

Abstract
The vasodilator signals regulating muscle blood flow during exercise are unclear. We tested the hypothesis that in young adults leg muscle vasodilation during steady-state exercise would be reduced independently by sequential pharmacological inhibition of nitric oxide synthase (NOS) and cyclooxygenase (COX) with NG-nitro-l-arginine methyl ester (l-NAME) and ketorolac, respectively. We tested a second hypothesis that NOS and COX inhibition would increase leg oxygen consumption (V̇o2) based on the reported inhibition of mitochondrial respiration by nitric oxide. In 13 young adults, we measured heart rate (ECG), blood pressure (femoral venous and arterial catheters), blood gases, and venous oxygen saturation (indwelling femoral venous oximeter) during prolonged (25 min) steady-state dynamic knee extension exercise (60 kick/min, 19 W). Leg blood flow (LBF) was determined by Doppler ultrasound of the femoral artery. Whole body V̇o2 was measured, and leg V̇o2 was calculated from blood gases and LBF. Resting intra-arterial infusions of acetylcholine (ACh) and nitroprusside (NTP) tested inhibitor efficacy. Leg vascular conductance (LVC) to ACh was reduced up to 53 ± 4% by l-NAME + ketorolac infusion, and the LVC responses to NTP were unaltered. Exercise increased LVC from 4 ± 1 to 33.1 ± 2 ml·min−1·mmHg−1 and tended to decrease after l-NAME infusion (31 ± 2 ml·min−1·mmHg−1, P = 0.09). With subsequent administration of ketorolac LVC decreased to 29.6 ± 2 ml·min−1·mmHg−1 (P = 0.02; n = 9). While exercise continued, LVC returned to control values (33 ± 2 ml·min−1·mmHg−1) within 3 min, suggesting involvement of additional vasodilator mechanisms. In four additional subjects, LVC tended to decrease with l-NAME infusion alone (P = 0.08) but did not demonstrate the transient recovery. Whole body and leg V̇o2 increased with exercise but were not altered by l-NAME or l-NAME + ketorolac. These data indicate a modest role for NOS- and COX-mediated vasodilation in the leg of exercising humans during prolonged steady-state exercise, which can be restored acutely. Furthermore, NOS and COX do not appear to influence muscle V̇o2 in untrained healthy young adults.
Keywords: femoral blood flow, nitric oxide, vascular control
exercise hyperemia appears to be regulated by a complex interaction of metabolic, physical, neural, and vascular signals. The importance of local factors such as nitric oxide (NO) and prostaglandins (PGs) derived from nitric oxide synthase (NOS) and cyclooxygenase (COX), respectively, is unclear. Contrasting evidence exists both for and against a role for NO and PGs in regulating exercise hyperemia (3, 7, 8, 10, 14, 16, 26, 31, 36, 37). For example, during forearm exercise NOS inhibition can rapidly reduce hyperemia by ∼20%, and COX inhibition reduces blood flow by 12% (26). However, oral COX inhibitors do not reduce the active hyperemia during forearm exercise in young subjects (32) or during leg exercise in middle-aged men (14). During leg exercise, the effect of NOS inhibition seems to be more modest [0–10% reduction in leg vascular conductance (LVC)], and effects on hyperemia are more apparent during inhibition of a second system (i.e., COX pathway) (3, 16, 23). Thus it appears that the magnitude and interaction of local factors contributing to exercise hyperemia may be influenced by factors including age, fitness, or the vascular bed and muscle group being examined (i.e., arm vs. leg).
The above findings support the concept that local production of NO and PGs [and potentially endothelium-derived hyperpolarizing factor (EDHF)] interact to compensate for the loss of any one vasodilator signal (redundancy). This may further explain why inhibitors administered orally or enzyme inhibition before exercise has not been shown to consistently alter exercise hyperemia in humans. Further evidence for the redundant vasodilator concept is found in the exercising forearm, where COX inhibition led to a transient reduction (only 2–3 min) in blood flow by ∼12% before forearm blood flow was restored (26). The identity of the redundant signal(s), as well as the complex signaling interactions between the products of the NOS and COX enzyme pathways, remain speculative.
Since exercise hyperemia is tightly coupled to metabolism, an idea that has emerged suggests that NO also directly reduces mitochondrial respiration (2, 27, 28). If this is the case, NOS inhibition would increase muscle oxygen consumption (V̇o2) and perhaps stimulate an increase in blood flow to support the increased O2 demand. Thus the “vascular role” of NO might be masked by the “metabolic role” of NO as a modulator of muscle mitochondrial metabolism. This dual role of NO remains controversial, and several studies in humans do not support the idea that NOS inhibition alters leg V̇o2 (4, 7, 20, 23). Recent work during leg exercise suggests that NOS inhibition has the opposite effect on V̇o2; leg V̇o2 was reduced by ∼16%, but only when NOS inhibition was coupled with COX inhibition (16, 17). However, those results directly contrast with findings in canine models suggesting that loss of NO leads to increased hindlimb V̇o2 at rest and during treadmill exercise (27, 28). Thus the idea that NO (and/or PGs) can directly modify muscle V̇o2 in a way that might influence blood flow may be species dependent and remains an open question.
Thus the present study was designed to test the hypothesis that NO and PG blockade independently reduce leg vasodilation during continuous steady-state dynamic exercise. We also tested the hypothesis that NOS inhibition would result in increased V̇o2 in the exercising leg. These two hypotheses were examined by measuring leg blood flow (LBF) and blood gases from rest to exercise (∼19 W) with subsequent infusion of NOS and COX inhibitors in the working leg to ensure their delivery to the active parts of the contracting muscles. We also measured whole body and whole leg V̇o2 and measured deep venous oxygen saturation continuously with an indwelling oximeter to provide insight into the acute effects of NOS and COX inhibitors on the time course of oxygen delivery and utilization in the working leg.
METHODS
Subjects
Fourteen young healthy subjects (12 men and 2 women) were recruited. Subjects were nonobese [body mass index (BMI) < 30 kg/m2], free of cardiovascular disease, nonsmoking, and normotensive. All procedures were approved by the Mayo Institutional Review Board, and all subjects provided informed consent before participation. Subjects were taking no medications other than daily vitamins. Subjects reported normal daily activity, but none was participating in regular physical training. All subjects refrained from caffeine and exercise for at least 24 h and from NSAIDs for at least 48 h. Women were studied in the early follicular phase of the menstrual cycle. Because of a technical malfunction with blood flow measurements data from one woman were excluded. Her hemodynamic responses [e.g., cardiac output, blood pressure (BP), heart rate (HR)] were similar to those of the remaining subjects but were not included in the analysis.
Subject Monitoring
HR was measured by three-lead ECG. Arterial blood pressure (BP) was measured via femoral catheter.
Screening Visit
At least 1 wk before the study day, subjects became familiarized with the leg ergometer exercise on a screening visit. After a dual-energy X-ray absorptiometry (DEXA) scan to determine leg volume, peak V̇o2 (V̇o2peak) and peak leg ergometer workload were determined with a graded maximal knee extension exercise protocol consisting of 1- to 2-min workload increments. HR was determined by ECG, and changes in cardiac output and BP were measured noninvasively with model flow methodology on a Finometer (Finapres Medical Systems).
