

Abstract
Propagation of ryanodine receptor (RyR2)-derived Ca2+ signals to the mitochondrial matrix supports oxidative ATP production or facilitates mitochondrial apoptosis in cardiac muscle. Ca2+ transfer likely occurs locally at focal associations of the sarcoplasmic reticulum (SR) and mitochondria, which are secured by tethers. The outer mitochondrial membrane and inner mitochondrial membrane (OMM and IMM, respectively) also form tight focal contacts (contact points) that are enriched in voltage-dependent anion channels, the gates of OMM for Ca2+. Contact points could offer the shortest Ca2+ transfer route to the matrix; however, their alignment with the SR-OMM associations remains unclear. Here, in rat heart we have studied the distribution of mitochondria-associated SR in submitochondrial membrane fractions and evaluated the colocalization of SR-OMM associations with contact points using transmission electron microscopy. In a sucrose gradient designed for OMM purification, biochemical assays revealed lighter fractions enriched in OMM only and heavier fractions containing OMM, IMM, and SR markers. Pure OMM fractions were enriched in mitofusin 2, an ∼80 kDa mitochondrial fusion protein and SR-mitochondrial tether candidate, whereas in fractions of OMM + IMM + SR, a lighter (∼50 kDa) band detected by antibodies raised against the NH2 terminus of mitofusin 2 was dominating. Transmission electron microscopy revealed mandatory presence of contact points at the junctional SR-mitochondrial interface versus a random presence along matching SR-free OMM segments. For each SR-mitochondrial junction at least one tether was attached to contact points. These data establish the contact points as anchorage sites for the SR-mitochondrial physical coupling. Close coupling of the SR, OMM, and IMM is likely to provide a favorable spatial arrangement for local ryanodine receptor-mitochondrial Ca2+ signaling.
Keywords: Ca2+ signaling, cardiac muscle, tethering, mitofusin 2
microdomain signaling and local solute or lipid exchange have been emerging as fundamental interorganellar communication mechanisms that support diverse cellular functions. A prominent example is the local delivery of ryanodine receptor (RyR2)-derived Ca2+ signals to the mitochondria. This mechanism can provide feedforward support for the oxidative ATP production and, in certain stress conditions, promote mitochondrial permeabilization and apoptosis in the cardiac muscle (4, 15–17, 23, 27, 28). The platforms for these local interactions are focal associations between the organelles that are fastened by sturdy protein tethers in a variety of nonmuscle cell types (12, 13, 21, 32). Our earlier work has been the first to demonstrate that sarcoplasmic reticulum (SR) fragments remain associated with mitochondria in mitochondrial fractions of cardiac muscle and can locally deliver Ca2+ to the matrix (19). Furthermore, actual tethers connecting the SR and the outer mitochondrial membrane (OMM) have been recently visualized in mouse heart via both conventional transmission electron microscopy (TEM) (8) and electron tomography (20). The molecular nature of the tethers is still to be elucidated. Based on the heterogeneous appearance of the tethers (12), it is likely that more than one protein species contribute to fastening SR/endoplasmic reticulum (SR/ER) to the mitochondria. Out of a handful of proteins that have been proposed to form SR/ER-mitochondrial tethers (reviewed in Ref. 22) mitofusin 2 (MFN2), a small GTPase known to reside in the OMM and being essential for mitochondrial fusion, is highly expressed in the cardiac muscle (16). MFN2 has been reported to also be expressed in the ER, participating in the shaping of the ER as well as forming ER-mitochondrial tethers via homodimerization or heterodimerization with MFN1 in the OMM (13). Delivery of molecules from the cytosol to the mitochondrial matrix requires passing through two membrane boundaries, the OMM and the inner mitochondrial membrane (IMM). The mitochondrial import of matrix and IMM proteins takes place at locations where OMM and IMM membranes are closely anchored together (so called mitochondrial contact points or contact sites) (9) and the OMM/IMM-resident protein import complexes (translocases of the OMM adn IMM, TOM and TIM23, respectively) can directly interact (25). However, it has not been clarified whether similar localization preference existed for the delivery of metal cations to the matrix. A possible advantage of such a scenario could be the enhanced efficacy of Ca2+ delivery from high [Ca2+] microdomains to the low-affinity Ca2+ uptake sites (mitochondrial Ca2+ uniporter) in the IMM. Enrichment at the contact points of voltage-dependent anion channels (VDAC), the main pathways for Ca2+ to cross the OMM, supports this notion (10). Favored localization to the contact points of mitochondria-associated ER membranes contributing to phospholipid biosynthesis has been reported before in the liver (2). However, in the cardiac muscle, preferential localization of mitochondria-coupled SR Ca2+ sources to the regions of contact points is still to be tested.
