

Abstract
Neuronal modulation of the sinoatrial node (SAN) plays a crucial role in the initiation and maintenance of atrial arrhythmias (AF), although the exact mechanisms remain unclear. We used a computer model of a rabbit right atrium (RA) with a heterogeneous SAN and detailed ionic current descriptions for atrial and SAN myocytes to explore reentry initiation associated with autonomic activity. Heterogeneous acetylcholine (ACh)-dependent ionic responses along with L-type Ca current (ICa,L) upregulation were incorporated in the SAN only. During control, activation was typical with the leading pacemaker site located close to the superior vena cava or the intercaval region. With cholinergic stimulation, activation patterns frequently included caudal shifts of the leading pacemaker site and occasional double breakouts. The model became increasingly arrhythmogenic for the ACh concentration >20 nM and for large ICa,L conductance. Reentries obtained included counterclockwise rotors in the free wall, clockwise reentry circulating between the SAN and free wall, and typical flutter. The SAN was the cause of reentry with a common leading sequence of events: a bradycardic beat with shifting in the caudal direction, followed by a premature beat or unidirectional block within the SAN. Electrotonic loading, and not just overdrive pacing, squelches competing pacemaker sites in the SAN. Cholinergic stimulation concomitant with ICa,L upregulation shifts leading pacemaker site and can lead to reentry. A heterogeneous response to autonomic innervation, a large myocardial load, and an extensive SAN in the intercaval region are required for neurally induced SAN-triggered reentry.
Keywords: electrophysiology, reentry, autonomic innervation
atrial fibrillation (af) is the most common and diverse cardiac arrhythmia (21). Experimental observations have illustrated the crucial role that neuronal mechanisms have in its initiation and maintenance (33). Atrial arrhythmias can be induced in dogs by localized electrical stimuli applied to nerve branches of the thoracic vagosympathetic complex (31) or mediastinal nerves (2, 31). Experiments show that acetylcholine (ACh) can trigger reentry and AF in the right atrium (RA) and that adrenergic stimulation facilitates both AF initiation and maintenance (33). Atropine completely abolishes bradycardia and subsequent tachyarrhythmias, indicating that cholinergic efferents play a predominant role in induction (33).
Different studies describe a similar sequence of events for arrhythmias originating in the RA that were induced by neural stimulation: cycle length (CL) prolongation, often with leading pacemaker site (LPS) shifting, followed by an atrial premature beat (APB), and subsequent tachyarrhythmia (1, 2, 31, 33). The escape beat initiating the tachyarrhythmia is primarily located in either the intercaval region or an area underlying a major ganglionated plexus (2). However, the electrophysiologic conditions necessary that give rise to the preceding observations and reentry remain unelucidated. Given the extensive size and strong heterogeneity of the SAN, we propose that it is these properties that can lead to the SAN itself triggering reentry.
Tissue simulations have studied SAN (10) and AF initiation under different ACh distributions and L-type Ca current (ICa,L) upregulation (19, 22, 37). Recent SAN modeling studies have been limited to one dimension or a thin radial slice (8, 30), and thus the inferior/superior LPS shifting has not been a target of study. Furthermore, extension of these one-dimensional (1D) gradient models to three-dimensional (3D) is ambiguous, as it can be performed by either rotation or translation. Rotation is suggested by the models of Zhang et al. (40) and Cloherty et al. (10), while translation is inline with the work of Dobrzynski et al. (14). Since it is experimentally very difficult to determine the origin of an isolated SAN cell, the intrinsic heterogeneity pattern remains controversial. How the pattern affects SAN function and arrhythmogenesis has not been studied in a 3D model. Thus despite modeling and much experimental work, the behavior of the neurally modulated SAN in arrhythmia initiation remains unclear.
The purpose of this study was build a 3D mathematical SAN model that incorporated known vertical and horizontal gradients in ionic and coupling properties to understand how SAN heterogeneity can initiate reentry in the RA under neural stimulation, as well as determine the neural excitation patterns that trigger reentry. We constructed a detailed rabbit RA computer model with an embedded heterogeneous SAN. We incorporated vagally mediated ACh effects, ICa,L upregulation that resembled adrenergic stimulation, and conductance variation within the SAN. We methodically examine and expose how these factors cause LPS shifting and APBs, ultimately leading to reentry, providing clear mechanisms. By amalgamating known electrophysiological data, we are able to reproduce and explain a large set of experimentally observed organ-level behavior.
METHODS
Ionic models.
The Kurata ionic model that appropriately represents effects of channel blockers and incorporates Ca2+ handling (24–27) was used for central, intermediate, and peripheral action potentials (APs) in the SAN region. Membrane ionic flow include the following components:
| (1) |
-The region outside the SAN used the lapine Lindblad model (28) and was homogeneous since arrhythmia maintenance was not the object of the study but only arrhythmia initiation.
RA and SAN geometry.
To provide the basic atrial structure, a human finite element mesh obtained with MRI was reduced in size to match that of a rabbit. It comprised 315,986 nodes and 1,065,333 tetrahedral elements with an average length of 100 μM. A rabbit SAN and block zone, as determined by 3D computer reconstructions (14) (Fig. 1A), was superimposed on the mesh using the crista terminalis (CT) as one boundary. The CT was considered peripheral as proposed in histological (14, 36) and electrophysiological studies (20) and was not explicitly modeled.
Fig. 1.
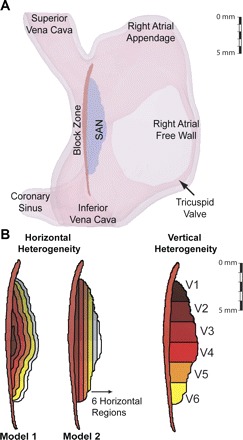
A: right atrium (RA) geometry indicating the sinoatrial node (SAN), septal, and the block zone. B: isolated SAN showing regions used to define gradients in ionic parameters and conductivity. Two different models used for horizontal gradients are shown at left. Color indicates region with numbering beginning with darker colors. Note that not all vertical regions overlap with all horizontal regions.
SAN heterogeneity.