Instrumentation
A 20-gauge, 10-cm catheter was placed in the femoral artery of the right leg under aseptic conditions after local anesthesia (2% lidocaine) for administration of local vasoactive drugs. The arterial catheter was connected to a pressure transducer and continuously flushed at 3 ml/h with heparinized saline. A side port system permitted concurrent administration of study drugs and measurement of arterial BP (6). For venous blood samples an 18-gauge flexible single-lumen catheter was inserted into the femoral vein 2 cm below the inguinal ligament, advanced 14 cm proximal, and secured in place. This venous catheter was equipped with an oximeter [Presep central venous oximetry catheter (ScVO2), Edwards Life Sciences, Irvine, CA] on the tip, allowing continuous monitoring of venous blood oxygen saturations.
Study Visit
Figure 1 demonstrates the timeline for the experimental session. On the study day, subjects arrived 1 h after a light meal. Catheters were placed in the right leg (exercising) for measures of arterial BP, venous oxygen saturation, arterial drug infusion, and arterial and venous blood sampling (for blood gases and catecholamines). After a 30-min rest period, supine subjects received arterial infusions of acetylcholine (ACh; 2 and 8 μg·100 ml leg volume−1·min−1) and nitroprusside (NTP; 0.5 and 2 μg·100 ml leg volume−1·min−1) in a random order for 5 min per dose. Each dose was separated by 5–10 min for drug washout. Subjects then moved to the leg ergometer for the exercise protocol. An absolute workload of 19 W was chosen based on pilot work that demonstrated that untrained adults could maintain this effort without fatigue while achieving high LBF.
Fig. 1.
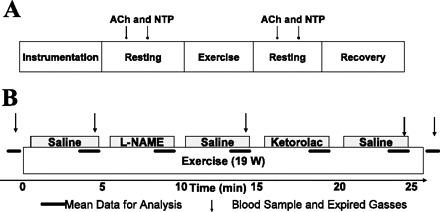
Experimental timeline. A: after ∼60 min of instrumentation subjects rested quietly in supine position for 30 min before drug infusion [acetylcholine (ACh) or nitroprusside (NTP)]. Subjects were then seated in the leg ergometer for at least 5 min before starting continuous, dynamic 1-leg knee extensor exercise. After exercise, subjects lay supine for repeated ACh and NTP infusions. Catheters were then removed, and subjects recovered for 2 h while catheter sites were monitored for healing. B: data were collected specifically during minutes 4 and 5 of each 5-min portion of the exercise bout. Gray boxes indicate drug infusion during each 5-min period. Horizontal bars indicate times for key data collection, and arrows indicate blood sampling. l-NAME, NG-nitro-l-arginine methyl ester.
Protocol 1 (n = 9).
After a 5-min baseline, subjects performed 25 min of continuous leg kicking exercise (60 kicks/min, 19 W). The arterial infusions during each 5-min portion of exercise were, in order, 1) saline, 2) NG-nitro-l-arginine methyl ester (l-NAME), 3) saline, 4) ketorolac, and 5) saline. Expired gases were collected in meteorological balloons and analyzed to calculate whole body V̇o2. Arterial and venous blood gases drawn at 5-min intervals (Fig. 1) were used with LBF measures to calculate leg V̇o2. Subjects then returned to the supine position to repeat the arterial infusions of ACh and NTP. All instrumentation was removed, and subjects rested quietly in the Clinical Research Unit for 2 h with pressure on the catheter sites.
Protocol 2 (n = 4).
All measures were identical to those in protocol 1; however, subjects performed only 15 min of exercise and received arterial infusions during each 5-min portion of exercise, in order: 1) saline, 2) l-NAME, and 3) saline. Whole body V̇o2 was not measured in protocol 2.
Rationale for 25-min Protocol
We chose a 25-min protocol rather than a 20-min protocol [used in forearm blood flow studies (25, 26)] for several key reasons. First, we wanted to be certain that steady-state leg hemodynamics were achieved between 3 and 5 min. Previous forearm exercise studies in our laboratory demonstrated that 3 min was sufficient time to achieve steady-state exercise. Second, we needed to include sufficient time to draw blood samples at key intervals to address the oxygen consumption hypothesis. Third, based on the transient blood flow changes with ketorolac in the forearm, we decided to proceed more conservatively during leg exercise to ensure that potential transient changes in blood flow were observed.
Blood Sampling
Arterial and venous blood samples (3 ml each) were collected anaerobically in heparinized plastic syringes, placed on ice, and analyzed within 15 min with an IL-1620 blood gas analyzer. All blood gas measurements were adjusted to 37°C. Plasma catecholamines (norepinephrine and epinephrine) were measured with high-performance liquid chromatography.
Leg Exercise
Subjects performed “leg kicking” exercise on a modified bike used for single-leg knee extension exercise to isolate the quadriceps muscle (1). Force was continuously measured while subjects kicked in time to a metronome set at 1 Hz.
Leg Blood Flow
LBF was measured by Doppler ultrasound. A high-resolution 7- to 4-MHz transducer (GE Vivid 7, Milwaukee, WI) was used to measure mean blood velocity and vessel diameter of the right common femoral artery, distal to the inguinal ligament and above the bifurcation into the superficial and profunda femoral branch. For velocity measurements, the artery was insonated at a constant angle of 60° with the sample volume adjusted to cover the width of the artery. Velocity measurements were taken during the fourth and fifth minutes of each 5-min section of exercise. A commercial interface unit (Multigon Industries, Yonkers, NY) processed the angle-corrected, intensity-weighted Doppler audio information from the GE Vivid ultrasound system into a flow velocity signal that was sampled in real time by PowerLab (AD Instruments, Colorado Springs, CO) at 1 kHz. Postprocessing with PowerLab's Chart application package yielded mean blood velocities. High-resolution diameter measurements were obtained by perpendicular insonation of the femoral artery. Common femoral artery diameter was determined from a mean of five diastolic diameters recorded during the last 15 s of minute 4 in each 5-min period. Previous leg exercise studies indicated that femoral artery diameter is similar in systole and diastole and stable during exercise (22). LBF was calculated as the product of femoral artery cross-sectional area and mean blood velocity.
Whole Leg Drug Administration
To normalize drug doses, each subject's total fat-free leg volume was calculated from whole body DEXA (GE Lunar) scans. All study drugs were dissolved in saline and infused at rates of 2–3 ml/min directly into the femoral artery catheter. l-NAME was infused at 200 μg · 100 ml fat-free leg volume−1 · min−1 for 5 min to inhibit NO synthesis. This local (leg) intra-arterial dose of l-NAME is 2.5-fold greater (on a per kg basis) than the systemic dose (intravenous) of this compound known to reduce NOS activity in human muscle by 60–70% and remain at high levels in the circulation for at least 90 min after infusion stops (7). Ketorolac was infused at 30 μg · 100 ml fat-free leg volume−1 · min−1 for 5 min to block the COX enzyme. Ketorolac works via the same mechanisms of action as the more commonly used indomethacin; in fact, ketorolac is equally or more efficacious at COX inhibition (35). This dose was based on previous work in the exercising forearm and results in a whole body dose (∼15 mg) delivered locally to ∼2 kg of quadriceps muscle. We designed our study to follow NOS inhibition with COX inhibition since previous work in the leg suggests that COX inhibition alone has no effect on LBF (16). ACh was infused at 2 and 8 μg · 100 ml fat-free leg volume−1 · min−1 at rest to determine endothelial responsiveness. Each dose was infused for up to 5 min to attain a stable vasodilatory response. NTP was infused at 0.5 and 2 μg · 100 ml fat-free leg volume−1 · min−1 at rest to determine vascular smooth muscle responsiveness.