In this study we have aimed to clarify this point in rat heart via biochemical analysis of submitochondrial fractions obtained using osmotic disruption, sonication, and sucrose density gradient separation as well as TEM analysis of cardiac muscle samples.
MATERIALS AND METHODS
Chemicals.
Chemicals were purchased from Fisher or Sigma unless specified otherwise. Reagents for preparation of TEM specimens were from Electron Microscopy Sciences. Primary antibodies with sources were as follows: rabbit polyclonal anti-calsequestrin was from Millipore/Upstate (Catalog No. 06-382); mouse monoclonal anti-human Complex III, ubiquinol cytochrome C oxidoreductase, was from Molecular Probes (Invitrogen; Catalog No. 459220); anti-MFN2 antibodies are described in results; rabbit polyclonal anti-prohibitin was from Abcam (Catalog No. ab28172); mouse monoclonal anti-sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA2) was from Thermo Scientific/Pierce (Catalog No. MA3-919); rabbit polyclonal anti-VDAC was from Thermo Scientific/Pierce (Catalog No. PA1-954A).
Animals.
Male Sprague-Dawley rats of 350–500 g body wt were used in the study. All the procedures were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and the protocols were approved by the Institutional Animal Care and Use Committee.
Cells and culture.
Mouse embryonic fibroblasts (MEF) derived from wild-type and MFN2 knockout mice were kind gifts from David Chan and cultured as described previously (11). Cell lysate was prepared using premade radioimmunoprecipitation assay buffer (Sigma) supplemented with protease inhibitors and 1 mM PMSF.
Fractionation of rat heart mitochondria.
Crude mitochondria (10,000 g pellet) were isolated from the homogenates of two rat heart ventricles as described previously (19). Submitochondrial fractions from the crude mitochondria were purified using an OMM purification protocol (5, 7) modified as follows. After a washing step, the mitochondrial (10,000 g) pellet was resuspended and incubated in a hyposmotic phosphate buffer (swelling medium, 10 mM NaH2PO4/Na2HPO4, pH 7.4) for 20 min. Subsequently, a shrinking medium (60% sucrose) was added to the suspension (29% volume). After 15 min of shrinking, the mitochondrial membranes were disrupted by ultrasonication (Branson 102C CE Converter, 5 × 30 s of irradiation at 21% amplitude, 1-min break between each run). The sonicated material was spun down at 6,500 g for 10 min. The pellet containing unbroken mitochondria was discarded, and the supernatant containing a mixture of mitochondrial membrane vesicles was loaded on top of a discontinuous sucrose gradient (30%, 40%, 50%, and 60%; Fig. 2) to separate submitochondrial membrane fractions (ultracentrifuge at 200,000 g for 240 min). The entire procedure was performed at 4°C.
Fig. 2.

Purification of outer mitochondrial membrane (OMM) and OMM-inner mitochondrial membrane (IMM) contacts from rat heart mitochondria. Mitochondria isolated from rat heart homogenates were subfractionated on a discontinuous sucrose gradient as the scheme shows at left (with sucrose concentrations shown as percent weight per volume). Protein concentration profile (bottom) and normalized (to the maximum, max) activities (top) of monoaminooxidase (MAO), an OMM resident, and succinate dehydrogenase (SDH), an IMM localized enzyme in the isolated subfractions, are shown. Data are means ± range from 2 independent preparations (each from 2 hearts).
Fluorescence wide field imaging.
Mitochondrial [Ca2+] signals and NADH responses were carried out as described previously (19).
Enzyme assays.