The SAN is heterogeneous with AP properties and cell size changing from the center to periphery (20). We used a gradient SAN model to recreate the experimentally measured heterogeneity in intrinsic pacemaker activity between the center and periphery in individual cells and in the intact SAN (8, 10, 39). Two gradient distributions were implemented as have been suggested by earlier work: model 1, where peripheral cells were predominant and encircled the SAN in a 3D model, an extension of 1D models implemented by Zhang et al. (40) and Cloherty et al. (10); and model 2, where central cells are located in the intercaval region and peripheral cells are located on the epicardial surface of the CT as determined by Dobrzynski et al. (14) (Fig. 1B). Both gradients were implemented as six zones distributed horizontally from the center to the CT. Cell capacitance was increased with an exponential dependence on distance to the center. The gNa, gCa,L, gto, gsus, gKr, gKs, and gf were altered as functions of capacitance (Cm; Ref. 40). Values varied between the extrema assigned in the Kurata model (27). Full details are given in the online Supplement Material (Supplemental Material for this article is available online at the Am J Physiol Heart Circ Physiol website).
SAN electrical activity is also heterogeneous from inferior to superior regions. Vertically, there is a gradient in action potential duration (APD) and CL along with a marked loss of excitability in the inferior region (7). This heterogeneity was implemented by decreasing gKr in the caudal direction as suggested by Boyett et al. (7). Poor conduction in the block zone (4) has been explained by a lack or reduced number of myocytes and by reduced excitability (14, 16). The block zone was modeled by removing ICa,L and by lowering electrical coupling (6).
SAN conductivity.
Experiments regarding gap junction distribution within the SAN suggest that central coupling is poor with mainly connexin (Cx) 45 present, believed to protect central intrinsic pacemaker activity from the suppressive influence of the surrounding hyperpolarized muscle (14, 36). These studies also showed a gradient in electrical coupling from the center, where Cx43 is absent, to the periphery, where Cx43 is present. This gradient and the lower SAN conductivity could also result from a lower gap junction density and more fibroblasts (9). We used two uniform conductivity regions: a low conductivity zone comprising horizontal regions 1–3 and a high conductivity zone in contact with the RA comprising comprising regions 4–6. This was a mix of the uniform conductivity profile favoring frequency entrainment of SAN cells and the linear profile favoring propagation to the myocardium (10). Conductances for control in these regions were set to 125 and 250 mS/m, respectively, while myocardial conductivity was set to 250 mS/m, which produced a conduction velocity of 62 cm/s, close to the planar wave velocity for the rabbit atria of 30–80 cm/s (40).
ACh stimulation.
Experiments have classified SAN response into two types based on ACh input, either bath ACh application or transient/periodic vagal nerve stimulation. Specifically, IK,ACh, ICa,L, If, INa, Ib,Na, IK, and INaK are involved in the parasympathetic modulation of the SAN (12). Direct modulation of IK,ACh and indirect modulation of ICa,L and If are mainly responsible for bath ACh applications {high ACh concentration ([ACh]); Ref. 41}, while INa and Ib,Na along with the currents affected during bath stimulation are affected during vagally released ACh stimulation (low [ACh]; Refs. 12, 41).
We used the Zhang ACh model (41), where [ACh] activates IK,ACh directly, inhibits ICa,L by cAMP modulation, and inhibits If by shifting its activation curve to more positive voltages depending on [cAMP] (13). Figure 2A shows simulated APs for different cell types and [ACh]s. Spontaneous activity stopped at very high [ACh] (1,000 nM). Figure 2B shows the heterogeneity in SAN AP properties for all regions. APD decreased and CL increased in all cell models in a [ACh]-dependent manner. ACh effects were not implemented outside of the SAN.
Fig. 2.
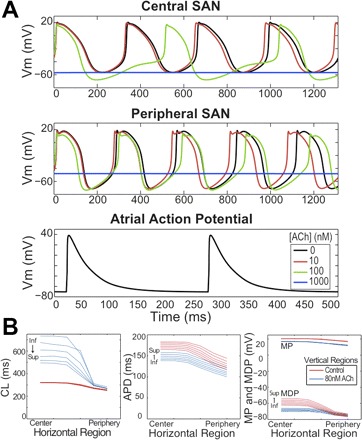
SAN and RA isolated cell action potentials. A: top to bottom: intrinsic action potentials (APs) from the central and peripheral SAN and RA free wall under various ACh concentrations ([ACh]). Note that [ACh] was not introduced into the free wall. B: intrinsic SAN AP properties under 0 (red) and 80 (blue) nM [ACh]. From left to right, cycle length (CL), action potential duration (APD), minimum diastolic potential (MDP), and maximum potential (MP) as functions of position within the SAN. Labels on each group of curves indicate inferior (Inf.) and superior (Sup.) regions. For orientation purposes, the crista terminalis would run along the right side of the SAN from superior vena cava to inferior vena cava. Vm, transmembrane voltage.
LPS shifting is believed to be due to a differential sensitivity to neurotransmitters and heterogeneity of ionic channels (41), although uneven cholinergic and adrenergic innervation can also produce this effect (11). Functionally, these factors are inseparable. Shifting of the LPS was induced by a differential sensitivity to [ACh]s within the SAN and by [ACh] gradients along the horizontal and vertical directions. [ACh] gradients were maximal in central and superior regions and minimal in peripheral and inferior regions (3, 16).
Choice of model parameters.
It should be noted that failure to include heterogeneity and gradients in the SAN yielded a nonfunctioning model. Improper choices for conductivities and cellular properties can result in a blocked SAN, wherein impulses fail to leave the SAN, or a quiescent SAN wherein automaticity is compromised due to large electrotonic loading. In the latter case, oscillations of sufficient magnitude fail to manifest or do so irregularly. Thus the parameter range for the model was constrained by having to implement experimentally measured gradients to reproduce experimentally recorded behavior. See Supplemental Material for a sensitivity analysis. APD was measured from the point of maximum ∂Vm/∂t to 90% repolarization.
Computational techniques and simulation protocols.
Electrical activity was calculated by the monodomain formulation:
| (2) |
where σi is conductivity (S/m), Cm is membrane capacitance (μF/cm2), and Iion is total membrane ionic current (μF/cm2); these parameters varied depending on the SAN region. The β is the surface-to-volume-ratio (μm−1), and Vm is the transmembrane voltage (mV).