Time Control Studies
Five additional subjects were studied without invasive monitoring in an identical exercise protocol to assess the potential for drift in hemodynamic variables over 25 min of steady-state exercise. Using the same equipment, we measured HR, BP, and cardiac output (Finometer), whole body V̇o2, and LBF. To mimic the time course of the main study, subjects laid supine for 30 min after instrumentation, followed by 10 min seated in the leg ergometer. Subjects performed single-leg exercise at 19 W (1 Hz) for 25 min, followed by 5-min seated recovery and 30-min supine recovery. The 30-min supine intervals were used to compare basal LBF used in analysis of ACh and NTP infusions before and after l-NAME and ketorolac drug infusions and tested for baseline effects of NOS and COX inhibition on LBF.
Data Collection and Statistical Analysis
All key study variables (HR, arterial BP, venous BP, venous saturation, leg ergometer workload, and blood velocity) were collected at 1 kHz on a Powerlab system. For analysis, 20- to 30-s sections of data were averaged for each minute of exercise, and the last 2 min of each 5-min stage (steady state) were averaged for comparisons. LVC was calculated as LVC = LBF/leg BP, where BP = leg arterial pressure − venous pressure, and expressed in milliliters per minute per millimeter of mercury.
The primary analysis was to test whether subjects' LBF or LVC changed in response to l-NAME and ketorolac infusion; therefore, hemodynamic and blood flow data were analyzed by repeated-measures ANOVA (SPSS version 13). All data are expressed as means ± SE. Significance for all comparisons was set a priori at P < 0.05; trends were considered at P < 0.10.
RESULTS
Subject Characteristics
Subjects were 27 ± 5 yr old, 180 ± 2 cm tall, weighed 86 ± 4 kg, and had a BMI of 26.3 ± 0.6 kg/m2. Subject V̇o2peak for single-leg ergometer exercise was 1.3 ± 0.2 l O2/min, and peak workload was 64 ± 6 W. Steady-state submaximal exercise (19.0 ± 0.3 W) corresponded to 33 ± 3% of peak work. The mean leg fat-free volume (inguinal line to ankle) by DEXA was 8.7 ± 0.5 liters, resulting in l-NAME and ketorolac infusions of 87 ± 5 and 13 ± 1 mg, respectively, over the 5-min infusions.
Screen Day Hemodynamics
Steady-state submaximal exercise was assessed during minutes 3–5 of leg exercise. The workload averaged 19.0 ± 0.2 W. HR increased from 73 ± 6 to 90 ± 5 beats per minute (bpm), mean arterial pressure (MAP) increased from 86 ± 6 to 102 ± 6 mmHg, and cardiac output increased from 7.7 ± 0.5 to 11.2 ± 0.7 l/min, an increase of 3.5 ± 0.7 l/min. Whole body V̇o2 increased from 259 ± 16 to 647 ± 61 ml O2/min (n = 7) for exercise.
Study Day Hemodynamics
Table 1 summarizes hemodynamic variables from rest, exercise, and recovery. Exercise increased HR from 77 ± 5 to 96 ± 6 bpm, MAP increased from 97 ± 4 to 108 ± 4 mmHg, and femoral artery diameter did not change significantly with exercise or drug infusion from resting measurements (9.6 ± 0.4 mm). LBF increased from 0.4 ± 0.04 to 3.6 ± 0.2 l/min, an increase of 3.2 ± 0.2 l/min (Fig. 2A). LVC increased from 4 ± 1 to 33 ± 2 ml · min−1 · mmHg−1 (Fig. 2B). Similar responses were observed in protocol 2 (n = 4) (P = 0.66, 0.46, and 0.91 for MAP, HR, and LBF, respectively). Table 2 summarizes key blood values during the same time intervals of rest, exercise, drug infusion, and recovery from both protocols.
Table 1.
Hemodynamics
| Baseline | Exercise | l-NAME | Post-l-NAME | Ketorolac | Postketorolac | Recovery | |
|---|---|---|---|---|---|---|---|
| Protocol 1: l-NAME + ketorolac | |||||||
| HR, beats/min | 77 ± 5* | 96 ± 6 | 98 ± 5 | 97 ± 6 | 89 ± 6 | 91 ± 4 | 76 ± 6 |
| MAP, mmHg | 97 ± 4* | 108 ± 4 | 113 ± 4 | 114 ± 4 | 116 ± 4 | 112 ± 5 | 101 ± 3 |
| MVP, mmHg | 2 ± 1 | 5 ± 1 | 4 ± 1 | 4 ± 1 | 3 ± 1 | 3 ± 1 | 1 ± 1 |
| Femoral artery diameter, cm | 0.96 ± 0.04 | 0.97 ± 0.04 | 0.97 ± 0.04 | 0.97 ± 0.03 | 0.98 ± 0.04 | 0.97 ± 0.03 | 0.95 ± 0.05 |
| LBF, l/min | 0.4 ± 0.0* | 3.6 ± 0.2 | 3.5 ± 0.2 | 3.5 ± 0.2 | 3.4 ± 0.2 | 3.7 ± 0.3 | 0.4 ± 0.0* |
| LVC, l·min−1·mmHg−1 | 4.0 ± 0.6* | 33.1 ± 2.0 | 31.3 ± 2.1 | 31.0 ± 2.1 | 29.6 ± 1.7* | 33.0 ± 2.0 | 4.4 ± 0.3* |
| Venous O2 saturation, % | 60 ± 2* | 45 ± 4 | 40 ± 4 | 37 ± 2 | 36 ± 3 | 34 ± 3 | 46 ± 4 |
| Protocol 2: l-NAME only | |||||||
| HR, beats/min | 64 ± 6* | 94 ± 10 | 100 ± 5 | 93 ± 11 | N/A | N/A | 77 ± 9 |
| MAP, mmHg | 105 ± 3* | 117 ± 5 | 122 ± 4 | 123 ± 4 | N/A | N/A | 115 ± 4 |
| MVP, mmHg | 2 ± 2 | 4 ± 1 | 4 ± 1 | 5 ± 1 | N/A | N/A | 1 ± 1 |
| Femoral artery diameter, cm | 0.99 ± 0.03 | 0.98 ± 0.03 | 0.98 ± 0.04 | 0.98 ± 0.04 | N/A | N/A | 0.98 ± 0.03 |
| LBF, l/min | 0.2 ± 0.0* | 3.3 ± 0.4 | 3.2 ± 0.4 | 3.2 ± 0.2 | N/A | N/A | 0.4 ± 0.0* |
| LVC, l·min−1·mmHg−1 | 2.3 ± 0.1* | 29.6 ± 5.0 | 26.4 ± 4.0 | 25.8 ± 2.0 | N/A | N/A | 3.4 ± 0.7* |
| Venous O2 saturation, % | 50 ± 2 | 27 ± 3 | 22 ± 2 | 22 ± 2 | N/A | N/A | 39 ± 3* |
Values are means ± SE from the 4th and 5th minutes of each 5-min portion of the exercise bout. Heart rate (HR), mean arterial pressure (MAP), and leg blood flow (LBF) increased with exercise but did not change significantly with drug infusion. Leg vascular conductance (LVC) increased with exercise and decreased below control exercise values after ketorolac infusion. Femoral venous saturation decreased with exercise and decreased significantly after NG-nitro-l-arginine methyl ester (l-NAME) + ketorolac infusion. MVP, mean venous pressure; N/A, not applicable. Similar patterns were observed for protocol 2 (n = 4).
