue and surrounding ethanol. Similar positive speckling was not observed in any samples of the acute model that we scanned.
DISCUSSION
The major findings of this study are that the shape, size, and composition of atherosclerotic plaques are different between mice and humans and, as a result, the relative distribution of solid mechanical stresses in mice does not result in local maxima on a thin fibrous cap over a lipid-rich necrotic core, as is the case in humans. Therefore, our results demonstrate that atherosclerotic lesions in mice and humans have significant morphological and mechanical differences. Given the differences in genes, vessel diameter, posture (which implicitly may affect the blood flow patterns that modulate lesion progression), time course, and location of lesions, a distinction between mice and humans is not unreasonable to expect. However, the consequences of these dissimilarities suggest that other characteristics of lesions, such as the propensity for plaque rupture, may be different as well. As a link between lesion morphology and mechanical environment has been established, we felt that a direct comparison of the mechanical differences between human and murine plaques was prudent (21).
Our mechanical analysis is based on an established technique for calculating the relative distribution of stresses from histology specimens (10). We selected Von Mises stress as our metric because it approximates the magnitude of the local stress tensor at any given point and because it has been associated with inflammatory processes that may underlie the biological basis of plaque rupture (10). Here, we showed that the relative distribution of solid mechanical stresses in mice is such that the highest stresses are on the media and adventitia rather than on the thin fibrous cap of atherosclerotic lesions. In contrast, our results demonstrate that human plaques experience these highest stresses on the fibrous cap or shoulders of the lesion, in line with current understanding of the mechanics of plaque rupture (19). The nature of our model is a relative analysis of the wall stress distribution, so we have no way of knowing if these localized concentrations of high stresses are of sufficient magnitude to disrupt the tissue. What the relative distribution of stresses can tell us, however, is that because stresses are highest on media and adventitia, these mice would be susceptible to whole vessel aortic or carotid rupture before any intraplaque rupture resulting from lumenal blood pressure might occur. Such catastrophic vessel bursting has not, to our knowledge, been reported in these mouse models. Therefore, spontaneous plaque rupture, if it is possible in mice, seems unlikely to be driven solely by the overall mechanical environment.
From a purely mechanical perspective, the fact that stresses are locally the highest on the thin, plaque-free portions of the wall in mice is not unexpected. The punctate distribution of plaques in mice, which in our models tended to be primarily focal, almost like lipid-rich growths adhered to the surface of otherwise healthy media and adventitia, is a key difference between mouse and human lesions. The highly focal distribution of plaques in mice was especially noticeable at the brachiocephalic bifurcation from the aortic arch, a site where many investigators have searched for evidence of murine plaque rupture. Lesions in the brachiocephalic artery were always located on the upstream side of the bifurcation (where low and oscillatory shear stress occur), whereas the downstream wall never formed neointima (28). Maximum stresses were distributed on the healthy, thin wall and were of much lower magnitude within the lesion itself. Whereas in humans, some amount of fibrotic neointimal hyperplasia is frequently observed on all portions of the wall (100% of the human coronary lesions in this study had complete circumferential hyperplasia), our mouse models did not recapitulate this feature of the lesions, which likely influences the distribution of mechanical stresses.
The timescale of plaque formation may be a plausible explanation for this phenomenon. In humans, plaques, particularly those of the TCFA phenotype, develop over a period of 40–70 yr, orders of magnitude longer than the lifespan of even our chronic mouse model of atherosclerosis. In humans, this gradual plaque formation results in “Glagov remodeling,” wherein lesions remodel outward and preserve lumen diameter until the plaque burden has reached a certain threshold, at which point the plaque begins to encroach upon the lumen (9). In many of our mouse plaque specimens, which were pressure perfused with a vascular casting compound in an attempt to histologically preserve the relative in vivo conformation of the vessel, we could closely circumscribe a circle around the EEL (Fig. 11). The circular media with lesions protruding into the lumen suggests that Glagov remodeling may not be recapitulated in our mouse models, which is further evidence that mouse plaques are morphologically and mechanically different from human plaques.
Fig. 11.
