

Abstract
Mutations that result in the loss of the protein dysferlin result in defective muscle membrane repair and cause either a form of limb girdle muscular dystrophy (type 2B) or Miyoshi myopathy. Most patients are compound heterozygotes, often carrying one allele with a nonsense mutation. Using dysferlin-deficient mouse and human myocytes, we demonstrated that membrane blebbing in skeletal muscle myotubes in response to hypotonic shock requires dysferlin. Based on this, we developed an in vitro assay to assess rescue of dysferlin function in skeletal muscle myotubes. This blebbing assay may be useful for drug discovery/validation for dysferlin deficiency. With this assay, we demonstrate that the nonsense suppression drug, ataluren (PTC124), is able to induce read-through of the premature stop codon in a patient with a R1905X mutation in dysferlin and produce sufficient functional dysferlin (∼15% of normal levels) to rescue myotube membrane blebbing. Thus ataluren is a potential therapeutic for dysferlin-deficient patients harboring nonsense mutations.
Keywords: Miyoshi myopathy, limb girdle muscular dystrophy type 2B, PTC124, ataluren, myotube cultures, A/J mice, hypotonic shock
miyoshi myopathy and limb girdle muscular dystrophy 2B (LGMD2B) are caused by autosomal recessive inheritance of mutations in the DYSF gene, leading to the absence of dysferlin (4, 12, 14). Dysferlin is a member of the ferlin family of proteins, which are all thought to be involved in the process of vesicle fusions (2, 13). Dysferlin has been shown to be involved in muscle membrane repair and is critical for vesicle fusion in response to calcium influx, which seal breaks in the muscle plasma membrane (sarcolemma). Loss of dysferlin is thought to cause muscular dystrophy due to the inability to rapidly repair membrane breaks, causing activation of muscle breakdown.
Generally, Miyoshi/LGMD2B patients are compound heterozygotes, with both missense and nonsense mutations being common (12, 17). Carriers with one wild-type allele are asymptomatic (10). A significant number of Miyoshi patients harbor a nonsense mutation, and at least one nonsense allele (R1905X) is prevalent in specific populations due to founder effects (22). Thus the subset of Miyoshi/LGMD2B patients harboring the R1905X allele are candidates for nonsense suppression therapy.
This potential utility of nonsense suppression as a therapeutic approach has been demonstrated in the mouse model of Duchenne muscular dystrophy (DMD) using small molecules to allow read-through of nonsense mutations in dystrophin to correct the disease phenotype (3, 24). One of those studies (24) documented the discovery of ataluren (PTC124), an investigational drug that is currently being evaluated in patients with DMD and cystic fibrosis (11). The goal of the studies presented here was to evaluate whether PTC124 could be a possible therapeutic for a Miyoshi/LGMD2B patients harboring a nonsense mutation.
Mice with dysferlin deficiency have become accepted models for LGMD2B/Miyoshi myopathy and have been utilized to understand the progression of this disease. Two examples of naturally occurring mutations have been identified in the SJL/J and A/J mouse (5, 9), and gene targeting has produced two additional murine models (2, 9). In all animal models, the absence of dysferlin causes a defect in membrane repair, which is evident upon physical or chemical disruption of the sarcolemma. However, none of these mouse models carries nonsense mutations and, therefore, cannot be used to evaluate nonsense suppression drugs.
The only current source of skeletal muscle with nonsense mutations in dysferlin is the patients with Miyoshi/LGMD2B. Accordingly, we sought to develop an assay for dysferlin function that uses cultured myotubes derived from patient muscle biopsies. We felt, however, that to treat primary muscle cells with ataluren and then analyze for production of full-length dysferlin would not necessarily validate ataluren as a potential therapeutic. Dysferlin has a number of putative calcium-binding domains (C2 domains) that are predicted to be critical for its function. Many of the known missense mutations result in inactivation of dysferlin and disease (1, 12, 17). The most common of the dysferlin nonsense mutations (R1905X) is found in the last of the C2 domains (17).