Significantly different from 5-min steady-state exercise values (P < 0.05).
Fig. 2.
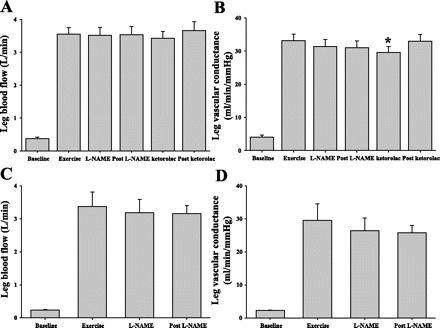
Leg hemodynamics with exercise during nitric oxide synthase (NOS) and cyclooxygenase (COX) inhibition. Leg blood flow did not change with either NG-nitro-l-arginine methyl ester (l-NAME) + ketorolac (A) or l-NAME alone (C). When normalized for blood pressure, leg vascular conductance (LVC) decreased slightly with l-NAME infusion (B, P = 0.09; D, P = 0.08). Addition of ketorolac significantly reduced LVC (B, P = 0.02), which returned to control exercise levels shortly after ketorolac infusion. *Statistically different from control exercise (P = 0.02).
Table 2.
Arterial and venous blood gases
| Baseline | Steady-State Exercise | Post-l-NAME | Postketorolac | Recovery | |
|---|---|---|---|---|---|
| Protocol 1: l-NAME + ketorolac | |||||
| Po2, mmHg | |||||
| a | 97 ± 3 | 106 ± 3 | 104 ± 3 | 103 ± 2 | 98 |
| v | 33 ± 1* | 28 ± 1 | 24 ± 1 | 23 ± 1 | 31 ± 2* |
| Pco2, mmHg | |||||
| a | 39 ± 1 | 38 ± 2 | 38 ± 1 | 37 ± 2 | 37 ± 1 |
| v | 47 ± 2* | 56 ± 3 | 56 ± 2 | 57 ± 1 | 49 ± 3* |
| pH | |||||
| a | 7.42 ± 0 | 7.41 ± 0 | 7.41 ± 0 | 7.42 ± 0 | 7.41 ± 0 |
| v | 7.38 ± 0 | 7.33 ± 0 | 7.34 ± 0 | 7.33 ± 0 | 7.36 ± 0 |
| HCO3−, mM | |||||
| a | 25 ± 1 | 23 ± 1 | 24 ± 1 | 23 ± 1 | 23 ± 1 |
| v | 28 ± 1 | 29 ± 1 | 29 ± 1 | 29 ± 1 | 26 ± 1 |
| Hb, g/dl | |||||
| a | 14.6 ± 1 | 15.1 ± 1 | 15.4 ± 1 | 15.0 ± 1 | 14.5 ± 1 |
| v | 14.8 ± 1 | 15.3 ± 1 | 15.1 ± 1 | 15.2 ± 1 | 14.7 ± 0 |
| O2 saturation, % | |||||
| a | 96 ± 0 | 97 ± 0 | 97 ± 0 | 97 ± 0 | 96 ± 0 |
| v | 58 ± 2* | 45 ± 3 | 35 ± 2 | 32 ± 2* | 52 ± 5* |
| O2 content, ml/dl | |||||
| a | 19.8 ± 1 | 20.7 ± 1 | 21.1 ± 1 | 20.5 ± 1 | 19.7 ± 1 |
| v | 12.0 ± 1* | 9.5 ± 1 | 7.3 ± 0 | 6.8 ± 0 | 10.5 ± 1 |
| Leg V̇o2, ml/min | 30 ± 5* | 441 ± 45 | 489 ± 37 | 512 ± 39 | 43 ± 9* |
| Protocol 2: l-NAME only | |||||
| Po2, mmHg | |||||
| a | 89 ± 4 | 108 ± 6 | 108 ± 7 | N/A | 91 ± 2 |
| v | 30 ± 1* | 24 ± 1 | 22 ± 1 | N/A | 28 ± 2* |
| Pco2, mmHg | |||||
| a | 42 ± 1 | 38 ± 0 | 37 ± 1 | N/A | 39 ± 1 |
| v | 49 ± 1* | 66 ± 3 | 64 ± 2 | N/A | 52 ± 3* |
| pH | |||||
| a | 7.40 ± 0 | 7.36 ± 0 | 7.41 ± 0 | N/A | 7.41 ± 0 |
| v | 7.38 ± 0 | 7.27 ± 0 | 7.30 ± 0 | N/A | 7.35 ± 0 |
| HCO3−, mM | |||||
| a | 25 ± 0 | 22 ± 2 | 23 ± 0 | N/A | 24 ± 0 |
| v | 28 ± 0 | 30 ± 0 | 30 ± 0 | N/A | 28 ± 1 |
| Hb, g/dl | |||||
| a | 16 ± 0 | 15.7 ± 1 | 16.3 ± 0 | N/A | 15.8 ± 0 |
| v | 15.9 ± 0 | 16.5 ± 0 | 16.5 ± 0 | N/A | 16.0 ± 0 |
| O2 saturation, % | |||||
| a | 94 ± 1 | 96 ± 1 | 96 ± 1 | N/A | 96 ± 0 |
| v | 52 ± 3* | 33 ± 4 | 28 ± 4 | N/A | 47 ± 5* |
| O2 content, ml/dl | |||||
| a | 21.1 ± 0 | 21.2 ± 1 | 22.0 ± 0 | N/A | 21.2 ± 0 |
| v | 11.6 ± 1* | 7.6 ± 1 | 6.5 ± 1 | N/A | 10.7 ± 1 |
| Leg V̇o2, ml/min | 23 ± 1* | 471 ± 102 | 494 ± 55 | N/A | 43 ± 12* |
Blood gas and oxygen consumption values are means ± SE from the 5th minute of each 5-min portion of the exercise bout. Femoral venous saturation decreased with exercise and was significantly reduced after l-NAME and combined l-NAME + ketorolac infusion. Similar patterns were seen in protocol 2 (n = 4), except that saturation was not significantly reduced after l-NAME. V̇o2, oxygen consumption; a and v, arterial and venous samples, respectively.
Statistically different from 5-min steady-state exercise values (P < 0.05).
Effects of NOS and Subsequent COX Inhibition
In response to drug infusion, MAP and HR did not change significantly from steady-state exercise values (P = 0.68 and 0.80, respectively). LBF did not change significantly during exercise with l-NAME or ketorolac infusion (P = 0.97; Fig. 2A), and LVC did not statistically decrease in response to l-NAME (from 33 ± 2 to 31 ± 2 ml · min−1 · mmHg−1, P = 0.09). However, LVC decreased significantly with ketorolac infusion (29.6 ± 2 ml · min−1 · mmHg−1, P = 0.02). Within 3 min after ketorolac infusion (minute 23 of exercise) LVC returned to control (no drug) steady-state exercise levels (33 ± 2 ml · min−1 · mmHg−1; Fig. 2B) without a change in MAP. Similar responses during l-NAME infusion were observed in protocol 2 (n = 4), where LBF did not decrease significantly with l-NAME infusion and LVC decreased slightly, but not statistically, from 29.6 ± 5 to 26 ± 2 ml · min−1 · mmHg−1 (P = 0.08, Fig. 2D). Leg vascular resistance (LVR) followed the inverse trends as LVC, but these changes were not statistically significant (P = 0.11 and 0.45 for protocols 1 and 2, respectively). Table 1 summarizes key hemodynamic values during the same time intervals of rest, exercise, drug infusion, and recovery from both protocols.