Monoaminooxidase activity was measured using kynuramine as fluorescent substrate that displays a decrease in fluorescence (315 nm excitation, 390 nm emission) upon oxidation by monoaminooxidase (MAO) in a phosphate buffer (24). In brief, the reaction buffer was composed of 100 mM K2HPO4/KH2PO4 (pH 7.4). A sample (200 μl) was added to 400 μl reaction buffer and incubated for 5 min at 35°C, and the reaction was then started by the addition of 250 μM kynuramine (Sigma). With the use of a multi-wavelength excitation, dual emission spectrofluorometer (Delta RAM, PTI), the slope of the decrease in kynuramine fluorescence was determined and considered proportional with the MAO enzyme activity after correction to the sample protein concentration. Hexokinase activity was measured based upon the reduction of NAD+ through a coupled reaction with glucose-6-phosphate dehydrogenase. The assay buffer contained 6 mM ATP, 2.8 mM NADP, 9.3 mM MgCl2, 0.64 units/ml glucose-6-phosphate dehydrogenase, 2.5 μg/ml F1/F0 ATP-ase blocker oligomycin, 3 μM respiratory complex I blocker rotenone, and 100 nM respiratory complex III blocker antimycin A. The slopes of NADPH fluorescence increase after addition of 10 mM glucose were determined and normalized to the protein concentration in the sample. Succinate dehydrogenase (SDH) assay measures the electron transfer from succinate (substrate) to ferricytochrome c catalyzed by SDH and respiratory complex III. Cytochrome c has a sharp absorption band at 550 nm in the reduced (ferrous) state that becomes weaker upon oxidation (ferricytochrome c). The reaction buffer was a phosphate buffer (0.1 M K2HPO4/KH2PO4, pH 7.4) supplemented with 3 mM succinate, 3 μM rotenone, and 0.3 mM KCN (to block complex IV). The reaction was started by the addition of 50 μM ferricytochrome c, and the absorbance at 550 nm was recorded in a spectrophotometer (Uvikon). The slopes of increase in the absorbance were determined and normalized to the protein concentration in the sample. For all enzyme assays, relative enzyme activities were expressed as the percentage of the maximum (the fraction with the highest activity).
Protein assays were done using a biuret-based reagent kit from Bio-Rad (Bio-Rad DC Protein Assay) according to the manufacturer's protocol.
Western blots were carried out using standard protocols, Bio-Rad hardware, and precast gels (Ready Gel from Bio-Rad). Secondary antibodies were either conjugated with horseradish peroxidase and visualized by the chemiluminescent substrate SuperSignal West Pico (from Pierce/Thermo Scientific) or with an infrared fluorophore (IRDye 800CW from LI-COR) and scanned using the LI-COR Odyssey infrared imaging system (Fig. 6).
Fig. 6.
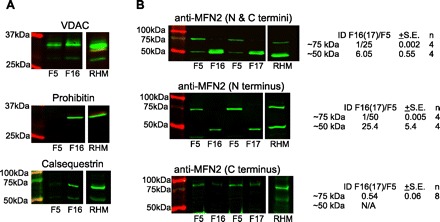
Determination of mitofusin 2 (MFN2) protein in the light and heavy submitochondrial fractions. Light and heavy fractions close to the 2 protein peaks (fractions 5 and 16–17, respectively, from gradients separated to 22 to 23 fractions in total) were compared in their MFN2 immunoreactivity via Western blot. Reactions were visualized using the infrared fluorescence LI-COR detection system. A: VDAC (∼31 kDa), prohibitin (∼29 kDa), and calsequestrin (∼60 kDa) antibodies were used to confirm the membrane composition of the fractions (note the omnipresence of VDAC and the enrichment of prohibitin and calsequestrin bands in fraction 16). The crude RHM was used for reference (positive control). B: MFN2 (∼80 kDa) immunoreactive bands detected by 3 different antibodies recognizing either the COOH terminus (bottom), NH2 terminus (middle), or both (top) in the subfractions shown in A and in fractions 5 and 17 (F5, F17) from another preparation. Note the lack or decreased presence of the ∼75 kDa band in the heavy fractions and at the same time the appearance of a lower MW band (∼50 kDa). The double-banding in F16/17 when using antibody against the N and C termini of MFN2 (top) is most likely due to a cross-reaction, and it is not an artifact since it also appears in mitochondrial preparations of rat skeletal muscle (not shown). The numbers on the right show the integrated fluorescence intensity [integrated density (ID)] of F16 relative to F5 for the ∼75 kDa and ∼50 kDa bands.
Electron microscopy.