While, in many regions of proteins, a random substitution of one amino acid will not lead to loss of function, it is clear that incorrect substitutions at active sites of proteins can potentially disrupt function (18). Dysferlin has a number of putative calcium-binding domains (C2 domains) that are predicted to be critical for its function (6). To test whether ataluren is a potential therapeutic for dysferlin patients, we wanted to develop an assay to monitor whether dysferlin protein produced by treating cells with ataluren would have functional activity. Such an assay would not only determine whether ataluren should be evaluated in dysferlin patients harboring nonsense mutations, but also could be used as a rapid screen of small molecules to monitor whether they rescue dysferlin function. At present, it is currently not possible to monitor whether potential therapeutics have activity, because there is no simple and reliable assay. While dye penetration in response to focal laser injury has been validated as an assay for rapid membrane repair that is mediated by dysferlin (2, 13), this assay would be difficult to implement in a high-throughput manner that would require expensive equipment. Therefore, the development of new in vitro functions for dysferlin function would be a major advance for the field.
One potential biomarker that can be monitored in an assay is blebbing. Plasma membrane blebbing has been reported in cells undergoing apoptosis or oncosis, and it is considered as a prominent feature of cell injury (21). Cultured myotubes develop expanding membrane blebs in response to a wide variety of treatments, including toxic agents such as fluvastatin and pravastatin (19), doxorubicin (20), or in response to hypotonic osmotic shock (16). These blebs grow and eventually collapse as a result of disassociation of the plasma membrane from the underlying cytoskeleton. Membrane leakage can be detected by efflux of cytosolic enzymes, a rapid increase in cytosolic calcium concentration, and activation of calpains (23).
We postulated that the formation of membrane blebs in skeletal muscle cells in response to hypotonic shock would require rapid fusion of membrane vesicles to increase membrane surface area. Rapid fusion of membrane vesicles is involved in membrane repair in response to mechanical injury (2, 13) and has been demonstrated to be a dysferlin-dependent process activated by calcium influx. In this study, we demonstrate membrane blebbing in skeletal muscle myotubes in response to hypotonic shock is also a dysferlin-dependent process. We then use the presence or absence of membrane blebbing in cultured myotubes in response to hypotonic shock as a new in vitro assay for dysferlin function. Using this assay, we demonstrate that ataluren can rescue dysferlin function in myotubes derived from a patient with a R1905X nonsense mutation.
MATERIALS AND METHODS
Primary skeletal muscle cultures.
Quadriceps femoris muscles from neonatal wild type (C57) or dysferlin-deficient (A/J) mice were minced and then trypsinized in Ca2+, Mg2+-free Hanks balanced salt solution that contained 0.25% trypsin (Gibco BRL). The trypsinized muscles were pelleted down by centrifugation and resuspended in the differentiation medium, which contained 70% DMEM (Gibco BRL), 20% horse serum (Hyclone), 8% chick embryo extract (custom made), 1% l-glutamine (Invitrogen), and 1% penicillin-streptomycin (Invitrogen). After filtering through a 45-μm Nitex mesh (Sefar America), cells were plated on collagen-coated (rat tail collagen type I, BD Bioscience) Aclar slips (Ted Pella) in culture dishes. Differentiation was allowed for 7 days for maturation of C57 and A/J myotubes.
C2C12 cell line culture was conducted by first plating the cells on collagen-coated Aclar slips in the growth medium that contained 79% DMEM, 20% fetal bovine serum (Hyclone), 1% l-glutamine, and 1% penicillin-streptomycin. After reaching 80% confluency, the cells were then switched into the differentiation medium, which contained 90% DMEM, 10% horse serum, and 1% penicillin-streptomycin. Differentiation was allowed for 5 days for maturation of C2C12 myotubes.
Human muscle biopsies were obtained from an unaffected human and a compound heterozygotic patient with a R1905X mutation contributing to dysferlin deficiency. A left forearm extensor was biopsied under Massachusetts General Hospital protocol number MGH2005-P-000647/1, and this protocol has been reviewed and approved by the Massachusetts General Hospital Internal Review Board. Human satellite cells were then obtained and frozen for future culture experiments using the explant culture technique described by Decary et al. (7). Myotube culture was initiated by plating the thawed frozen cells on collagen-coated Aclar slips in the growth medium that contained 20% fetal bovine serum and 1% penicillin-streptomycin in Ham's F10 (Invitrogen). After reaching to 80% confluency, the cells were then switched to the differentiation medium, which contained 97% DMEM, 0.001% insulin (Sigma), 0.01% apo-transferrin (Sigma), and 1% penicillin-streptomycin. Differentiation was allowed for 10 days for maturation of human myotubes.