Whole Body and Leg Oxygen Consumption
In protocol 1, whole body V̇o2 increased from 289 ± 15 to 709 ± 46 ml O2/min during exercise and leg V̇o2 increased from 30 ± 3 to 441 ± 45 ml O2/min. Neither whole body nor leg V̇o2 changed with l-NAME or ketorolac infusions (P = 0.5–0.8; Fig. 3, Table 2).
Fig. 3.
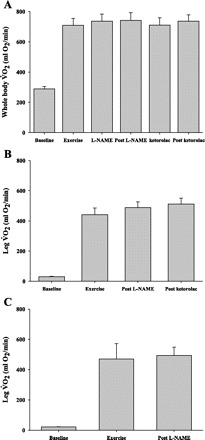
Whole body and leg oxygen consumption (V̇o2). Whole body V̇o2 increased with exercise (protocol 1) but did not change with drug infusion (A). Leg V̇o2 increased with exercise but did not change with drug infusion in protocol 1 (B) or protocol 2 (C).
Deep Venous Oxygen Saturation
The femoral venous catheter contained an indwelling oximeter that provided continuous monitoring of leg venous oxygen saturation. Venous blood oxygen saturation decreased from resting values during exercise (P = 0.01; Fig. 4A). Compared with the same time points used to calculate other hemodynamic data, infusion of l-NAME appeared to reduce saturation by ∼7%, but this was not statistically significant (P = 0.15), nor was it altered by ketorolac infusion (P = 0.2). In protocol 2, a similar pattern emerged with exercise decreasing saturation (P < 0.01), and l-NAME infusion showed a trend (P = 0.1, Fig. 4B) for decreased venous saturation. Venous blood sampling for blood gas measures indicated a similar pattern for Hb oxygen saturation. Specifically in protocol 1, venous Hb oxygen saturation tended to decrease after l-NAME (P = 0.07) but was statistically reduced after combined l-NAME + ketorolac infusion (P = 0.01). In protocol 2, the reduction in venous oxygen saturation (∼6%) was not as apparent (P = 0.2; Fig. 4B). Importantly, no transient reductions in deep venous saturation were observed at the onset of either drug, indicating that there was not an acute drop in flow followed by rapid recovery to steady-state values.
Fig. 4.
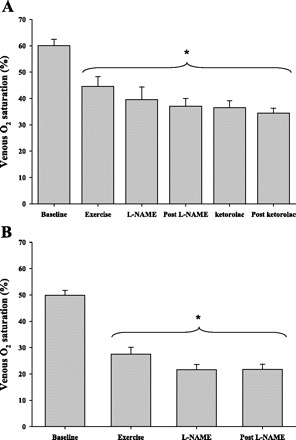
Continuous monitoring of femoral venous oxygen saturation during exercise. A: venous saturation from indwelling oximeter decreased from rest to exercise (P = 0.01) but did not significantly change during drug infusion (l-NAME + ketorolac) (P = 0.2). B: a similar pattern was observed during l-NAME-only infusion (P = 0.11). *Statistically different from baseline (nonexercising) values (P < 0.05).
Plasma Catecholamines
In protocol 1, arterial norepinephrine and epinephrine values did not significantly change from rest to exercise or with l-NAME + ketorolac infusion (Table 3). In protocol 2, arterial norepinephrine and epinephrine values did not significantly change from rest to exercise or with l-NAME infusion (Table 4).
Table 3.
Catecholamine values in protocol 1 (l-NAME + ketorolac)
| Baseline | Steady-State Exercise | Post-l-NAME | Postketorolac | Recovery | |
|---|---|---|---|---|---|
| Norepinephrine | |||||
| a | 227 ± 19 | 290 ± 28 | 272 ± 27 | 219 ± 24 | 205 ± 23 |
| v | 261 ± 25 | 311 ± 29 | 283 ± 36 | 229 ± 22 | 237 ± 27 |
| Epinephrine | |||||
| a | 61 ± 12 | 77 ± 17 | 73 ± 16 | 75 ± 17 | 73 ± 17 |
| v | 26 ± 3 | 66 ± 15 | 60 ± 14 | 57 ± 15 | 48 ± 18 |
Catecholamine values are means ± SE from the 5th minutes of 5-min portions of the exercise bout. Neither exercise nor drug infusions statistically changed arterial or venous norepinephrine or epinephrine (P = 0.1–0.98).
Table 4.
Catecholamine values in protocol 2 (l-NAME only)
| Baseline | Steady-State Exercise | End Exercise | Recovery | |
|---|---|---|---|---|
| Norepinephrine | ||||
| a | 196 ± 1 | 261 ± 29 | 228 ± 33 | 163 ± 21 |
| v | 229 ± 45 | 275 ± 34 | 230 ± 42 | 185 ± 14 |
| Epinephrine | ||||
| a | 58 ± 20 | 86 ± 36 | 61 ± 10 | 42 ± 12 |
| v | 19 ± 3 | 62 ± 16 | 48 ± 9 | 12 ± 3 |
Catecholamine values are means ± SE from the 5th minutes of 5-min portions of the exercise bout. Neither exercise nor drug infusions statistically changed arterial or venous norepinephrine or epinephrine (P = 0.45–0.55).
Endothelium-Dependent and -Independent Vasodilation
LBF and LVC increased in response to femoral arterial infusion of ACh in a dose-dependent fashion. Administration of l-NAME + ketorolac infusion blunted ACh-mediated vasodilation by 53 ± 4% (P = 0.01) and 18 ± 7% for the low- and high-ACh concentration infusions, respectively (Fig. 5A). Similar responses were obtained when analyzing data as LBF (not shown).
Fig. 5.
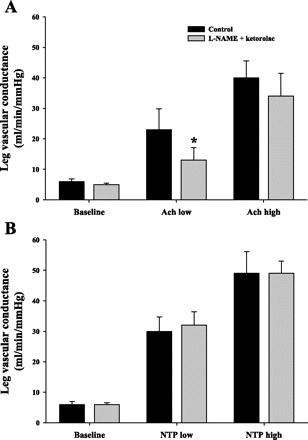
Leg vasodilation responses to endothelium-dependent and -independent agonist infusion. Control responses before exercise protocol and responses following exercise protocol after l-NAME + ketorolac infusion are shown. l-NAME + ketorolac reduced vasodilation to ACh (A, P = 0.01) but not to NTP (B, P = 0.6). *Statistically different from control (P < 0.05).
LBF and LVC increased in response to femoral arterial infusion of NTP in a dose-dependent fashion. There was no effect of l-NAME + ketorolac infusion on the increase in LVC compared with control (saline) (Fig. 5B). Similar responses were obtained when analyzing data as LBF (not shown).
Time Control Experiment
V̇o2, LBF, HR, MAP, and cardiac output all increased from rest to exercise in five time control subjects. Data are summarized in Table 5. The increase in oxygen consumption was very similar to that in the main protocol. HR tended to increase during exercise (P = 0.06). There were no significant changes in MAP (P = 0.55), LBF (P = 0.98), LVC (P = 0.91), LVR (P = 0.8), or whole body V̇o2 (P = 0.87) throughout 25 min of leg exercise.
Table 5.