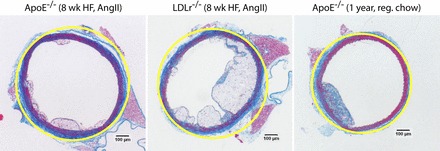
Glagov remodeling does not occur in some mouse plaques. The minimum bounding circle circumscribed around the external elastic lamina (yellow line) in pressure-fixed mouse aortas very closely approximates the geometry of the media. Atherosclerotic lesions in these vessels protrude inward from the media into the lumen rather than remodeling outward to preserve the shape and diameter of the lumen, in contrast with the Glagov theory of remodeling in humans (9).
Another noteworthy morphological difference with mechanical implications between mice and men is that of the fibrous cap. A current criterion for a TCFA in human arteries is a fibrous cap thinner than 65 μm. In mice, whose vessels and plaques are much smaller than in humans, almost every fibrous cap is below this threshold, and yet plaque rupture is certainly not as prevalent as in humans. Although peak stresses occasionally occurred on the surface of murine lesions (6.4%), these almost never coincided with the presence of a thin fibrous cap over a lipid-rich region of the plaque. In humans, current understanding of the mechanics of plaque rupture is that peak stresses on the thin fibrous cap or its shoulders lead to tissue disruption (19). Our mechanical model recapitulated this finding, with 100% of human TCFA specimens demonstrating a local maximum of stress on the fibrous cap or its shoulders. In mice, however, local stress concentrations did not appear to be associated with the presence of a thin fibrous cap. We observed numerous thin fibrous caps in mice without a local maximum of stress on that site, and the few stress concentrations we observed that were adjacent to the lumen occurred on relatively “thick” caps. Additionally, none of the murine plaques with superficial stress concentrations were brachiocephalic plaques, suggesting that if spontaneous plaque rupture can occur in mice, this may not be the best candidate site. Buried fibrous caps, which some have proposed are evidence of prior rupture that has healed, were present in many of our murine specimens (for example, see Fig. 5, top left) (18). Because histology is simply a snapshot in time, we cannot comment on the history of these plaques beyond observing that some buried fibrous caps are indeed present and that they did not seem to be host for local maxima of stress either.
In addition to differences between mice and humans, we observed differences in morphology and composition among our mouse models. Fibrous caps experiencing a local maximum of stress were different between our two young mouse models, with both the absolute and relative thickness lower in LDLR−/− mice, perhaps due to a different rate of fibrotic processes. Plaques from LDLR−/− mice contained more lipid and less fibrous tissue than either of their ApoE−/− counterparts, suggesting that fibrotic processes may differ between these genetically distinct mice.
Although we considered both acute and chronic models of plaque formation in mice, lesions shared quantitative morphological similarities among all three models. For all four morphological metrics we considered, brachiocephalic plaques were similar to other models' brachiocephalic plaques, and, other than the amount of the wall covered by lesion, descending aortic plaques were morpholologically similar to other models' descending aortic plaques. In the descending aorta, plaques in older ApoE−/− mice cover less of the wall than their younger counterparts. Considering that some rupture studies investigate murine brachiocephalic plaques because of purported similarity to human coronary plaques, the difference in magnitude of morphological metrics between mice and humans is striking.
Calcification is another morphological feature potentially tied to plaque rupture (20). That we saw extensive calcification (Fig. 9) only in our older mice and not in either of our 8-wk mouse models suggests that the formation of macrocalcification only occurs on longer timescales (such as that of humans, where plaques form over decades). We did not include the ascending aortas of mice in our mechanical analysis because of the extra complication of modeling the valve leaflets and because there is little evidence of purported plaque rupture in mice at this site, but there we observed tiny, punctate calcifications (we avoid definitively calling them microcalcifications because no established definition of the phenomenon exists that can be applied to mice) with both histology and micro-CT. In humans, valve calcification is regarded as a different pathophysiology as atherosclerosis, so it is unclear if the presence of these tiny calcifications in both the valve leaflet and around the lipid-laden cells on the leaflet are relevant for our understanding of plaque rupture. We did include the effects of calcifications in our modeling technique, but we saw only evidence of macrocalcifications in our particular sections. A recent study by Maldonado et al. (20) observed microcalcifications in 9 of 92 specimens, so given our sample size of 11 human coronary arteries, the absence of microcalcificiations is not unexpected.