Recombinant adenovirus and PTC124 treatment.
Recombinant adenovirus vectors expressing full-length human dysferlin (Ad-dysferlin) were generated as previously described (8). Cultured myotubes were infected with Ad-dysferlin at a multiplicity of infection of 100 for 24 h, and then cultured for an additional 7 days.
Ataluren (10 μg/ml) was added to cultures when they were switched to differentiation medium. This concentration was chosen because it gave optimal read-though in previous studies with human DMD patient myotubes (24). Drug was maintained for 7–10 days as the myotubes formed.
Immunofluorescence.
Myotubes destined for immunofluoresence evaluation were cultured on Aclar (Electron Microscopy Sciences, Fort Washington, PA, catalog no. 10501-25). They were fixed with 2% formaldehyde in PBS for 5 min at room temperature and heated in 10 mM citrate buffer (pH 6.0) in boiling water bath for 10 min. Immunofluorescence staining then followed. Briefly, the cells were incubated with a monoclonal anti-dysferlin antibody (Neomarkers, 1:20) and a polyclonal anti-myosin antibody (a gift from Dr. Howard Holtzer, 1:50) for 1.5 h. This was followed by incubation with secondary antibodies [Alexa Fluor 488 goat anti-mouse IgG (Invitrogen 1:100) and Alexa Fluor 568 goat anti-rabbit IgG (Invitrogen 1:100)] for 1 h. Finally, the cells were mounted using Vectashield (Vector Laboratories), and imaging was performed on a Leica TCS SL confocal microscope system using a ×63 oil immersed objective.
Western blots.
Cell samples were homogenized with lysis buffer containing 50 mM Tris·Cl (pH 8.0), 5 mM EDTA (pH 8.0), 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.5% deoxycholate, 50 mM DTT, and a protease inhibitor cocktail (Sigma). The protein homogenates were separated using 4–15% gradient gel and then transferred to a nitrocellulose membrane. After blocking in 5% nonfat milk in Tween-20 Tris-base sodium, immunodetection was performed by incubating with the primary anti-dysferlin (Neomarkers, 1:40) and anti-α-sarcoglycan (Abcam, 1:50) antibodies overnight at 4°C. Subsequently, the membrane was washed and incubated with horseradish peroxidase-conjugated goat anti-mouse IgG (GE Healthcare, 1:1,000) for 1 h at room temperature. For detection, the membranes were visualized by an enhanced chemiluminescence system (Pierce) and exposed to Hyblot CL autoradiograhic film.
Membrane blebbing in living myotubes.
Myotubes were maintained in an open heating RC-30 chamber (Warner Instruments) filled with normal differentiation medium at 37°C. For creating hypotonic osmotic shock, the diluted differentiation medium at different concentrations was pumped to the chamber at the speed of 0.206 ml/min. We ascertained that dilution of the media with 25% water was optimal to observe blebbing of human myotubes, while 50% dilution gave the best results for mouse myotubes. These dilutions were used to generate the data presented herein. If less osmotic shock was used, fewer blebs were observed, while, in the case of greater shock, the myotubes rapidly lysed. In case of Ca2+-chelating experiments, EGTA (using 0.5 M stock) was directly added to the dish to the desired final concentration immediately after dilution. Blebbing was observed with phase contrast imaging, performed on a Leica DMR microscope using a ×40 objective.
RESULTS
The initial goal of this study was to develop a new and simple way to assay dysferlin function in cultured myotubes. The first step was to test the hypothesis that the process of muscle membrane blebbing is a dysferlin-dependent process triggered by the entry of extracellular calcium. We first compared hypotonic shock-induced membrane blebbing in myotubes of mouse C2C12 muscle cell line, with and without the addition of 20 mM EGTA to the media to chelate extracellular calcium. As shown in Fig. 1, blebbing occurred within a few seconds, but only in the presence of extracellular calcium (absence of EGTA). Note that, to better see the membrane blebs, the contrast of all phase contrast images has been adjusted. In some cases, this has rendered the blebs dark, as in Fig. 1, but in most cases they appear light in contrast.
Fig. 1.