Rat hearts were perfusion-fixed as follows. The animals were euthanized via decapitation, and the heart was immediately removed and quickly secured to the cannula of a Langendorff-type perfusion setup by the ascending aorta. The initial perfusate was a modified Ringer's solution containing (in mM) 121 NaCl, 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, and 2 CaCl2, pH 7.4 supplemented with 0.1% procaine, oxygenized, and chilled to 4°C. The heart was still beating at the start of perfusion. After 5 min perfusion (at <10 ml/min flow rate maintained with peristaltic pump) the initial perfusate was switched to the ice-cold fixative composed of 2.5% glutaraldehyde in 0.1 M Na-cacodylate buffer (pH 7.4) for 5 min. The apical quarter of the prefixed heart was removed by a transversal (perpendicular to the baso-apical axis) cut and, in turn, 3 to 4 ∼1-mm-thick slices were made parallel to the first cut. From the left ventricular frontal wall segment of each slice 2–4 ∼ 1 mm3 pieces were cut and incubated in the fixative for another 2 h at 4°C. After that, samples were stained en block with partially reduced osmium tetroxide (0.8% potassium-ferrocyanide and 2% osmium tetroxide in 0.1 M sodium cacodylate buffer overnight at 4°C) and, in turn, with 1% uranyl acetate (1 h at 4°C). Stained samples were dehydrated on an acetone dilution series and embedded to Durcupan or Spurr's resin according to manufacturer's (Elecron Microscopy Sciences) suggestions. Thin sections (60–100 nm) were cut from the embedded specimens, mounted on electron microscopy grids, and examined using a FEI Tecnai 12 TEM equipped with a phosphor plate [Advanced Microscopy Techniques (AMT)] and Hamamatsu Orca 8Mpx digital camera.
RESULTS AND DISCUSSION
RyR2-dependent Ca2+ delivery from physically bound SR segments to the mitochondria and consequent activation of Ca2+-sensitive matrix dehydrogenases.
When the ventricular cardiac muscle is mechanically homogenized and membrane fractions are isolated via differential centrifugation, the bulk of the SR requires 50–100,000 g force to sediment. However, a smaller population of SR vesicles sediments in the mitochondrial fraction at 10,000 g because of their association with the mitochondria. As we have shown earlier, these mitochondria-bound SR segments can deliver Ca2+ to the mitochondrial matrix via RyR2-mediated Ca2+ release (19). This, in turn, can activate Ca2+-sensitive matrix dehydrogenases that feed reducing equivalents to the electron transport chain resulting in enhanced oxidative ATP production. This pathway in the muscle may work as a feedforward energy-regulatory mechanism in the excitation-contraction coupling. Figure 1 (top) shows representative traces of simultaneously recorded [Ca2+]c and [Ca2+]m in cover glass-attached organelles from rat heart mitochondrial fraction (10,000 g pellet). Responses were triggered by the RyR activator caffeine followed by the addition of a Ca2+ pulse (20 μM CaCl2 that raised [Ca2+] in the cytosolic buffer, [Ca2+]c to ∼7 μM). As expected, RyR2-mediated Ca2+ release is barely detectable in the bulk cytosolic buffer (Fig. 1A, thin line), owing to its dilution in an effectively infinite buffer volume. However, the [Ca2+] in the mitochondrial matrix ([Ca2+]m) elevation is considerable, with 5–40% of the [Ca2+]m rise caused by the reference Ca2+ pulse in 66 out of 100 mitochondria (see histogram in Fig. 1A). The [Ca2+]m rise occurring in the absence of a bulk [Ca2+]c rise indicates that the RyR2-mediated Ca2+ release was locally transferred to the mitochondria (19). From parallel experiments, recording of NAD(P)H autofluorescence is shown in Fig. 1B. Caffeine evoked a substantial increase in the NAD(P)H autofluorescence, comparable in size with that provoked by the reference Ca2+ pulse (see histogram). Notably, a very similar fraction (∼1/3) of the mitochondria displayed negligible [Ca2+]m and NADH responses, which fraction probably corresponded to the mitochondria lacking appended SR. Thus, extending our earlier data, a large portion of the mitochondria mechanically isolated from cardiac muscle preserves their connections to SR domains, from which they can receive calcium signals and respond by enhanced production of reducing equivalents and eventually ATP. The highly efficient local Ca2+ transfer in spite of the RyR2 being positioned to the junctional SR (jSR) surface facing away from the OMM (18) (Fig. 7) indicates an optimal arrangement of the Ca2+ path between the two organelles.