Time control study hemodynamic variables
| Leg-Kicking Exercise |
|||||||
|---|---|---|---|---|---|---|---|
| Supine Rest | Seated Baseline | 5 min | 10 min | 15 min | 20 min | 25 min | |
| HR, beats/min | 57 ± 2 | 61 ± 3 | 71 ± 2* | 72 ± 2* | 77 ± 2* | 76 ± 1* | 78 ± 2* |
| MAP, mmHg | 75 ± 3 | 80 ± 3 | 89 ± 4* | 84 ± 4* | 83 ± 4* | 82 ± 3* | 82 ± 2* |
| Cardiac output, l/min | 7 ± 1 | 7 ± 1 | 9 ± 1* | 9 ± 1* | 9 ± 1* | 9 ± 1* | 9 ± 1* |
| Femoral artery diameter, cm | 0.94 ± 0.04 | 1.00 ± 0.04 | 1.00 ± 0.03 | 1.00 ± 0.02 | 1.01 ± 0.03 | 1.01 ± 0.03 | 1.02 ± 0.03 |
| LBF, l/min | 0.4 ± 0.06 | 0.2 ± 0.02 | 1.9 ± 0.2* | 1.9 ± 0.2* | 2.0 ± 0.2* | 2.1 ± 0.3* | 2.0 ± 0.2* |
| LVC, ml·min−1·mmHg−1 | 4.8 ± 0.8 | 2.5 ± 0.2 | 22.1 ± 2.7* | 23.2 ± 2.9* | 25.1 ± 3.2* | 25.8 ± 3.3* | 24.8 ± 2.7* |
| Whole body V̇o2, ml/min | 315 ± 18 | 312 ± 15 | 674 ± 32* | 691 ± 40* | 708 ± 37* | 715 ± 37* | 727 ± 40* |
Hemodynamic values are means ± SE from the 4th and 5th minutes of 5-min portions of the time control exercise bout. Subjects (n = 5) were supine for 30 min before 5 min of seated rest (seated baseline). After the 25-min leg kicking exercise, subjects recovered seated for 5 min and for another 30 min supine (supine recovery). Exercise increased HR, MAP, cardiac output, LBF, LVC, and whole body oxygen consumption (P < 0.05). However, none of these variables changed during 25 min of leg exercise (P = 0.54–0.95) except HR (P = 0.06).
Statistically different from supine rest (P < 0.05).
DISCUSSION
We tested the hypotheses that NO and PG blockade independently reduce leg vasodilation and that NOS or NOS + COX inhibition would result in an increased V̇o2 in the exercising leg. While previous work has examined the roles of NOS and COX during the onset of leg exercise, this investigation is the first to examine their roles during prolonged, low-intensity steady-state leg exercise in humans. In this context, these data are important since factors regulating steady-state exercise blood flow may be different from those factors initiating exercise hyperemia (29). With this model, the primary novel findings were that 1) the combination of NOS and COX inhibition, administered during exercise, transiently attenuated LVC, which was restored to steady-state values within 3 min of exercise, and 2) neither NOS administration alone nor combined NOS and COX inhibition alters leg muscle V̇o2 during prolonged (>10 min) steady-state exercise. Furthermore, NOS and COX may play smaller roles in exercise vasodilation during prolonged low-intensity exercise, indicating that previously “silent” factors replace NOS and COX signaling in regulating steady-state exercise hyperemia.
Our results, combined with previously published data in humans, indicate that the role of NO in regulating leg vasodilation during exercise is modest and, at best, accounts for <10% of the overall response. For example, some studies in humans report no effect (4, 7, 9, 13, 23) of NOS inhibition on LBF, but NO-mediated vasodilation appears to play a modest role when expressed as LVC (7, 23). Data from the present study support this idea, where we observed a trend for an 8–10% decrease in LVC during intra-arterial l-NAME administration during leg exercise (Fig. 2). An important confounding factor when investigating a role for NO in leg and whole body exercise studies is that NOS inhibition can lead to a baroreceptor-mediated sympathetic withdrawal due to a drug-induced increase in arterial pressure. This sympathetic withdrawal may mask at least a portion of the vasoconstriction in the leg vasculature due to NOS inhibition (30). Thus previous reports (7, 23) and the present data may be underestimating a role for NO-mediated vasodilation during leg exercise. Our time control studies (Table 5) support this interpretation, and the modest changes in arterial pressure observed during the drug infusion suggest that baroreflex-mediated changes in sympathetic outflow could have influenced the overall LBF response. Despite the systemic effect of NOS inhibition (7, 23), our data are consistent with the majority of published reports demonstrating a modest role for NO in leg exercise vasodilation when inhibited alone.
PG production increases with exercise (37), which can be inhibited with COX inhibitors like indomethacin (14, 37) or ketorolac. Despite the effective inhibition of COX products, studies report no effect of COX inhibition alone on leg exercise hyperemia (16). Our findings suggest that LVC was significantly reduced when ketorolac was administered in combination with NOS inhibition (Fig. 2B), supporting the concept that the role for PGs in vascular control during moderate leg exercise may be masked by NO-mediated vasodilation (3, 16). Additionally, at least some portion of PG-mediated vasodilation is, in fact, NO dependent (19). This makes it difficult to quantify the “normal” contribution of PGs to the leg exercise hyperemia, since neither NOS nor COX inhibition alone reduced LBF (3, 16). Prior work also suggests a potential intensity-dependent interaction between NO and PGs, where combined inhibition of NOS and COX products reduces LBF at 30-W leg exercise but not at 15 W (3). This intensity-dependent NOS-COX interaction might explain why we observed a significant, albeit smaller, effect of combined inhibition, as our workload was between 15 and 30 W. Furthermore, the vascular effects of combined NOS and COX inhibition in the present study are less (∼10%) than those in recent reports (∼33%; Refs. 16, 17, 20). It is possible that the aerobic fitness level of the study population might influence the magnitude of NO and PG contribution to exercise hyperemia. That is, in the present report, maximum leg ergometer workload was 64 W, which was considerably lower than that published recently (92 W) (16). Given the reports of maximum workloads in truly sedentary adults of 35–40 W (21), it seems that the level of training in published leg exercise studies may vary widely and may affect the relative contribution of vasodilator signals.
When combined with our present data, available information suggests that NOS and COX play a larger role at the onset of exercise and that their contribution diminishes as duration of exercise increases. The concept of time-dependent mechanisms regulating exercise hyperemia is supported by work demonstrating that prolonged treadmill exercise alters sympathetic restraint of LBF compared with the initial response (5). Along these lines, the largest reported contribution for NO and PGs comes from studies that use very short bouts of exercise, typically 5–10 min or less (3, 16, 17, 20). Our study design differed in that we administered l-NAME during steady-state leg exercise (after the first 5 min) and followed with infusion of ketorolac during minutes 15–20 of continuous steady-state exercise. It was at this point where we observed a transient reduction in LVC with combined NOS and COX inhibition. These findings emphasize the idea that the relative contribution of vascular control mechanisms may differ not only with exercise intensity but also as exercise progresses within a given exercise intensity. In this context, it would seem reasonable to speculate that the importance of EDHF and/or adenosine in regulating blood flow to the exercising muscles might increase as the contribution of NOS and COX decrease. Along these lines, it is important to emphasize that key vasoactive signals, other than NO and PGs, are responsible for the majority (>65%) of leg vasodilation during exercise no matter the experimental design and/or exercise model. In the present report, these signals appear to account for at least 90% of the vasodilation during 20–25 min of dynamic exercise.