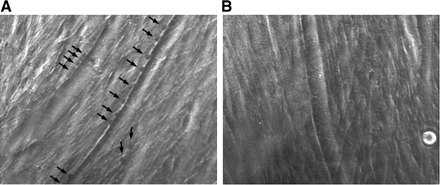
Membrane blebbing require extracellular calcium. A: C2C12 myotube with membrane blebs (dark) indicated by arrows. These blebs were formed after 14 s of exposure to hypotonic solution (50% dilution of media by water). B: C2C12 myotubes 14 s after exposure to the same hypotonic solution as in A, but with EGTA added to chelate extracellular calcium, no membrane blebs formed. This figure can be viewed in real time in Supplemental Movie 1. (The online version of this article contains supplemental data.)
We next compared blebbing in primary muscle cell cultures from C57 mice (Fig. 2A), which produce dysferlin (Figs. 3A and 4A) to those from A/J mice (Fig. 2B), which are dysferlin deficient. Blebbing was seen only in the dysferlin-producing myotubes. We then demonstrated that expression of full-length human dysferlin using recombinant adenoviral infection (Figs. 3A and 4A) could rescue blebbing in the A/J myotubes (Fig. 2C). Thus membrane blebbing in mouse skeletal muscle myotubes clearly relies on dysferlin to promote rapid vesicle fusion.
Fig. 2.
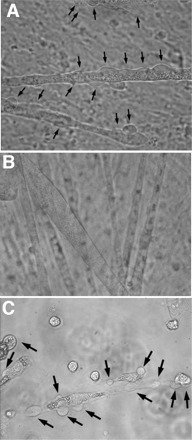
Membrane blebbing requires dysferlin. A: primary C57 mouse myotube after 10 s of exposure to hypotonic solution (50% dilution of media by water) demonstrating membrane blebs. Membrane blebs are indicated by arrows. (Again note that contrast was adjusted to optimize visualization of blebs. In this case, the blebs appear light.) B: primary A/J mouse myotube exposed for 10 s to hypotonic solution. No blebbing is seen. C: primary A/J mouse myotube that is expressing full-length human dysferlin (infection with adenovirus expressing dysferlin) exposed for 10 s to hypotonic solution. In this case, blebbing is easily visualized, demonstrating that dysferlin is sufficient to rescue blebbing function.
Fig. 3.
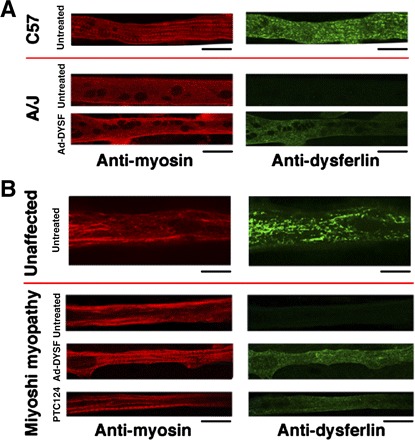
Detection of dysferlin expression in myotubes by immunofluoresence. A: shown are C57 and A/J myotubes stained with either anti-myosin (red) or anti-dysferlin (green) antibodies. Note that A/J myotubes express no detectable dysferlin, unless infected with the recombinant adenovirus that expresses full-length human dysferlin (Ad-DYSF). B: shown are myotubes from an unaffected human and from a Miyoshi patient. Note that, as in the A/J mouse, the Miyoshi myotubes do not express dysferlin, unless infected with recombinant adenovirus. In the case of the Miyoshi myotubes, treatment with PTC124 also results in dysferlin expression. All scale bars represent 20 μm.
Fig. 4.
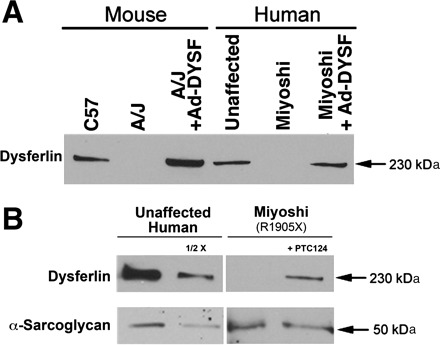
Western blots to assess dysferlin expression. A: comparison of dysferlin expression in mouse and human myotubes. Note, as in Fig. 3, no dysferlin expression can be detected in A/J and Miyoshi myotubes, unless they are infected with recombinant adenovirus expressing full-length human dysferlin (Ad-DYSF). B: treatment of Miyoshi myotubes with PTC124 results in ∼15% of the levels of dysferlin, as found in unaffected myotubes (based on densitometry). Normalization was performed using the levels of α-sarcoglycan expressed in unaffected and Miyoshi myotubes.