Initial conditions for myocytes were set by pacing for 10 s at a CL of 250 ms. To allow the system to reach steady state, the SAN under control conditions was simulated for 10 s. Six cases without ACh and upregulation of gCa,L, gKr, and gKs that resembles adrenergic effects (13) were explored (control conditions) for each SAN gradient model: three levels of peripheral gCa,L (50, 100, and 150%) and two levels of peripheral gKr and gKs (100 and 150%), with nominal levels as given by Zhang (40). Simulations ran for 5 s to stabilize transients. To implement cholinergic effects, maximum [ACh] (20, 40, and 80 nM) was varied in three patterns: a homogeneous [ACh] resembling bath ACh (12), an [ACh] gradient in the inferior direction with maximal ACh in superior regions, and in the inferior and peripheral directions with maximal ACh in central and superior regions, resembling vagal stimulation (3, 16). Finally, adrenergic effects were combined with cholinergic effects for a total of 289 simulations. Details are given in the online Supplement Material.
RESULTS
Activation patterns under control conditions.
Activation maps during sinus rhythm registered typical monofocal activation patterns, with the LPS in the superior SAN near the superior vena cava (SVC) and in the central SAN (Fig. 3; online Supplemental Movie Control-LPS). With a superior LPS, activation rapidly propagated over the superior free wall and appendage, in agreement with experiments (35). With a central LPS, activation spread radially along the inferior and superior SAN, reaching the free wall and subsequently the appendage (16). The last region activated was the area below the coronary sinus at the tricuspid valve, 73.33 ± 1.17 ms for a superior LPS and 74.66 ± 1.86 ms for a central LPS, which agrees well with experiments (6, 7). Conduction velocities around the LPS, measured during the first 20 ms of activation in the vertical direction (toward the SVC), were 7.64 and 9.07 cm/s for a superior and central LPS, respectively (see Fig. 3) Horizontally (toward the free wall), the corresponding velocities were 3.63 and 2.9 cm/s. These values were in close agreement with experiments (16). APD maps of these two patterns (Fig. 3) show that the LPS has the longest APD as is expected. For the superior LPS, the central region also has a long APD, indicating a competing site that was squelched.
Fig. 3.
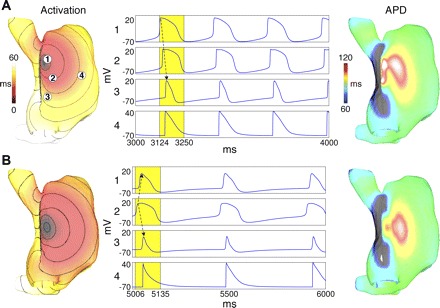
Activation maps (left), transmembrane voltages (middle), and APD maps (right) for different leading pacemaker sites (LPSs). A: superior LPS. B: central LPS. Voltage traces were obtained at locations labeled in A. Activation and APD maps were computed over yellow windows. The 10-ms isochrones are displayed on activation maps with black indicating inexcitable regions. Arrows in time traces indicate the sequence of propagation within the SAN.
Gradients within the SAN for different levels of gCa,L, gKr, and gKs were compared with nominal gradient values. We observed superior or typical LPS (Fig. 3A) in four simulations and a central LPS in two. The initial breakout originated in the first three central regions of the SAN for both cases. The superior LPS was the result of the gKr vertical gradient, and the central LPS resulted from low peripheral gCa,L (50%), which counteracted the gKr gradient. Regions of inexcitability or poor activation were observed, being the inferior and superior SAN for a central LPS (Fig. 3), and a small region slightly distal to a superior LPS, as observed experimentally (7). These unexcited regions were functional in nature: when the SAN was excited, a sudden, large increase in myocardial electrotonic load drew current from the SAN, producing hyperpolarization and preventing depolarization in near threshold cells.
Effects of cholinergic stimulation on LPS.
Cholinergic stimulation induced bradycardic beats, caudally shifting the LPS by 5.77 ± 0.26 mm (see Fig. 4A). as observed in vivo (2, 31) and in vitro (16, 35). Less frequently, vagal stimulation induced double breakouts (Fig. 4B and Table 1), with simultaneous LPSs adjacent to the SVC and inferior vena cava, also observed experimentally (16, 31). A higher ACh dependence in the superior SAN led to larger APD and cycle length reductions there than in the inferior region. Since ICa,L was the main depolarizing current, a larger gCa,L in the periphery helped propagation into the myocardium. Thus inferior LPSs were most frequently associated with high [ACh] and a large gCa,L gradient (Table 1). Double breakouts were more likely for intermediate [ACh] (40 nM), since this was not high enough to abolish the superior LPS. A high peripheral gCa,L also facilitated double breakout, by increasing excitability and diminishing ACh effects in superior regions. (See online Supplemental Movie Double-LPS.)
Fig. 4.
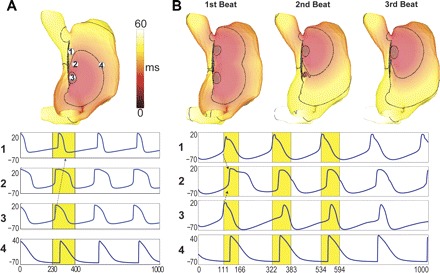
Activation maps (top) and transmembrane voltages (bottom) during bath vagal stimulation. A. LPS caudal shift for 80 nM [ACh]. B: double breakout for 40 nM [ACh]. Voltage traces were obtained at locations labeled in A. Activation maps were computed over yellow windows. Arrows in time traces indicate the sequence of propagation within the SAN. 20-ms isochrones are drawn on activation maps.
Table 1.
Percentages of LPS activation patterns for SAN gradient model 1 (concentric) under [ACh]bath, heterogeneous cholinergic stimulation, and variation
| [ACh], nM |
ACh Regions |
Peripheral (gCa,L), % |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Pattern | 80 | 40 | 20 | 1–3 | 1–4 | 1–5 | 150 | 100 | 50 |
| Caudal shift | 92.9 | 82.3 | 61.5 | 84.9 | 89.3 | 64.8 | 73.3 | 82.4 | 85.7 |
| Double breakout | 6.06 | 9.73 | 8.79 | 8.22 | 4.10 | 13.0 | 13.71 | 5.88 | 2.60 |
| No effect | 1.01 | 8.00 | 29.7 | 6.85 | 6.56 | 22.2 | 12.9 | 11.8 | 11.7 |
LPS, leading pacemaker site; SAN, sinoatrial node; [ACh], acetylcholine concentration.