Fig. 1.
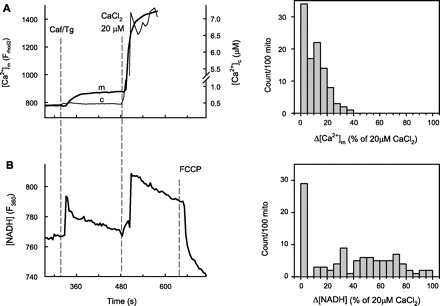
Sarcoplasmic reticulum (SR)-derived Ca2+ signal reception and matrix dehydrogenase activation are preserved in isolated rat heart mitochondria (mito). A: [Ca2+]m and [Ca2+]c (measured with rhod2 accumulated to the mitochondrial matrix and fura 2 added to the cytosolic buffer, respectively) responses were simultaneously imaged in the mitochondrial fractions (10,000 g pellet) of rat heart homogenates. The stimulation protocol was sequential addition of caffeine [Caf; 10 mM, that evokes maximum ryanodine receptor (RyR2)-mediated Ca2+ release from the SR] and a reference Ca2+ pulse (20 μM CaCl2) that elevated [Ca2+]c to ∼7 μM. To maximize RyR2-mediated Ca2+ mobilization from the SR, caffeine was applied together with the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) inhibitor thapsigargin (Tg; 10 μM). B: in parallel experiments, the changes in NAD(P)H autofluorescence were recorded. The traces of [Ca2+]m and NAD(P)H are means of ∼100 mitochondria. The histograms at right show the distribution of responses to caffeine stimulation normalized to the reference Ca2+ pulse amongst 100 randomly selected mitochondrial spots on the field. Mitochondria not showing response to either caffeine or the Ca2+ reference pulse were excluded from the analysis.
Fig. 7.

Functional consequences of the alignment of jSR-OMM junctions with mitochondrial contact points. The scheme illustrates the potential advantages of aligning the OMM-anchored jSR with mitochondrial contact points with regard to the efficacy of local Ca2+ transfer to the mitochondrial matrix. The top depicts a nonaligned scenario, whereas the bottom shows the aligned arrangement. MCU, mitochondrial Ca2+ uniporter; PT, permeability transition.
Purification of IMM-free and IMM-associated OMM from rat heart mitochondria.
The possible association of SR-mitochondrial anchorage sites to the OMM-IMM contact points was examined first in mitochondrial fractions of rat heart homogenates. To isolate OMM segments not connected to the IMM from those that are IMM connected, the organelles were disrupted by means of osmotic shock followed by sonication and the resulting membrane fragments were separated on a discontinuous sucrose density gradient. Ten fractions were collected from the five gradient levels (see scheme in Fig. 2), and from each fraction enzyme activities of MAO (OMM resident) and SDH (IMM resident) were determined. The protein concentration profile of the collected fractions showed a bimodal distribution, the first peak coinciding with the interphase between the sample and 30% sucrose and the second peak with the 40% sucrose fraction (Fig. 2). In the initial fractions, above the first (30%) sucrose layer, MAO activities were progressively increasing (from fraction 1 to 4), whereas SDH activity was barely detectable (fractions 1-3). SDH activity started to increase in fraction 4 and remained high in most of the heavier fractions (Fig. 2). This separation of OMM- and IMM-bound enzyme activities in the lighter (1–3) fractions implied that they were enriched in pure (IMM-free) OMM. Further supporting a differential distribution of OMM and IMM on the sucrose gradient, SDS-PAGE protein profiles resolved on 4–15% silver-stained gradient gels also revealed remarkable differences between light and heavy fractions (Fig. 3A). Notably, based on their distribution in the light and heavy fractions, the protein bands clearly could be divided to three groups: present in light fractions only, heavy fractions only, and those present in both. Most likely, the first group represented proteins confined to the pure (IMM-free) OMM, the second one comprised IMM and contact point proteins, and the third group represented OMM proteins residing in both IMM-free and contact point-forming OMM regions. Western blot (Fig. 3B) analysis of OMM-resident VDAC and the IMM resident proteins prohibitin and respiratory complex III (CIII) further confirmed the differences in the light and heavy fractions. In line with the notion that VDAC is enriched in the mitochondrial contact points (3, 31), the VDAC bands in the fractions containing both IMM and OMM were stronger than those in the pure OMM fractions (n = 3).