Our findings agree with and build on previous studies investigating the solid mechanics of murine blood vessels from Broisat et al. (4) and Vengrenyuk et al. (32). Vengrenyuk et al. (32) investigated how fibrous cap thickness affected stress magnitude in ApoE−/− mouse plaques, although the threshold magnitude for rupture to occur is not known. More recently, Broisat et al. (4) studied the solid mechanics of brachiocephalic plaques in female ApoE−/− mice of varying ages and found that the distribution of solid mechanical stresses was not favorable for plaque rupture to occur. These previous studies provided initial evidence that spontaneous plaque rupture is mechanically unlikely in mice and established a need for expanded evaluation of the solid mechanics of commonly studied atherosclerotic mouse models with direct comparisons to human plaques.
However, these prior studies did not use identical mechanical modeling or histological embedding techniques, and thus it is difficult to compare results from the acute model of atherosclerosis studied by Broisat et al. (4) with the chronic model studied by Vengrenyuk et al. (32). Because many assumptions go into mechanical modeling, it is important to validate the findings against existing knowledge, which is why we included human TCFA specimens in the present study. These other studies calculated specific magnitudes of mechanical stresses, but we only focused on the relative distribution of stresses because of the known limitations of our technique for calculating such quantities.
Our study investigated both ends of the spectrum of atherosclerotic mouse models, both chronic and acute, in a manner such that a range of murine plaque morphology and mechanics could be compared with human coronary plaques. Although this is not a comprehensive investigation of all possible permutations of atherosclerotic mouse models, it represents a telling cross-section of the spectrum of mouse models. Our rationale for selecting such extreme models of lesion formation was that if mice with the largest plaques don't appear to experience spontaneous plaque rupture, rupture seems even less likely in more moderate models. We considered a different set of animal models than either previous study on mechanical modeling of murine plaques. We focused exclusively on male mice since sex is known to affect plaque phenotype in mice as well as humans, and we included hypertension, a common comorbidity of atherosclerosis (30, 34). We included the LDLR−/− strain, often studied but not previously considered in mechanical studies, and we considered plaques from the descending thoracic aorta in addition to the brachiocephalic artery, a site not included in previous analyses (38). In this study, we considered all of these features to analyze a different subset of mice and compare them directly with humans, and our results in both mice and humans are consistent with the findings of prior studies.
A recent study by Ohayon et al. (22) investigated the role of species-specific material properties in computational solid mechanical simulations. They found that using human material properties in simulations of murine plaques led to an overestimation of stresses inside the plaque. Although their mechanical modeling approach and geometry generation technique differed from ours, this finding suggests that we may be overestimating the frequency of local maxima outside the media and adventitia in mice shown in Fig. 4. This strengthens our conclusion that the relative distribution of stresses is fundamentally different between mice and humans.
It is interesting to note that lesion morphology was variable both between and within each of our experimental groups. Even though we used inbred mouse strains given identical treatments to induce atherosclerosis, the exact extent, shape, and location of lesions varied from mouse to mouse. In humans, the etiology of lesions was even more variable, since so many physiological and lifestyle factors influence plaque formation. Therefore, we expect lesion-to-lesion variability to be even higher in humans than in mice.
One limitation to our present study is that our human and mouse tissues were histologically prepared differently. Both our embedding and staining protocols differed because the tissues came from different sources. Human coronary arteries were obtained from an existing tissue bank: already formalin fixed, paraffin embedded, and stained with Movat's pentachrome. This technique provides high-quality preservation of vessel composition, but some morphological distortion occurs during tissue processing and sectioning. To overcome such processing artifacts in our murine data set, we embedded tissue in MMA and perfused the lumen with a vascular casting compound. A consequence of this technique is that Movat's pentachrome staining is impossible: the vascular casting compound, transparent in Masson's trichrome staining, swells during Movat's staining and absorbs the black elastic/nuclear stain (visible in Fig. 7). In many sections, it obscures the surrounding tissue, making analysis impossible. Rather than discard the majority of our murine data set where this occurred, we opted to stain with Masson's trichrome after demonstrating that the stain did not affect our mechanical analysis.