APs for the LPS shift in Fig. 4A show that RA excitation drew significant current from the SAN, causing hyperpolarization in superior SAN regions (solid arrow in A1) immediately preceding depolarization. This is similar to the process generating inexcitable regions; however, here, the regions activated. Double activations only occurred for one beat (Fig. 4B). Ensuing double breakouts failed because subsequent superior LPSs activated slightly sooner and upon reaching the free wall drew current that squelched the emerging inferior LPS. Similar hyperpolarization spikes were observed after double breakouts in the inferior SAN (solid arrow in Fig. 4B3). Such dual breakout patterns led to propagation in distinct directions with shorter activation times, averaging (64.73 ± 2.60 ms), almost 10 ms shorter than sinus. The third and following beats were monofocal superior LPSs as seen experimentally (1).
Homogeneous [ACh] did not cause LPS shifting, but CL increased with increased [ACh], 64.63% for 80 nM, 14.53% for 20 nM, and 7.08% for 10 nM, similar to previous studies (23, 41). Spontaneous CL also depended on the particular ACh gradient applied. Increases in CL of 54.95, 37.20, and 8.85% were obtained for five, four, and three vertical regions of ACh stimulation, respectively, with a maximum [ACh] of 80 nM. Vagal stimulation also induced hyperpolarization (16, 23). On average, minimum diastolic potential decreased by 8% in superior, 15% in central, and 3% in inferior SAN cells. The maximum increase was observed in central regions where [ACh]s were stronger.
Types of reentry.
Reentry occurred with ACh stimulation after APBs escaped the SAN or when a wavefront within the SAN was blocked by refractory tissue. Reentry initiated by APBs was elicited when an escape beat of short coupling interval reached the myocardium. A delay in the wavefront exiting the SAN induced a spiral wave in front of the SAN region where the APB was located (Fig. 5B, 1,255–1,505 ms). This mechanism generated counterclockwise reentry for inferior APBs and clockwise reentry for superior APBs (Figs. 5 and 6 and online Supplemental Movies Counter-Clockwise-Reentry and Clockwise-Reentry). Reentry initiated by unidirectional block after LPS shifting, was elicited when refractory tissue and ACh prevented and attenuated RA excitation in the inferior SAN causing the wavefront to displace to the superior SAN, from where the RA was activated. If the free wall was initially activated, clockwise reentry was generated (Fig. 6), but if activation reached the appendage first, atrial flutter was obtained (see online Supplemental Movie Flutter). LPS shifting was required for unidirectional block, since a shifted beat encountered tissue not fully recovered in the inferior SAN. Otherwise, with a stable LPS, the beat encountered recovered tissue upon exiting the SAN, resulting in sinus activity.
Fig. 5.
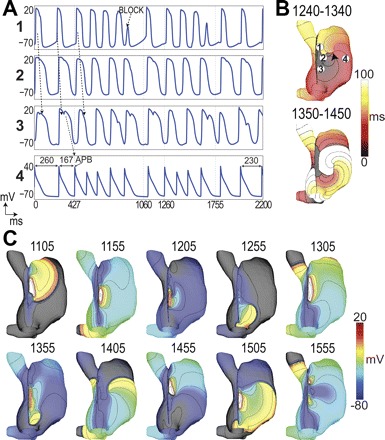
Counterclockwise reentry after vagal stimulation. A: voltage traces were obtained at locations labeled in B upper. An atrial premature beat (APB) occurs leading to conduction block labeled in A1. Arrows indicate direction of propagation. Cycle lengths are indicated in A4 to show the initiation and termination of the reentry. B: activation maps for 2 cycles of reentry over the periods (ms) indicated. C: voltage maps taken at the times indicated in ms since ACh application. 10-mV isopotentials are drawn.
Fig. 6.
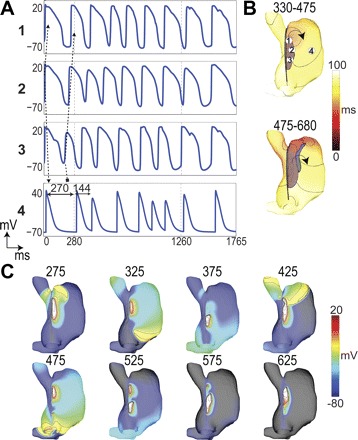
Clockwise reentry after vagal stimulation. A: voltage traces were obtained at locations labeled in B. An unidirectional block from 3 to 4 leads to reentry. Arrows show sequence of activation. Cycle lengths in trace 4 (myocardium) show the onset of the reentry. B: activation maps for 2 cycles of reentry over the periods (in ms) indicated. C: voltage maps taken at the times indicated with 10-mV isopotential lines displayed.
Figure 5 shows APs and activations for two counterclockwise reentry episodes induced after a sustained 80 nM [ACh] was applied superiorly and no ACh inferiorly, with high gCa,L. The first episode had five reentry cycles lasting 670 ms, and the second had four cycles lasting 495 ms. Initially, waves circulated from the inferior to superior SAN, (Fig. 5B, 1,240–1,340 ms), but during the final stage, the spiral wave detached from the SAN, into the free wall (Fig. 5B, 1,350–1,450 ms), until it annihilated itself after collision with refractory tissue near the appendage. An initial bradycardic beat in this episode originated the superior LPS, shown in the first AP of Fig. 5A, followed by a shorter beat (Fig. 5A4) that was ended by an APB originating in the inferior SAN (Fig. 5A3). This APB initiated tachycardia, with reentrant activity overriding the pacemaker function. in accordance with previous studies (31, 33) (see online Supplemental Movie Counter-Clockwise-Reentry).
Figure 6 shows clockwise reentry after sustained 80 nM cholinergic stimulation in the superior and central SAN and 20 nM in the inferior and peripheral SAN, with a high gCa,L gradient. This episode consisted of six cycles lasting 980 ms. The wavefront circulated from the superior SAN and SVC to the free wall and back to the inferior SAN until refractory tissue in the inferior SAN terminated reentry. An initial bradycardic beat from the SAN center in this episode is shown in Fig. 6A2, followed by a beat that initiated in the inferior SAN that was unidirectionally blocked (Fig. 6A3). The remaining activity moved cranially within the SAN and activated the RA from the superior SAN causing reentry (Fig. 6, A4 and C, 275 ms). This type of reentry was also obtained by superior SAN APBs after ACh (see online Supplemental Movie Clockwise-Reentry).
Vulnerability to reentry.