If NO inhibited mitochondrial function (27, 28), the effect of NOS inhibition on LBF during exercise might have been masked by a compensatory increase in V̇o2 by the exercising leg. Thus we tested a second hypothesis, that inhibition of NO or PGs would increase leg V̇o2. Contrary to our hypothesis, our data do not support a role for either NO or combined NO and PGs in altering whole body or leg V̇o2 (Fig. 3). Our NOS inhibition data are consistent with previous reports in humans (4, 7, 13, 23) as well as whole body V̇o2 in running swine (15); however, they contrast with reports of increased V̇o2 seen in exercising dogs (27, 28). Recent human studies also report that marked reductions in LBF during combined NOS and COX inhibition are accompanied by 13–17% reductions in V̇o2 (16, 17). It is interesting that NOS (7, 9) or COX (16) inhibition alone did not change leg V̇o2, and the reduced leg V̇o2 during double blockade was paralleled by reductions in LBF (∼33%) (16, 17). Therefore it is possible that the reduced leg muscle metabolism at least partially drove the reductions in blood flow (O2 delivery). Taken together, the available human studies suggest that inhibition of NOS does not increase V̇o2 during exercise, and our findings demonstrate that combined COX and NOS inhibition does not alter V̇o2 in the exercising leg.
Experimental Considerations
In addition to possible baroreflex reductions in sympathetic nervous outflow with NOS inhibition, it is possible that insufficient inhibition of NOS and COX may have contributed to our results. Consistent with reports in several human studies using similar drug doses (8, 11, 12, 18, 24, 33, 34), we significantly reduced the LVC response to intra-arterial ACh but not NTP (Fig. 5). Furthermore, the dose of l-NAME infused in the present study was based on whole body intravenous doses that reduce NOS activity by almost 70% (7). Since we infused a similar dose directly into the working leg, we likely provided a much higher regional dose to the exercising limb. Graphically, the acute reductions in deep venous oxygen saturation (Fig. 4) suggest that l-NAME had an almost immediate effect on distribution of blood flow within the leg, leading to increased oxygen extraction. In terms of an effective ketorolac dose, doses of COX inhibitor in the range of 13 ± 1 mg markedly reduce plasma levels of PG metabolites (14, 37), and we administered twice the dose infused in previous leg exercise studies that demonstrate an exercise effect (16).
Using LVR for data analysis suggested that resistance changed in an inverse fashion compared with LVC (Table 1), but our conclusions are the same. Given the linear relationship of LVC, LBF, and vessel radius with conductance and the inverse hyperbolic relationship of resistance, a relatively large change in LBF could occur with only a small change in LVR, which would mask modest changes in vascular diameter (vasoconstriction or vasodilation). Thus we chose to focus on conductance measures as LVC creates a more sensitive measure for vascular caliber changes; especially in studies where drug effects are small, these changes can be missed by using LVR.
Our data also highlight the marked variability in basic LBF responses to 19-W knee extensor exercise. For example, the LBF response in our time control studies was ∼2 l/min, compared with 3.5 l/min in protocol 1. These results are consistent with studies in which the LBF response ranges from 2.5 to 4.5 l/min (1). Furthermore, in all protocols, the estimated rise in cardiac output (Finometer) very closely matched the rise in LBF. Finally, the blood sampling, combined with our LBF measures to calculate leg V̇o2, matched very well with the whole body increase in V̇o2. Taken together, we are confident our measures of LBF are accurate in untrained humans.
Perspectives
The roles of NOS and COX in human exercise hyperemia appear modest; however, a number of questions remain. These include whether the contributions of NO and PGs are influenced by limb studied (i.e., leg vs. arm model), fitness, sex, and aging, along with exercise intensity and duration. For example, NO and PGs appear to signal redundantly in the leg (Refs. 3, 16, 17 and present data), whereas the compensatory vasodilator signal following COX inhibition appears NOS independent in the forearm (26). However, in no case does blockade of these factors alone, or in combination, reduce blood flow or vascular conductance dramatically (3, 7, 16, 17, 20, 23, 25, 26). These observations indicate that 1) NOS and/or COX signaling is not obligatory for the majority of the exercise hyperemia response under almost every circumstance that has been studied or 2) redundant control mechanisms make this topic difficult to address in a definitive way. The latter possibility might also reflect the limited pharmacological tools available to study such a complex phenomenon in humans.
Conclusions
The novel findings from the present study suggest that combined products of NOS and COX play a relatively small role in vasodilation during prolonged steady-state leg exercise. A major new finding is that COX inhibition, when combined with NOS inhibition, caused a transient reduction in LVC, suggesting that vasodilation was restored by an alternative signaling pathway. Our results differ from reports in subjects with greater maximum workloads, suggesting that aerobic fitness/training may alter the contributions of NOS and COX to exercise vasodilation. When combined with previous reports, our exercise model raises the possibility that NOS and COX enzymes may contribute more to exercise hyperemia at the onset of exercise (<10 min) and their contribution diminishes as exercise progresses. Finally, our results demonstrate that NOS or COX products do not influence leg V̇o2 in untrained subjects exercising at modest intensities.
GRANTS
This research was supported by Clinical and Translational Science Award (CTSA) Grant RR-024150, National Institutes of Health Grants HL-46493 (M. J. Joyner), HL-78019 (B. W. Wilkins), HL-091397 (W. G. Schrage), and RR-017520 (J. H. Eisenach), and the Caywood professorship via the Mayo Foundation (M. J. Joyner).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
ACKNOWLEDGMENTS
We express our thanks to the subjects for their time, to Pamela Engrav for administrative organization, and to Karen P. Krucker, Shelly K. Roberts, Tasha Pike, Dr. Christine A. Kenyon, and Madhuri Somaraju for technical assistance.