To overcome morphological distortion in the human specimens, cross-sections were digitally preinflated. This technique resulted in more circular lumens as would be found in vivo and eliminated stress and strain concentrations at sites of steep bending not associated with features of the plaque, presumed to be tissue processing artifacts. After 10 iterations of this preinflation technique, stress and strain concentrations at sites of presumed histology artifacts disappeared, but stress and strain concentrations located at sites associated with plaque morphology (on the thin fibrous cap, plaque shoulders near the lipid core, etc.) remained. Sensitivity analysis revealed that results were unaffected after ∼10 preinflation iterations.
We further discuss our mechanical model and its assumptions in the appendix. Although our modeling technique determines only relative levels of stress, the results are still useful for understanding the mechanical environment in murine plaques. Additional steps, such as estimation of initial stress or a full three-dimensional reconstruction of the lesions, while elegant, would add considerable complexity and carry their own set of assumptions and limitations. In this study, we isolated the role of Von Mises stress in plaque rupture, although additional factors, such as fluid shear stress and hydrostatic pressure, likely play a role. Here, we specifically wanted to investigate the role of solid mechanics in plaque rupture. Our lesion-specific, histology-based analysis of the morphology and mechanics of human and murine atherosclerotic plaques indicates that solid mechanics may not predispose mice to plaque rupture in mice in the same way as they do in humans. Additional morphological metrics could have been considered in addition to those included in this study, but those we measured provide clear evidence that the relative shape and composition of murine lesions are unique from human lesions.
In the literature, researchers not infrequently interpret plaque morphology in mice as a transformation to a “more vulnerable phenotype.” Our results suggest that such terminology may be inappropriate. Geometric risk factors for plaque rupture in humans, such as thin fibrous caps and large necrotic cores, do not appear to be associated with a distribution of mechanical stresses likely to cause rupture in the mouse models we examined. Certainly, spontaneous plaque rupture in mice is controversial. This study suggests that, if spontaneous plaque rupture does occur in mice, then, unlike humans, it does so through a different mechanism than mechanics.
GRANTS
This material is based on work supported by an American Heart Association Predoctoral Fellowship (to I. Campbell), by National Science Foundation Grant DGE-0644493 (to I. Campbell), and by National Institutes of Health (NIH) Grant R01-HL-70531. D. Weiss and W. R. Taylor received grants from NIH.
DISCLOSURES
R. Virmani consults and/or receives honoraria from the following: Abbott Vascular, Arsenal Medical, Atrium Medical Corporation, Biosensors International, GlaxoSmithKline, Lutonix, Medtronic AVE, Terumo, and W. L. Gore.
AUTHOR CONTRIBUTIONS
Author contributions: I.C.C., D.W., R.V., A.V., R.P.V., J.N.O., and W.R.T. conception and design of research; I.C.C. and D.W. performed experiments; I.C.C., J.D.S., R.V., A.V., J.N.O., and W.R.T. analyzed data; I.C.C., D.W., J.D.S., R.V., A.V., R.P.V., J.N.O., and W.R.T. interpreted results of experiments; I.C.C. prepared figures; I.C.C. drafted manuscript; I.C.C., D.W., J.D.S., R.V., A.V., R.P.V., J.N.O., and W.R.T. edited and revised manuscript; I.C.C., D.W., J.D.S., R.V., A.V., R.P.V., J.N.O., and W.R.T. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Angela Lin for assistance with the micro-CT and Hong Yi (Robert P. Apkarian Integrated Electron Microscopy Core, Emory University) for assistance with the scanning electron microscopy.
APPENDIX
Tissue handling protocol.
To ensure that our lesion-specific model was based on accurate histology data, we developed a novel vessel harvesting technique that uses vascular casting and plastic resin embedding to maximize the accuracy by which we capture lesion morphology in its in vivo conformation. Even when a mouse aorta is formalin fixed under pressure, the vessel remains compliant and collapses as soon as pressure is released, presenting a significant problem for investigators attempting to preserve the lumen geometry. Failure to preserve the lumen in its circular in vivo conformation will lead to calculation of high strains at sites where the vessel collapses, making it impossible to distinguish the mechanics of the vessel from the mechanics of the histology artifact. By perfusing our mice with vascular casting compound, we were able to minimize such artifacts by preserving the lumen in its pressurized, circular conformation.