Reentry inducibility depends mostly on the parameters assigned to the SAN region as summarized in Fig. 7. Probabilities for APBs were proportional to those for reentry. For both gradient models, APBs increased with an increase in gCa,L. [ACh] >20 nM was necessary for a higher probability of reentry. Increasing gKr and gKs to 150% had an antiarrhythmic effect, diminishing the number of APBs. Conductivity within the SAN had no significant effect on arrhythmia formation but on intrinsic pacemaker rate. With increased conductivity, electrotonic load on pacemaker cells increased, decreasing pacemaker rate. The number of APBs and reentry cycles were approximately two and four times higher, respectively, for SAN model 2. Reentry within the SAN resulted from APBs caused by APBs in most of the cases. APBs that started inferiorly gave rise to counterclockwise reentry, the opposite sense to reentry initiated by superior APBs. Figure 7C shows that the LPS was more stable for SAN model 1 than for model 2 during control conditions, shifting 0.27 and 1.73 mm from beat to beat, respectively. After ACh application, LPS shifting for model 1 was less pronounced (5.77 ± 0.26 mm) and more stable than for model 2 (6.69 ± 1.14 mm). An increased number of central cells as in model 2 increased the probability of APBs and caused more prominent shifting and instability, which were directly related to the occurrence of reentry.
Fig. 7.
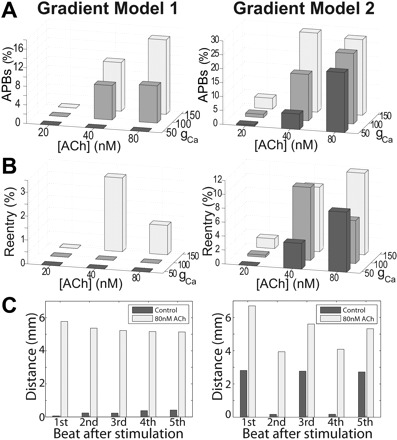
Comparison of SAN gradient models. A: occurrence of APBs for different maximum [ACh]s and gCa,L. B: occurrence of reentry for different [ACh]s and gCa,L. C: LPS shifting in control and after 80 nM [ACh]. Distance is measured from the cranial end of the SAN complex.
DISCUSSION
This work presents the first realistic 3D geometrical SAN model with a complex distribution of ACh response that emulated parasympathetic stimulation (41) and complex gCa,L, gKr, and gKs upregulation that were related to sympathetic stimulation (12), along with a variation in electrical coupling. Previous SAN models to date have been 1D or a thin radial slice (8, 30) and are, thus, unable to capture the phenomenon studied herein. Building upon histological, anatomical, and mapping studies, our SAN model captured a broad array of experimentally observed behavior and offers comprehensive mechanistic insight into SAN function and neurally induced AF genesis. All activity derived from the automaticity of the SAN complex. We systematically examined effects of the aforementioned factors on LPS disturbances and reentry formation. We provide a novel mechanism for SAN triggered reentry due to the interaction of LPS shifting, myocardial loading leading to transient unidirectional block, and APB formation. We also show that an LPS emerges because of electrotonic loading, which suppresses competing sites, and not merely overdrive pacing wherein the fastest pacemaker triggers the others. If overdrive pacing were the only mechanism, SAN propagation velocity would be much higher.
As in experiments (2, 16, 31, 33), cholinergic concomitant with adrenergic stimulation promoted reentry. We identified the pacemaker complex itself as a possible source of reentry. The common sequence of events preceding reentry comprised a bradycardic beat with a LPS shift toward the inferior SAN, followed by an APB and/or unidirectional block within the SAN, and subsequent reentry or tachycardia. This was possibly owing to the extensive SAN and heterogeneous autonomic response implemented that led to LPS instability and APBs and/or unidirectional block. These reentry episodes could develop into AF with further APD heterogeneities (22, 37). When APBs were present, rotors of both chirality circulating between the SAN and free wall resulted, while unidirectional conduction block generated flutter or clockwise reentry. The most common types of reentry were 1) counterclockwise spirals in the free wall that had detached from the SAN, 2) clockwise reentry originating in the SAN and circulating between the SAN and free wall, and 3) flutter resulting from unidirectional block. We found that the model became arrhythmogenic when the LPS shifted with [ACh] >20 nM and gCa,L was increased. The concentric gradient (model 1) with fewer central cells was more stable and less arrhythmogenic, making it more plausible than model 2. While experiments have confirmed gradients, the exact gradient is difficult to measure because the large change in properties over small distances makes it difficult to know exactly where samples have been taken from.
Activation during control and ACh.
In control, activation maps revealed typical excitation patterns for both atrial substrates (Fig. 3A) with the LPS situated consistently in the intercaval region and activation spreading smoothly over the RA in 70–80 ms (34). In a simulation subset, immediately after ACh application, bradycardic impulses arose in the inferior central SAN but activation was monofocal. We found that the LPS caudal shift in the intercaval region (Fig. 3) was mainly in response to cholingeric effects but to a lesser degree in response to gCa,L increases.
We present here the first bifocal activation of the SAN obtained in a computer model (Fig. 4B) that has been reported in vitro and in vivo (1, 5, 16, 31). Some studies (29) have suggested that impulse propagation is generated by a relatively small group of primary pacemaker cells in the central intercaval SAN. SAN impulse initiation was dynamic, and multicentric with more than one focus, as suggested by Boineau et al. (5). In model 2 (Fig. 1B), where the numbers of central and peripheral cells were similar, the LPS was more dynamic and the occurrence of double breakouts was higher. Double breakouts were uncommon since the myocardial load drew large current that squelched competing emerging pacemaker sites. This may be an antiarrhythmic design to prevent the interaction of multiple pacemakers.
Although electrical coupling anisotropy was not implemented in our model, heterogeneity of cell properties generated a highly anisotropic spread of activation, with cranial propagation twice as fast as cross-CT. Conduction velocities obtained near the SAN center were very similar to those observed in electrical and optical mappings (7, 16). There was no conduction between the intercaval region and septum through the block zone. The septum was activated after SAN activity propagated around the SVC and inferior vena cava, as observed experimentally (6, 16). Thus AP differences within the SAN, and low coupling, may be the chief factors for anisotropic propagation in the central SAN.
We observed a low excitable zone in the inferior SAN during basal conditions during control for central and superior LPSs (black regions in activation maps of Fig. 3) as has been seen experimentally (1, 7). This zone was functional, created by current extraction from the SAN during RA excitation. The SAN produced sufficient current to excite the myocardium, and not a great excess, otherwise multiple LPSs would be much more common.