Next, we obtained a skeletal muscle biopsy from a Miyoshi patient with a R1905X mutation, performed explant culture, and generated a population of myogenic cells. These cells were then cultured to produce myotubes, as were myogenic cells obtained from an unaffected human biopsy. The unaffected human myotubes expressed dysferlin (Figs. 3B and 4A) and underwent membrane blebbing in response to hypotonic shock (Fig. 5A). As was the case with myotubes from the A/J mouse, the Miyoshi myotubes failed to form membrane blebs in the face of hypotonic shock (Fig. 5B) and were dysferlin deficient (Figs. 3B and 4A). Again, we were able to successfully rescue blebbing with adenoviral infection with a virus that expressed full-length human dysferlin (Figs. 3B, 4A, and 5C).
Fig. 5.
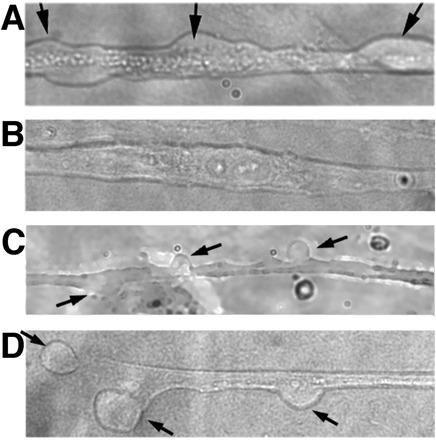
Membrane bleb formation in human myotubes requires dysferlin. A: shown is a primary human (unaffected) myotube after 10 s of exposure to hypotonic solution (25% dilution of media by water) demonstrating membranes blebs. Membrane blebs are indicated by arrows. B: primary myotube from a Miyoshi patient exposed for 10 s to hypotonic solution. No blebbing is seen. C: primary Miyoshi myotube that is expressing full-length human dysferlin (infection with adenovirus expressing dysferlin) exposed for 10 s to hypotonic solution. In this case, blebbing is easily visualized, demonstrating that dysferlin is sufficient to rescue blebbing function in Miyoshi myotubes. D: membrane blebs are seen in the myotubes derived from the Miyoshi patient carrying a R1905X mutation following PTC124 treatment. This rescue of blebbing by treatment with PTC124 demonstrates that the dysferlin produced via nonsense suppression is functional.
To test whether suppression of the premature stop codon at R1905X using ataluren could produce full-length dysferlin, we treated myotubes after fusion with 10 μg/ml PTC124. This level of drug was chosen based on the optimal concentration in human DMD cultures (24) and on limited dose-response studies on R1905X myotubes. As shown in Fig. 3B, dysferlin expression was detected by immunofluoresence following PTC124 treatment. Western blot analysis revealed that the treated Miyoshi myotubes expressed ∼15% of the level of dysferlin detected in unaffected human myotubes (Fig. 4B). We then performed blebbing experiments with the patient myotubes grown in the presence of 10 μg/ml PTC124. Blebbing was apparent (Fig. 5), indicating that the dysferlin produced was functional.
DISCUSSION
The major goal of this work presented here was to develop a simple in vitro assay for dysferlin function in skeletal muscle myotubes. We hypothesized and subsequently demonstrated that membrane blebbing induced by hypotonic shock is dependent on both calcium and dysferlin and thus can fulfill the need for a simple functional assay. A limitation of the assay is that, in our experience, the density of membrane blebs on myotubes was highly variable. Thus the assay likely cannot be used to ascertain if a given condition induces more blebbing. However, in the absence of dysferlin, there were never membrane blebs, and thus the presence or complete absence of blebs can be used to assess whether or not dysferlin function has been rescued, as we have done in this study. While quantification of blebbing has been previously reported, it was in the context of mature skeletal muscle fibers (16). The lack of synchrony in myotube formation, and therefore the gradient of differentiation status of myotubes in a culture, likely underlies the observed variability in the extent of blebbing. Nonetheless, the absence of any blebbing can be used as a monitor for loss of dysferlin function and restoration of blebbing as a monitor of functional rescue.