REFERENCES
- 1. Andersen P, Saltin B. Maximal perfusion of skeletal muscle in man. J Physiol 366: 233–249, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bernstein RD, Ochoa FY, Xu X, Forfia P, Shen W, Thompson CI, Hintze TH. Function and production of nitric oxide in the coronary circulation of the conscious dog during exercise. Circ Res 79: 840–848, 1996 [DOI] [PubMed] [Google Scholar]
- 3. Boushel R, Langberg H, Gemmer C, Olesen J, Crameri R, Scheede C, Sander M, Kjaer M. Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J Physiol 543: 691–698, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bradley SJ, Kingwell BA, McConell GK. Nitric oxide synthase inhibition reduces leg glucose uptake but not blood flow during dynamic exercise in humans. Diabetes 48: 1815–1821, 1999 [DOI] [PubMed] [Google Scholar]
- 5. DeLorey DS, Hamann JJ, Kluess HA, Clifford PS, Buckwalter JB. Alpha-adrenergic receptor-mediated restraint of skeletal muscle blood flow during prolonged exercise. J Appl Physiol 100: 1563–1568, 2006 [DOI] [PubMed] [Google Scholar]
- 6. Dietz NM, Rivera JM, Eggener ES, Fix RJ, Warner DO, Joyner MJ. Nitric oxide contributes to the rise in forearm blood flow during mental stress in humans. J Physiol 480: 361–368, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frandsen U, Bangsbo J, Sander M, Hoffner L, Betak A, Saltin B, Hellsten Y. Exercise-induced hyperaemia and leg oxygen uptake are not altered during effective inhibition of nitric oxide synthase with NG-nitro-l-arginine methyl ester in humans. J Physiol 531: 257–264, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gilligan DM, Panza JA, Kilcoyne CM, Waclawiw MA, Casino PR, Quyyumi AA. Contribution of endothelium-derived nitric oxide to exercise-induced vasodilation. Circulation 90: 2853–2858, 1994 [DOI] [PubMed] [Google Scholar]
- 9. Hillig T, Krustrup P, Fleming I, Osada T, Saltin B, Hellsten Y. Cytochrome P450 2C9 plays an important role in the regulation of exercise-induced skeletal muscle blood flow and oxygen uptake in humans. J Physiol 546: 307–314, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hirai T, Visneski MD, Kearns KJ, Zelis R, Musch TI. Effects of NO synthase inhibition on the muscular blood flow response to treadmill exercise in rats. J Appl Physiol 77: 1288–1293, 1994 [DOI] [PubMed] [Google Scholar]
- 11. Kamper AM, Paul LC, Blauw GJ. Prostaglandins are involved in acetylcholine- and 5-hydroxytryptamine-induced, nitric oxide-mediated vasodilatation in human forearm. J Cardiovasc Pharmacol 40: 922–929, 2002 [DOI] [PubMed] [Google Scholar]
- 12. Katz SD, Krum H. Acetylcholine-mediated vasodilation in the forearm circulation of patients with heart failure: indirect evidence for the role of endothelium-derived hyperpolarizing factor. Am J Cardiol 87: 1089–1092, 2001 [DOI] [PubMed] [Google Scholar]
- 13. Kingwell BA, Formosa M, Muhlmann M, Bradley SJ, McConell GK. Nitric oxide synthase inhibition reduces glucose uptake during exercise in individuals with type 2 diabetes more than in control subjects. Diabetes 51: 2572–2580, 2002 [DOI] [PubMed] [Google Scholar]
- 14. Lang CC, Chomsky DB, Butler J, Kapoor S, Wilson JR. Prostaglandin production contributes to exercise-induced vasodilation in heart failure. J Appl Physiol 83: 1933–1940, 1997 [DOI] [PubMed] [Google Scholar]
- 15. McAllister RM, Newcomer SC, Pope ER, Turk JR, Laughlin MH. Effects of chronic nitric oxide synthase inhibition on responses to acute exercise in swine. J Appl Physiol 104: 186–197, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mortensen SP, Gonzalez-Alonso J, Damsgaard R, Saltin B, Hellsten Y. Inhibition of nitric oxide and prostaglandins, but not endothelial-derived hyperpolarizing factors, reduces blood flow and aerobic energy turnover in the exercising human leg. J Physiol 581: 853–861, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mortensen SP, Nyberg M, Thaning P, Saltin B, Hellsten Y. Adenosine contributes to blood flow regulation in the exercising human leg by increasing prostaglandin and nitric oxide formation. Hypertension 53: 993–999, 2009 [DOI] [PubMed] [Google Scholar]
- 18. Newby DE, Boon NA, Webb DJ. Comparison of forearm vasodilatation to substance P and acetylcholine: contribution of nitric oxide. Clin Sci (Lond) 92: 133–138, 1997 [DOI] [PubMed] [Google Scholar]
- 19. Nicholson WT, Vaa B, Hesse C, Eisenach JH, Joyner MJ. Aging is associated with reduced prostacyclin-mediated dilation in the human forearm. Hypertension 53: 973–978, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nyberg M, Mortensen SP, Saltin B, Hellsten Y, Bangsbo J. Low blood flow at onset of moderate-intensity exercise does not limit muscle oxygen uptake. Am J Physiol Regul Integr Comp Physiol 298: R843–R848, 2010 [DOI] [PubMed] [Google Scholar]
- 21. Parker BA, Smithmyer SL, Pelberg JA, Mishkin AD, Herr MD, Proctor DN. Sex differences in leg vasodilation during graded knee extensor exercise in young adults. J Appl Physiol 103: 1583–1591, 2007 [DOI] [PubMed] [Google Scholar]
- 22. Radegran G. Ultrasound Doppler estimates of femoral artery blood flow during dynamic knee extensor exercise in humans. J Appl Physiol 83: 1383–1388, 1997 [DOI] [PubMed] [Google Scholar]
- 23. Radegran G, Saltin B. Nitric oxide in the regulation of vasomotor tone in human skeletal muscle. Am J Physiol Heart Circ Physiol 276: H1951–H1960, 1999 [DOI] [PubMed] [Google Scholar]
- 24. Schrage WG, Dietz NM, Eisenach JH, Joyner MJ. Agonist-dependent variability of contributions of nitric oxide and prostaglandins in human skeletal muscle. J Appl Physiol 98: 1251–1257, 2005 [DOI] [PubMed] [Google Scholar]
- 25. Schrage WG, Eisenach JH, Joyner MJ. Ageing reduces nitric-oxide- and prostaglandin-mediated vasodilatation in exercising humans. J Physiol 579: 227–236, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schrage WG, Joyner MJ, Dinenno FA. Local inhibition of nitric oxide and prostaglandins independently reduce forearm exercise hyperaemia in humans. J Physiol 557: 599–611, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shen W, Hintze TH, Wolin Nitric oxide MS An important signaling mechanism between vascular endothelium and parenchymal cells in the regulation of oxygen consumption. Circulation 92: 3505–3512, 1995 [DOI] [PubMed] [Google Scholar]
- 28. Shen W, Xu X, Ochoa M, Zhao G, Bernstein RD, Forfia P, Hintze TH. Endogenous nitric oxide in the control of skeletal muscle oxygen extraction during exercise. Acta Physiol Scand 168: 675–686, 2000 [DOI] [PubMed] [Google Scholar]
- 29. Shepherd JT. Circulation to skeletal muscle. Handbook of Physiology. The Cardiovascular System. Peripheral Circulation and Organ Blood Flow. Bethesda, MD: Am Physiol Soc, 1983, sect. 2, vol. III, pt. 1, chapt. 11, p. 319–370 [Google Scholar]
- 30. Sheriff DD, Nelson CD, Sundermann RK. Does autonomic blockade reveal a potent contribution of nitric oxide to locomotion-induced vasodilation? Am J Physiol Heart Circ Physiol 279: H726–H732, 2000 [DOI] [PubMed] [Google Scholar]
- 31. Shoemaker JK, Halliwill JR, Hughson RL, Joyner MJ. Contributions of acetylcholine and nitric oxide to forearm blood flow at exercise onset and recovery. Am J Physiol Heart Circ Physiol 273: H2388–H2395, 1997 [DOI] [PubMed] [Google Scholar]
- 32. Shoemaker JK, Naylor HL, Pozeg ZI, Hughson RL. Failure of prostaglandins to modulate the time course of blood flow during dynamic forearm exercise in humans. J Appl Physiol 81: 1516–1521, 1996 [DOI] [PubMed] [Google Scholar]
- 33. Taddei S, Galetta F, Virdis A, Ghiadoni L, Salvetti G, Franzoni F, Giusti C, Salvetti A. Physical activity prevents age-related impairment in nitric oxide availability in elderly athletes. Circulation 101: 2896–2901, 2000 [DOI] [PubMed] [Google Scholar]
- 34. Vallance P, Collier J, Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet 2: 997–1000, 1989 [DOI] [PubMed] [Google Scholar]
- 35. Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci USA 96: 7563–7568, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wilson JR, Kapoor S. Contribution of endothelium-derived relaxing factor to exercise-induced vasodilation in humans. J Appl Physiol 75: 2740–2744, 1993 [DOI] [PubMed] [Google Scholar]
- 37. Wilson JR, Kapoor SC. Contribution of prostaglandins to exercise-induced vasodilation in humans. Am J Physiol Heart Circ Physiol 265: H171–H175, 1993 [DOI] [PubMed] [Google Scholar]