Reentry and tachyarrhythmias.
Comparing activity during reentry induced by ACh stimulation with typical activation, rhythm increases and APBs were found. Earlier, Sharifov et al. (33) reported focal or reentrant rapid activity in a RA preparation after ACh administration through the SAN artery. This focal activity was located intercavally but only after the onset of tachycardia, with impulses tending to start from ectopic RA locations. APBs that started in the CT with a short coupling interval (138 ms) resulted in conduction block and reentrant excitation in the free wall. Later, Armour et al. (2) and Pagé et al. (31) found early breakouts in the free wall and CT and lines of block in the free wall. These epicardial breakouts were associated in endocardial mappings to locations near the SVC, CT, and interatrial septum by Armour et al. (2). The onset mechanism and activation pattern of counterclockwise reentry caused by APBs in the inferior SAN region with a coupling interval of 153 ± 34 ms (Fig. 5) were similar to the early breakouts in the free wall and CT observed by Sharifov et al. (33) and Armour et al (2). Armour and colleagues (2, 31) also found early epicardial breakouts located in Bachmann's bundle and the adjacent base of the appendage, similar to the APBs in the superior SAN region and unidirectional block in the inferior SAN region (Fig. 6) that produced clockwise reentry in this study.
Locations that underlie neurons in the dorsal component of RA ganglionated plexus raise the possibility of a neuronal element involved in arrhythmogenesis, similar to this work. Nerve stimulation induced changes in different SAN regions in electrical (23, 38), and optical recordings (16) demonstrate a neuronal component. Therefore, we conclude that heterogeneous cholinergic and adrenergic stimulation of a broad intercaval SAN model in the RA is more prone to leading pacemaker imbalances, producing APBs and unidirectional blocks that give rise to reentrant wavefronts.
Reentry mechanisms–SAN role.
Sharifov et al. (33) reported that tachycardia is modulated by β-adrenoreceptors and is suppressed if ACh effects are blocked by atropine, suggesting a mixed sympathetic and parasympathetic influence like that proposed by this model. Adrenergic tone enhanced by isoproterenol decreased the [ACh] needed and significantly prolonged AF; in addition, AF could not be induced by a very intense isoproterenol stimulation (33). For both atrial substrates, increases in gCa,L increased reentry and APB probabilities and an [ACh] >20 nM that induced a change in the leading pacemaker position were necessary for arrhythmia formation, irrespective of gCa,L. The effects of simultaneous sympathetic and vagal stimulation on RA refractoriness are not only additive but also synergistic. In our model, the parasympathetic tone tends to move the LPS inferiorly, but the increase in ICa,L makes the whole system more excitable. The end result is that pacemaking is not squelched in the superior region and the inferior starts to generate impulses, leading to interaction and reentry.
In pulmonary vein experiments, simultaneous adrenergic and cholinergic stimulation during rapid pacing facilitated the development of APBs and tachycardias by abbreviated APDs that elevated diastolic [Ca2+] and increased forward Na/Ca exchanger activity (32). In our work, increasing [ACh] decreased APD, and increasing L-type calcium channels increased the magnitude of the Ca2+ influx, resulting in APBs within the SAN where gCa,L was higher. IK,ACh activation also leads to hyperpolarization and CL increases, which generated strong rebound effects in the area of highest hyperpolarization, also helping APB formation. Some beats triggering reentry in the RA, which have been traditionally been assumed ectopic by investigators, may really have originated from the large SAN region.
Pagé et al. (31) investigated reentry by stimulating the vagosympathetic trunk that is mainly parasympathetic (10% sympathetic) and mediastinal nerves that have greater sympathetic effect. They found that mediastinal nerve stimulation occupied smaller areas of neurally induced repolarization changes, and more frequent reentrant activity in the free wall with lines of block. For stimulation of the vagosympathetic trunk, they found more frequent early activity foci with radial spread and dual breakouts. Depending on the type of nerve stimulated, different mechanisms were involved. This is corroborated by our work where a change in any of the parameters analyzed can spawn clockwise, counterclockwise, or flutter reentries.
Another factor contributing to arrhythmias was the transient region of inexcitability in the inferior or superior SAN region during control conditions (Fig. 3). Vinogradova et al. (38) suggested inexcitable regions serving as obstacles to create reentry. Here we show the regions may temporarily block activity but then recover to establish the reentrant circuit. These regions contributed to APB induction and LPS shifting. If an inexcitable region occurred in the inferior SAN, after ACh stimulation, the LPS shifted and activated the inferior SAN more easily because of the small region of recovered tissue.
Recently, Fedorov et al. (15, 17) found discrete exit points in the canine SAN but, in a study of murine hearts, did not note any (18). In our rabbit model, exit points were functional and anatomically distinct exit points were not necessary for proper physiological function. Hence, the SAN frequency was the same as the free wall. Furthermore, given that peripheral cells in our model have an intrinsically higher frequency, they will be the LPS unless sufficiently electrotonically loaded, which may not be possible with only a very limited number of discrete junctions. However, discrete exit points may be needed in larger hearts where electrotonic effects may not be sufficient to capture the whole SAN and squelch competing pacemaker sites. Regardless, we are able to replicate essential features of SAN behavior that is also seen in larger hearts. In these larger hearts, it is possible that extra exit points present in individuals may lead to reentry as we describe and that these exit points may only become functional under high autonomic tone. Discrete sites may also lead to reentry if activity originating from the exit site nearest the LPS reaches the other exit site and finds it recovered.
Study limitations.
Since ACh stimulation was limited only to SAN tissue, the duration and number of wavefronts during reentry were smaller. SAN reentry induction mechanisms would not be affected by incorporating neural effects in the atrium. Simulations with neurally induced APD heterogeneities in the atrium should be a next step in this study where a more comprehensive and complex cholinergic and adrenergic stimulation model can be investigated. This will also allow us to study maintenance of reentries.
The geometry was human, but was scaled to rabbit dimensions, and had the common mammalian morphology. All other parameters in the model were lapine. Pectinate muscles and a CT were not explicitly represented, although poor trans CT coupling was implemented.