A second consideration sometimes overlooked in histology-based models is the choice of embedding medium. Although frozen and paraffin sections both offer high compatibility with commercial antibodies for immunohistochemistry, both suffer from morphological distortion of the tissue during sectioning, as the vessel stretches axially in the cutting direction. By embedding our tissues in MMA resin and then cutting with a motorized diamond blade microtome, we could evenly apply pressure across the surface of the block to minimize sectioning artifacts. Additionally, because of the stiffness of MMA, we were able to cut calcified mouse plaques in many cases with minimal shattering or shredding of neighboring tissue. Unfortunately, unlike frozen sections, the MMA embedding process requires tissue dehydration through graded alcohols, so lipid (which shows as negative space in our sections) cannot be detected through stains like oil red O.
Description of modeling and assumptions.
Our computational model is based on an existing technique that we selected because of its ability to use high-resolution histology imagery to generate lesion-specific relative stress distributions (10). Because model geometry is based on tissue data pressure fixed in its diastolic conformation, we are able to recreate the mechanical environment of the vessel resulting from the incremental pressure of each cardiac pulse. The downside to this technique is that, because the tissue is embedded for histology, we cannot derive residual stresses and therefore cannot compute exact stress magnitudes. As the deformations considered in our model are small compared with those resulting from initial pressurization of the vessel, the relative distribution of stresses should be minimally affected by this missing prestress data. The relative distribution of stresses provides insights into the lack of plaque rupture in mice, and no published data on the stress magnitudes necessary to cause plaque rupture in mice exist, anyway.
Our model took advantage of several theoretical simplifications to exploit the properties of the system. Biological tissues are nonlinear, viscoelastic materials that are complex to represent theoretically (13). However, because material properties are known to be approximately bilinear and because we only simulated the pressure range between diastolic and systolic blood pressure, material properties for each tissue component could be represented with a linear elastic modulus (1). Higher-order hyperelastic models, commonly used in simulations computing exact stress magnitudes, are inappropriate for our scenario because data on the unloaded vessel are unavailable to us.
In our model, we simulated an incremental pressure of 40 mmHg normal to the lumen, equal to the difference in pressure between tissue fixed at 80 mmHg and a typical systolic blood pressure of 120 mmHg. Because the nonlinear material properties of arterial tissue are roughly bilinear, large deformations occur below 80 mmHg that yield very little stress (1). Above the breakpoint in these bilinear material properties, minimal deformation occurs, but large stresses accumulate. Thus, by fixing our tissue at 80 mmHg, we underestimate the overall magnitude of stress but input optimal starting geometry. Since the stress-strain relation is approximately linear in this region, simulating higher magnitudes of stress (for example, simulating an incremental pressure of 120 mmHg) will primarily affect the local stress magnitudes rather than distribution. Because we only considered stress distribution and not exact magnitudes in this study, we have minimized the limitation of missing patient-specific incremental stresses. Calculation of exact magnitudes of stress requires knowledge of residual stress as well as wall stresses from pressures below diastolic blood pressure. Estimation of residual stress requires destructive measurements of tissue, impossible for our banked data set. Estimation of wall stresses from pressures below diastole is possible with inverse method techniques, but the relative contribution of these stresses is expected to be small based on the approximately bilinear stress-strain relationship for these tissues. We can approximate the distribution of stresses in the wall from pulsatile blood pressure but not the exact magnitudes, which is why we only consider the relative distribution of stresses in the present study.
For each element of the mesh, we applied linear elastic material properties based on a constrained rule-of-mixtures approach (1, 14). Mechanical properties for each of the four primary tissue components were derived by Beattie et al. (1): lipid, 3.88 × 105 dyn/cm2; cellular, 2.45 × 106 dyn/cm2; fibrous, 1.82 × 107 dyn/cm2; and calcified, 1.07 × 108 dyn/cm2. Based on our segmented histology, we overlaid a fine-resolution mesh generated by Ansys software based on the contours of the lumen and outer edge of the adventitia. For each element of the mesh, the rule-of-mixtures approach dictates that the effective elastic modulus is equal to the weighted average of the elastic moduli of tissue components present in that element. As the mesh was extremely fine, the majority of elements represented only a single tissue type.