Transient hypotonic shock could also be used as the basis for high-throughput screens for drug discovery in the search for compounds that induce compensation for dysferlin deficiency. In this case, blebbing itself could be monitored in a high-content screen, or indicators of cytosolic calcium rise (21) or even indicators of calpain activation (23) could be used. Compounds that trigger blebbing by mechanisms other than osmotic shock or membrane injury (e.g., apoptosis or toxic agents) may or may not require dysferlin, but would be excluded from screens in that they would lead to blebbing before hypotonic challenge. Alternatively, entry of cytosolic enzyme contents into the media in response to transient hypotonic shock could be assessed, but this would not be as amenable to high-throughput screens as optical indicators.
We have demonstrated that PTC124 (ataluren) promotes read-through of the R1905X mutation in human dysferlin to allow for the accumulation of ∼15% of normal levels in cultured myotubes. This level of protein is being produced from mRNA from just one allele that is most likely degraded rapidly due to nonsense-mediated decay (15). Furthermore, use of the membrane blebbing assay developed and described here further demonstrated that dysferlin protein produced as a consequence of ataluren treatment was functional. The levels of dysferlin that are necessary to completely rescue its function in skeletal muscle is unknown, although it is known that haplo insufficiency is not associated with disease. We anticipate, however, that any significant level of functional dysferlin production is likely to slow disease progression in human Miyoshi/LGMD2B patients. Therefore, nonsense mutation patients, like those harboring R1905X mutations, are candidates for potential therapeutic trials with PTC124. Patients who are homozygotic for the mutation are particularly strong candidates. The blebbing assay developed here can also be used to monitor ataluren activity in patients harboring other nonsense mutations in the dysferlin gene.
DISCLOSURES
S. W. Peltz and E. M. Welch have equity interests in PTC Therapeutics. H. L. Sweeney receives more than $10,000 per year in consulting fees from PTC Therapeutics.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the Paul Wellstone Muscular Dystrophy Cooperative Research Center (U54 AR052646).
REFERENCES
- 1. Bansal D, Campbell KP. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol 14: 206–213, 2004 [DOI] [PubMed] [Google Scholar]
- 2. Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 423: 168–172, 2003 [DOI] [PubMed] [Google Scholar]
- 3. Barton-Davis ER, Cordier LL, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest 104: 375–381, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, Richard I, Marchand S, Bourg N, Argov Z, Sadeh M, Mahjneh I, Marconi G, Passos-Bueno MR, Moreira Ede S, Zatz M, Beckmann JS, Bushby K. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet 20: 37–42, 1998 [DOI] [PubMed] [Google Scholar]
- 5. Bittner RE, Anderson LV, Burkhardt E, Bashir R, Vafiadaki E, Ivanova S, Raffelsberger T, Maerk I, Höger H, Jung M, Karbasiyan M, Storch M, Lassmann H, Moss JA, Davison K, Harrison R, Bushby KM, Reis A. Dysferlin deletion in SJL mice (SJL-Dysf) defines a natural model for limb girdle muscular dystrophy 2B. Nat Genet 23: 141–142, 1999 [DOI] [PubMed] [Google Scholar]
- 6. Davis DB, Doherty KR, Delmonte AJ, McNally EM. Calcium-sensitive phospholipid binding properties of normal and mutant ferlin C2 domains. J Biol Chem 277: 22883–22888, 2002 [DOI] [PubMed] [Google Scholar]
- 7. Decary S, Mouly V, Hamida CB, Sautet A, Barbet JP, Butler-Browne GS. Replicative potential and telomere length in human skeletal muscle: implications for satellite cell-mediated gene therapy. Hum Gene Ther 8: 1429–1438, 1997 [DOI] [PubMed] [Google Scholar]