Reentry and tachycardias emerged after one or two bradycardic beats, not consistent with the involvement of a more slowly developing adrenergic influence, because adrenergic effects were modeled as simple gCa,L upregulation without considering a dynamic [cAMP] and other currents. Cholinergic and adrenergic stimulation share common cellular and neuronal pathways in antagonistic manners that make it difficult to model simultaneous activation. We have taken the approach to only model the final effect on ion transport and have investigated a broad range that covers all possible combinations. Furthermore, while vagal effects are not instantaneous experimentally, negative chronotropic effects manifest in <1 safter nerve stimulation onset.
There are discrepancies between AP morphology as produced by this study and those recorded experimentally. Given the high degree of heterogeneity in intrinsic AP properties and coupling found in situ occuring over very small distances, the model is unable to capture all subtleties necessary. However, basic cycle length and amplitude properties are maintained.
In conclusion, coupling in and around the SAN must be delicately balanced so that the SAN provides current periodically to excite the myocardium but, at the same time, is protected against itself and the myocardium from premature reexcitation. Gradients in ionic properties must exist to protect a pacemaking region from electrotonic suppression and allow for the excitation to leave the SAN. Under high autonomic tone, the broad, heterogeneous SAN region produces responses leading to severe LPS shifting, multiple pacemaker sites, APBs, and transient block that can lead to reentry. These mechanisms may account for the onset of some forms of paroxysmal or vagotonic AF, or flutter.
GRANTS
This work was supported by the Canadian Heart and Stroke Foundation–Alberta, NWT, and Nunavut.
DISCLOSURES
E. Vigmond is President of Cardiosolv, LLC. The company had no participation in the study.
Supplementary Material
REFERENCES
- 1. Abramochkin D, Kuzmin VS, Sukhova G, Rosenshtraukh L. Modulation of rabbit sinoatrial node activation sequence by acetylcholine and isoproterenol investigated with optical mapping technique. Acta Physiol 196: 385–394, 2009. [DOI] [PubMed] [Google Scholar]
- 2. Armour JA, Richer LP, Pagé P, Vinet A, Kus T, Vermeulen M, Nadeau R, Cardinal R. Origin and pharmacological response of atrial tachyarrhythmias induced by activation of mediastinal nerves in canines. Auton Neurosci 118: 68–78, 2005. [DOI] [PubMed] [Google Scholar]
- 3. Beau SL, Hand DE, Schuessler RB, Bromberg BI, Kwon B, Boineau JP, Saffitz JE. Relative densities of muscarinic cholinergic and beta-adrenergic receptors in the canine sinoatrial node and their relation to sites of pacemaker activity. Circ Res 77: 957–963, 1995. [DOI] [PubMed] [Google Scholar]
- 4. Bleeker WK, Mackaay AJ, Masson-Pévet M, Bouman LN, Becker AE. Functional and morphological organization of the rabbit sinus node. Circ Res 46: 11–22, 1980. [DOI] [PubMed] [Google Scholar]
- 5. Boineau JP, Canavan TE, Schuessler RB, Cain ME, Corr PB, Cox JL. Demonstration of a widely distributed atrial pacemaker complex in the human heart. Circulation 77: 1221–1237, 1988. [DOI] [PubMed] [Google Scholar]
- 6. Boyett MR, Honjo H, Kodama I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc Res 47: 658–687, 2000. [DOI] [PubMed] [Google Scholar]
- 7. Boyett MR, Honjo H, Yamamoto M, Nikmaram MR, Niwa R, Kodama I. Downward gradient in action potential duration along conduction path in and around the sinoatrial node. Am J Physiol Heart Circ Physiol 276: H686–H698, 1999. [DOI] [PubMed] [Google Scholar]
- 8. Butters TD, Aslanidi OV, Inada S, Boyett MR, Hancox JC, Lei M, Zhang H. Mechanistic links between Na+ channel (SCN5A) mutations and impaired cardiac pacemaking in sick sinus syndrome. Circ Res 107: 126–137, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Camelliti P, Green CR, LeGrice I, Kohl P. Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ Res 94: 828–835, 2004. [DOI] [PubMed] [Google Scholar]
- 10. Cloherty SL, Dokos S, Lovell NH. A comparison of 1-D models of cardiac pacemaker heterogeneity. IEEE Trans Biomed Eng 53: 164–177, 2006. [DOI] [PubMed] [Google Scholar]
- 11. Crick SJ, Sheppard MN, Ho SY, Anderson RH. Localisation and quantitation of autonomic innervation in the porcine heart I: conduction system. J Anat 195: 341–357, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Demir SS, Clark JW, Giles WR. Parasympathetic modulation of sinoatrial node pacemaker activity in rabbit heart: a unifying model. Am J Physiol Heart Circ Physiol 276: H2221–H2244, 1999. [DOI] [PubMed] [Google Scholar]
- 13. DiFrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 351: 145–147, 1991. [DOI] [PubMed] [Google Scholar]
- 14. Dobrzynski H, Li J, Tellez J, Greener ID, Nikolski VP, Wright SE, Parson SH, Jones SA, Lancaster MK, Yamamoto M, Honjo H, Takagishi Y, Kodama I, Efimov IR, Billeter R, Boyett MR. Computer three-dimensional reconstruction of the sinoatrial node. Circulation 111: 846–854, 2005. [DOI] [PubMed] [Google Scholar]
- 15. Fedorov VV, Chang R, Glukhov AV, Kostecki G, Janks D, Schuessler RB, Efimov IR. Complex interactions between the sinoatrial node and atrium during reentrant arrhythmias in the canine heart. Circulation 122: 782–789, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fedorov VV, Hucker WJ, Dobrzynski H, Rosenshtraukh LV, Efimov IR. Postganglionic nerve stimulation induces temporal inhibition of excitability in rabbit sinoatrial node. Am J Physiol Heart Circ Physiol 291: H612–H623, 2006. [DOI] [PubMed] [Google Scholar]