Additionally, biological tissues have been shown to exhibit anisotropy, particularly along fiber directions (14). Unfortunately, the atherosclerotic lesions that we simulated are extraordinarily complex and protrude nonuniformly into the lumen. Without information on the specific fiber directions within the lesion, anisotropy is nearly impossible to model correctly, particularly in the traditional cylindrical coordinate system by which most anisotropic material properties in the literature are described. The cross-sections of the blood vessels in our study are only approximately circular, and, therefore, a polar coordinate system cannot be easily adopted.
In this study, we derived our material properties from published values of elastic moduli for human coronary arteries. Recently, Tracqui et al. (29) measured tissue-specific mechanical properties for several components of atherosclerotic lesions in ApoE−/− mice, and these properties were used in the publication of Broisat et al. (4). However, Tracqui et al. did not publish values for the media or for calcifications, which are essential components of our computational model, and Broisat et al. used a hyperelastic model of the unloaded media that cannot be applied to our pressure-fixed tissue data. Similarly, Hayenga et al. (12) also measured material properties of murine atherosclerotic plaques but also did not differentiate between the different components of plaque that we identified in our model with histology. Rather than arbitrarily selecting material properties for the missing components from the literature, we decided to use an internally consistent data set of material properties all measured from identical atherosclerotic lesions. Although one might extrapolate that the material properties of mouse plaques are different from humans based on our results showing unique mechanical environments between organisms, we prioritized self-consistency of modeling parameters over use of animal-specific properties in only a portion of the model. The relative stress distribution is insensitive to small differences in elastic moduli as well as to large differences as long as the relative order of magnitude of the tissue components remains constant, and the order of magnitude between tissue components in our model is comparable with those measured by Tracqui et al. (36). Additionally, Ohayon et al. (22) recently found that the use of human material properties to calculate murine solid mechanics overestimates stresses inside of plaques. Therefore, because we used human material properties, our methods likely led to an overestimation of the frequency of local maxima of stress in mice.
Preliminary modeling revealed that human tissues suffered from tissue processing artifacts, likely related to paraffin embedding and sectioning. Von Mises stress and strain maps revealed stress and strain concentrations at sites of morphological distortion in the original histology, such as steeply curved edges of the lumen. These concentrations were not located at sites related to the composition of the plaque, such as at the edge of necrotic core or fibrous cap, and were instead at seemingly random locations. Because these probably resulted from tissue distortion during embedding and sectioning rather than from the innate biomechanics of human plaques, we “preinflated” these plaques in our computer model by applying a pressure of 40 mmHg normal to the lumen, allowing the tissue to deform, and then remeshing and repeating the process for a total of 10 iterations. All modeling protocols during the preinflation steps were identical to those used for the final inflation step for human tissues as well as for mouse tissues (which were not preinflated because they did not suffer from similar histology artifacts). Von Mises stress data from the 10th iteration was used for our final analysis for human specimens. To ensure that preinflation was not affecting our final results, sensitivity analysis was performed. This demonstrated that stress and strain concentration sites did not change after the 10th iteration and that the only sites that changed between the 1st and 10th iteration were those at sites of presumed tissue distortion.