- 8. He TC, Zhou S, Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95: 2509–2514, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ho M, Post CM, Donahue LR, Lidov HG, Bronson RT, Goolsby H, Watkins SC, Cox GA, Brown RH., Jr Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum Mol Genet 15: 1999–2010, 2004 [DOI] [PubMed] [Google Scholar]
- 10. Illarioshkin SN, Ivanova-Smolenskaya IA, Greenberg CR, Nylen E, Sukhorukov VS, Poleshchuk VV, Markova ED, Wrogemann K. Identical dysferlin mutation in limb-girdle muscular dystrophy type 2B and distal myopathy. Neurology 55: 1931–1933, 2000 [DOI] [PubMed] [Google Scholar]
- 11. Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, Nissim-Rafinia M, Blau H, Rivlin J, Aviram M, Elfring GL, Northcutt VJ, Miller LL, Kerem B, Wilschanski M. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet 372: 719–727, 2008 [DOI] [PubMed] [Google Scholar]
- 12. Klinge L, Aboumousa A, Eagle M, Hudson J, Sarkozy A, Vita G, Charlton R, Roberts M, Straub V, Barresi R, Lochmüller H, Bushby K. New aspects on patients affected by dysferlin deficient muscular dystrophy. J Neurol Neurosurg Psychiatry. 2009. [Epub] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lennon NJ, Kho A, Bacskai BJ, Perlmutter SL, Hyman BT, Brown RH. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J Biol Chem 278: 50466–50473, 2003 [DOI] [PubMed] [Google Scholar]
- 14. Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, Serrano C, Urtizberea JA, Hentati F, Hamida MB, Bohlega S, Culper EJ, Amato AA, Bossie K, Oeltjen J, Bejaoui K, McKenna-Yasek D, Hosler BA, Schurr E, Arahata K, de Jong PJ, Brown RH., Jr Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 20: 31–36, 1998 [DOI] [PubMed] [Google Scholar]
- 15. Mendell JT, Dietz HC. When the message goes awry: disease-producing mutations that influence mRNA content and performance. Cell 107: 411–414, 2001 [DOI] [PubMed] [Google Scholar]
- 16. Menke A, Jockusch H. Decreased osmotic stability of dystrophin-less muscle cells from the mdx mouse. Nature 349: 69–71, 1991 [DOI] [PubMed] [Google Scholar]
- 17. Nguyen K, Bassez G, Bernard R, Krahn M, Labelle V, Figarella-Branger D, Pouget J, Hammouda EH, Béroud C, Urtizberea A, Eymard B, Leturcq F, Lévy N. Dysferlin mutations in LGMD2B, Miyoshi myopathy, and atypical dysferlinopathies. Hum Mutat 26: 165, 2005 [DOI] [PubMed] [Google Scholar]
- 18. Rennell D, Bouvier SE, Hardy LW, Poteete AR. Systematic mutation of bacteriophage T4 lysozyme. J Mol Biol 222: 67–88, 1991 [DOI] [PubMed] [Google Scholar]
- 19. Sakamoto K, Honda T, Yokoya S, Waguri S, Kimura J. Rab-small GTPases are involved in fluvastatin and pravastatin-induced vacuolation in rat skeletal myofibers. FASEB J 21: 4087–4094, 2007 [DOI] [PubMed] [Google Scholar]
- 20. Sardão VA, Oliveira PJ, Holy J, Oliveira CR, Wallace KB. Morphological alterations induced by doxorubicin on H9c2 myoblasts: nuclear, mitochondrial, and cytoskeletal targets. Cell Biol Toxicol 25: 227–243, 2009 [DOI] [PubMed] [Google Scholar]
- 21. Trump BF, Berezesky IK, Chang SH, Phelps PC. The pathways of cell death: oncosis, apoptosis, and necrosis. Toxicol Pathol 25: 82–88, 1997 [DOI] [PubMed] [Google Scholar]
- 22. Vilchez JJ, Gallano P, Gallardo E, Lasa A, Rojas-García R, Freixas A, De Luna N, Calafell F, Sevilla T, Mayordomo F, Baiget M, Illa I. Identification of a novel founder mutation in the DYSF gene causing clinical variability in the Spanish population. Arch Neurol 62: 1256–1259, 2005 [DOI] [PubMed] [Google Scholar]
- 23. Villalobos JC, Mora R, Lomonte B, Gutiérrez JM, Angulo Y. Cytotoxicity induced in myotubes by a Lys49 phospholipase A2 homologue from the venom of the snake Bothrops asper: evidence of rapid plasma membrane damage and a dual role for extracellular calcium. Toxicol In Vitro 21: 1382–1389, 2007 [DOI] [PubMed] [Google Scholar]
- 24. Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL. PTC124 targets genetic disorders caused by nonsense mutations. Nature 446: 87–91, 2007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.