- 17. Fedorov VV, Schuessler RB, Hemphill M, Ambrosi CM, Chang R, Voloshina AS, Brown K, Hucker WJ, Efimov IR. Structural and functional evidence for discrete exit pathways that connect the canine sinoatrial node and atria. Circ Res 104: 915–923, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Glukhov AV, Fedorov VV, Anderson ME, Mohler PJ, Efimov IR. Functional anatomy of the murine sinus node: high-resolution optical mapping of ankyrin-B heterozygous mice. Am J Physiol Heart Circ Physiol 299: H482–H491, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gong Y, Xie F, Stein KM, Garfinkel A, Culianu CA, Lerman BB, Christini DJ. Mechanism underlying initiation of paroxysmal atrial flutter/atrial fibrillation by ectopic foci: a simulation study. Circulation 115: 2094–2102, 2007. [DOI] [PubMed] [Google Scholar]
- 20. Honjo H, Boyett MR, Kodama I, Toyama J. Correlation between electrical activity and the size of rabbit sino-atrial node cells. J Physiol 496: 795–808, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kannel WB, Wolf PA, Benjamin EJ, Levy D. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol 82: 2N–9N, 1998. [DOI] [PubMed] [Google Scholar]
- 22. Kneller J, Zou R, Vigmond EJ, Wang Z, Leon LJ, Nattel S. Cholinergic atrial fibrillation in a computer model of a two-dimensional sheet of canine atrial cells with realistic ionic properties. Circ Res 90: E73–E87, 2002. [DOI] [PubMed] [Google Scholar]
- 23. Kodama I, Boyett MR, Suzuki R, Honjo H, Toyama J. Regional differences in the response of the isolated sino-atrial node of the rabbit to vagal stimulation. J Physiol 495: 785–801, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kurata Y, Hisatome I, Imanishi S, Shibamoto T. Dynamical description of sinoatrial node pacemaking: improved mathematical model for primary pacemaker cell. Am J Physiol Heart Circ Physiol 283: H2074–H2101, 2002. [DOI] [PubMed] [Google Scholar]
- 25. Kurata Y, Hisatome I, Imanishi S, Shibamoto T. Roles of L-type Ca+ and delayed-rectifier K+ currents in sinoatrial node pacemaking: insights from stability and bifurcation analyses of a mathematical model. Am J Physiol Heart Circ Physiol 285: H2804–H2819, 2003. [DOI] [PubMed] [Google Scholar]
- 26. Kurata Y, Matsuda H, Hisatome I, Shibamoto T. Effects of pacemaker currents on creation and modulation of human ventricular pacemaker: theoretical study with application to biological pacemaker engineering. Am J Physiol Heart Circ Physiol 292: H701–H718, 2007. [DOI] [PubMed] [Google Scholar]
- 27. Kurata Y, Matsuda H, Hisatome I, Shibamoto T. Regional difference in dynamical property of sinoatrial node pacemaking: role of Na+ channel current. Biophys J 95: 951–977, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lindblad DS, Murphey CR, Clark JW, Giles WR. A model of the action potential and underlying membrane currents in a rabbit atrial cell. Am J Physiol Heart Circ Physiol 271: H1666–H1696, 1996. [DOI] [PubMed] [Google Scholar]
- 29. Opthof T, de Jonge B, Mackaay AJ, Bleeker WK, Masson-Pevet M, Jongsma HJ, Bouman LN. Functional and morphological organization of the guinea-pig sinoatrial node compared with the rabbit sinoatrial node. J Mol Cell Cardiol 17: 549–564, 1985. [DOI] [PubMed] [Google Scholar]
- 30. Oren RV, Clancy CE. Determinants of heterogeneity, excitation and conduction in the sinoatrial node: a model study. PLoS Comput Biol 6: e1001041, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pagé P, Armour JA, Yin Y, Vermeulen M, Nadeau R, Cardinal R. Differential effects of cervical vagosympathetic and mediastinal nerve activation on atrial arrhythmia formation in dogs. Auton Neurosci 128: 9–18, 2006. [DOI] [PubMed] [Google Scholar]
- 32. Patterson E, Lazzara R, Szabo B, Liu H, Tang D, Li YH, Scherlag BJ, Po SS. Sodium-calcium exchange initiated by the Ca+ transient: an arrhythmia trigger within pulmonary veins. J Am Coll Cardiol 47: 1196–1206, 2006. [DOI] [PubMed] [Google Scholar]
- 33. Sharifov OF, Fedorov VV, Beloshapko GG, Glukhov AV, Yushmanova AV, Rosenshtraukh LV. Roles of adrenergic and cholinergic stimulation in spontaneous atrial fibrillation in dogs. J Am Coll Cardiol 43: 483–490, 2004. [DOI] [PubMed] [Google Scholar]
- 34. Sharifov OF, Zaitsev AV, Rosenshtraukh LV, Kaliadin AY, Beloshapko GG, Yushmanova AV, Schuessler RB, Boineau JP. Spatial distribution and frequency dependence of arrhythmogenic vagal effects in canine atria. J Cardiovasc Electrophysiol 11: 1029–1042, 2000. [DOI] [PubMed] [Google Scholar]
- 35. Shibata N, Inada S, Mitsui K, Honjo H, Yamamoto M, Niwa R, Boyett MR, Kodama I. Pacemaker shift in the rabbit sinoatrial node in response to vagal nerve stimulation. Exp Physiol 86: 177–184, 2001. [DOI] [PubMed] [Google Scholar]
- 36. Verheule S, van Kempen MJ, Postma S, Rook MB, Jongsma HJ. Gap junctions in the rabbit sinoatrial node. Am J Physiol Heart Circ Physiol 280: H2103–H2115, 2001. [DOI] [PubMed] [Google Scholar]
- 37. Vigmond EJ, Tsoi V, Kuo S, Arevalo H, Kneller J, Nattel S, Trayanova N. The effect of vagally induced dispersion of action potential duration on atrial arrhythmogenesis. Heart Rhythm 1: 334–344, 2004. [DOI] [PubMed] [Google Scholar]
- 38. Vinogradova TM, Fedorov VV, Yuzyuk TN, Zaitsev AV, Rosenshtraukh LV. Local cholinergic suppression of pacemaker activity in the rabbit sinoatrial node. J Cardiovasc Pharmacol 32: 413–424, 1998. [DOI] [PubMed] [Google Scholar]
- 39. Zhang H, Holden AV, Boyett MR. Gradient model versus mosaic model of the sinoatrial node. Circulation 103: 584–588, 2001. [DOI] [PubMed] [Google Scholar]
- 40. Zhang H, Holden AV, Kodama I, Honjo H, Lei M, Varghese T, Boyett MR. Mathematical models of action potentials in the periphery and center of the rabbit sinoatrial node. Am J Physiol Heart Circ Physiol 279: H397–H421, 2000. [DOI] [PubMed] [Google Scholar]
- 41. Zhang H, Holden AV, Noble D, Boyett MR. Analysis of the chronotropic effect of acetylcholine on sinoatrial node cells. J Cardiovasc Electrophysiol 13: 465–474, 2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.