REFERENCES
- 1. Beattie D, Xu C, Vito R, Glagov S, Whang MC. Mechanical analysis of heterogeneous, atherosclerotic human aorta. J Biomech Eng 120: 602–607, 1998 [DOI] [PubMed] [Google Scholar]
- 2. Bentzon JF, Falk E. Atherosclerotic lesions in mouse and man: is it the same disease? Curr Opin Lipidol 21: 434–440, 2010 [DOI] [PubMed] [Google Scholar]
- 3. Bond AR, Jackson CL. The fat-fed apolipoprotein E knockout mouse brachiocephalic artery in the study of atherosclerotic plaque rupture. J Biomed Biotechnol 2011: 379069, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Broisat A, Toczek J, Mesnier N, Tracqui P, Ghezzi C, Ohayon J, Riou LM. Assessing low levels of mechanical stress in aortic atherosclerotic lesions from apolipoprotein E−/− mice–brief report. Arterioscler Thromb Vasc Biol 31: 1007–1010, 2011 [DOI] [PubMed] [Google Scholar]
- 5. Calara F, Silvestre M, Casanada F, Yuan N, Napoli C, Palinski W. Spontaneous plaque rupture and secondary thrombosis in apolipoprotein E-deficient and LDL receptor-deficient mice. J Pathol 195: 257–263, 2001 [DOI] [PubMed] [Google Scholar]
- 6. Cheng GC, Loree HM, Kamm RD, Fishbein MC, Lee RT. Distribution of circumferential stress in ruptured and stable atherosclerotic lesions. A structural analysis with histopathological correlation. Circulation 87: 1179–1187, 1993 [DOI] [PubMed] [Google Scholar]
- 7. Chodorowski A, Mattsson U, Langille M, Hamarneh G. Color lesion boundary detection using live wire. In: Proceedings of SPIE Medical Imaging: Image Processing. Bellingham, WA: SPIE, 2005, vol. 5747, p. 1589–1596 [Google Scholar]
- 8. Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest 105: 1605–1612, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med 316: 1371–1375, 1987 [DOI] [PubMed] [Google Scholar]
- 10. Hallow KM, Taylor WR, Rachev A, Vito RP. Markers of inflammation collocate with increased wall stress in human coronary arterial plaque. Biomech Model Mechanobiol 8: 473–486, 2009 [DOI] [PubMed] [Google Scholar]
- 11. Hansson GK, Heistad DD. Two views on plaque rupture. Arterioscler Thromb Vasc Biol 27: 697, 2007 [DOI] [PubMed] [Google Scholar]
- 12. Hayenga HN, Trache A, Trzeciakowski J, Humphrey JD. Regional atherosclerotic plaque properties in ApoE−/− mice quantified by atomic force, immunofluorescence, and light microscopy. J Vasc Res 48: 495–504, 2011 [DOI] [PubMed] [Google Scholar]
- 13. Holzapfel GA, Weizsacker HW. Biomechanical behavior of the arterial wall and its numerical characterization. Comput Biol Med 28: 377–392, 1998 [DOI] [PubMed] [Google Scholar]
- 14. Humphrey JD, Rajagopal KR. A constrained mixture model for growth and remodeling of soft tissues. Math Mod Meth Appl S 12: 407–430, 2002 [Google Scholar]
- 15. Jackson CL. Defining and defending murine models of plaque rupture. Arterioscler Thromb Vasc Biol 27: 973–977, 2007 [DOI] [PubMed] [Google Scholar]
- 16. Jawien J. The role of an experimental model of atherosclerosis: apoE-knockout mice in developing new drugs against atherogenesis. Curr Pharm Biotechnol; in press [PubMed] [Google Scholar]
- 17. Jin SX, Shen LH, Nie P, Yuan W, Hu LH, Li DD, Chen XJ, Zhang XK, He B. Endogenous renovascular hypertension combined with low shear stress induces plaque rupture in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 32: 2372–2379, 2012 [DOI] [PubMed] [Google Scholar]
- 18. Johnson J, Carson K, Williams H, Karanam S, Newby A, Angelini G, George S, Jackson C. Plaque rupture after short periods of fat feeding in the apolipoprotein E-knockout mouse: model characterization and effects of pravastatin treatment. Circulation 111: 1422–1430, 2005 [DOI] [PubMed] [Google Scholar]
- 19. Li ZY, Howarth S, Trivedi RA, UKI RAJM, Graves MJ, Brown A, Wang L, Gillard JH. Stress analysis of carotid plaque rupture based on in vivo high resolution MRI. J Biomech 39: 2611–2622, 2006 [DOI] [PubMed] [Google Scholar]
- 20. Maldonado N, Kelly-Arnold A, Vengrenyuk Y, Laudier D, Fallon JT, Virmani R, Cardoso L, Weinbaum S. A mechanistic analysis of the role of microcalcifications in atherosclerotic plaque stability: potential implications for plaque rupture. Am J Physiol Heart Circ Physiol 303: H619–H628, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ohayon J, Finet G, Gharib AM, Herzka DA, Tracqui P, Heroux J, Rioufol G, Kotys MS, Elagha A, Pettigrew RI. Necrotic core thickness and positive arterial remodeling index: emergent biomechanical factors for evaluating the risk of plaque rupture. Am J Physiol Heart Circ Physiol 295: H717–H727, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ohayon J, Mesnier N, Broisat A, Toczek J, Riou L, Tracqui P. Elucidating atherosclerotic vulnerable plaque rupture by modeling cross substitution of ApoE−/− mouse and human plaque components stiffnesses. Biomech Model Mechanobiol 11: 801–813, 2012 [DOI] [PubMed] [Google Scholar]
- 23. Rosenfeld ME, Polinsky P, Virmani R, Kauser K, Rubanyi G, Schwartz SM. Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler Thromb Vasc Biol 20: 2587–2592, 2000 [DOI] [PubMed] [Google Scholar]
- 24. Saraff K, Babamusta F, Cassis LA, Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 23: 1621–1626, 2003 [DOI] [PubMed] [Google Scholar]
- 25. Sasaki T, Kuzuya M, Nakamura K, Cheng XW, Shibata T, Sato K, Iguchi A. A simple method of plaque rupture induction in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 26: 1304–1309, 2006 [DOI] [PubMed] [Google Scholar]
- 26. Schwartz SM, Galis ZS, Rosenfeld ME, Falk E. Plaque rupture in humans and mice. Arterioscler Thromb Vasc Biol 27: 705–713, 2007 [DOI] [PubMed] [Google Scholar]
- 27. Shoelson B. ClusterImg. Natick, MA: The MathWorks, 2006 [Google Scholar]
- 28. Suo J, Ferrara DE, Sorescu D, Guldberg RE, Taylor WR, Giddens DP. Hemodynamic shear stresses in mouse aortas: implications for atherogenesis. Arterioscler Thromb Vasc Biol 27: 346–351, 2007 [DOI] [PubMed] [Google Scholar]
- 29. Tracqui P, Broisat A, Toczek J, Mesnier N, Ohayon J, Riou L. Mapping elasticity moduli of atherosclerotic plaque in situ via atomic force microscopy. J Struct Biol 174: 115–123, 2011 [DOI] [PubMed] [Google Scholar]
- 30. van Ree JH, van den Broek WJ, Dahlmans VE, Groot PH, Vidgeon-Hart M, Frants RR, Wieringa B, Havekes LM, Hofker MH. Diet-induced hypercholesterolemia and atherosclerosis in heterozygous apolipoprotein E-deficient mice. Atherosclerosis 111: 25–37, 1994 [DOI] [PubMed] [Google Scholar]
- 31. Vengrenyuk Y, Carlier S, Xanthos S, Cardoso L, Ganatos P, Virmani R, Einav S, Gilchrist L, Weinbaum S. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc Natl Acad Sci USA 103: 14678–14683, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vengrenyuk Y, Kaplan TJ, Cardoso L, Randolph GJ, Weinbaum S. Computational stress analysis of atherosclerotic plaques in ApoE knockout mice. Ann Biomed Eng 38: 738–747, 2010 [DOI] [PubMed] [Google Scholar]
- 33. Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol 47: C13–18, 2006 [DOI] [PubMed] [Google Scholar]
- 34. Weiss D, Kools JJ, Taylor WR. Angiotensin II-induced hypertension accelerates the development of atherosclerosis in apoE-deficient mice. Circulation 103: 448–454, 2001 [DOI] [PubMed] [Google Scholar]
- 35. Weiss D, Sorescu D, Taylor WR. Angiotensin II and atherosclerosis. Am J Cardiol 87: 25C–32C, 2001 [DOI] [PubMed] [Google Scholar]
- 36. Williamson SD, Lam Y, Younis HF, Huang H, Patel S, Kaazempur-Mofrad MR, Kamm RD. On the sensitivity of wall stresses in diseased arteries to variable material properties. J Biomech Eng 125: 147–155, 2003 [DOI] [PubMed] [Google Scholar]
- 37. Yezzi AJ, Jr, Prince JL. An Eulerian PDE approach for computing tissue thickness. IEEE Trans Med Imaging 22: 1332–1339, 2003 [DOI] [PubMed] [Google Scholar]
- 38. Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, Princen HM, Kooistra T. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol 27: 1706–1721, 2007 [DOI] [PubMed] [Google Scholar]