Fig. 3.
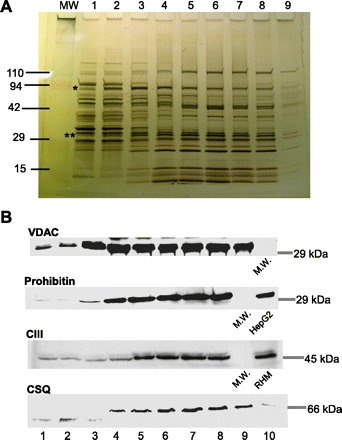
SDS-PAGE and Western blot analysis of proteins in the mitochondrial subfractions. A: silver nitrate-stained protein bands after running the mitochondrial subfractions (1–9) on a 4–15% gradient gel. The lane labeled MW was loaded with molecular weight markers. *Predicted bands for hexokinase (HK); **predicted bands for voltage-dependent anion channel (VDAC) with the adenine nucleotide translocator of IMM (ANT). B: representative Western blots of VDAC (OMM marker), prohibitin (IMM marker), respiratory complex III (CIII; IMM marker), and calsequestrin (CSQ; SR marker). RHM, rat heart mitochondrial fraction.
Thus, using a modified purification method originally established by Brdiczka and coworkers (1) (liver, brain, kidney) and applied to cardiac muscle by Beutner et al. (7), we have successfully isolated membrane fractions enriched in OMM but lacking IMM from those enriched in both OMM and IMM. In contrast with Brdiczka and coworkers (1) and Beutner et al. (7), pure IMM (OMM-free) fractions were missing from our gradient perhaps because, due to a milder disruption protocol, the IMM did not break up to sufficiently small fragments. However, obtaining pure IMM was not a goal in this project.
Copurification of the SR with IMM-associated OMM.
Next, we investigated the distribution of OMM-associated SR in the cardiac mitochondrial subfractions. To start, the relative abundance of calsequestrin 2 (CSQ) was tested with Western blot in the 10 subfractions of rat cardiac mitochondria. Just like the IMM markers (prohibitin, CIII), CSQ was missing from fractions 1-3 (Fig. 3B, bottom) placing CSQ to the protein group with preferred contact point (and/or IMM) location. To further enhance the separation among the different membrane fractions, we increased the number of fractions collected from the sucrose gradient. As shown in Fig. 4A, both MAO and the also OMM-associated hexokinase displayed high activities in the lighter fractions where IMM-resident SDH activity was still close to its minimum. Similar to SDH activity, the IMM-resident CIII immunoreactivity also was low in the light fractions and was concentrated in the heavier components. In the same gradient, the immunoreactivity of the SR-resident SERCA2 also appeared only in the heavy fractions, like the IMM markers (Fig. 4B). Thus the distribution of both CSQ and SERCA2 supports that SR is largely absent in the IMM-free fractions of the OMM. In light of these results, some of the protein bands that were stronger at the heavier fractions in the silver-stained gel (Fig. 3A) might have corresponded to SR resident proteins (e.g., the stronger bands at 110 kDa could correspond to SERCA).
Fig. 4.

Purification of OMM-associated SR from rat heart mitochondria. Submitochondrial fractions (20–24) were separated on a sucrose density gradient. The approximate mapping of the fractions to the sucrose density levels is illustrated by the scheme at left. A: bimodal protein profile is shown at the bottom; enzyme activities of HK (○; OMM marker), MAO (●; OMM marker), and SDH (squares; IMM marker) in the subfractions (normalized to maximum) are shown at top. Data are means ± SE from 3 preparations (HK, SDH) or means ± range from 2 preparations (MAO). B: representative Western blots of CIII (IMM marker) and SERCA2a from the submitochondrial fractions shown in A.
The data above suggest that SR fragments fastened to the mitochondria (8, 20) are likely to be linked to OMM regions that are also connected to the IMM: the OMM-IMM contact points. Because the OMM-bound SR retains its competence for RyR2-dependent local Ca2+ transfer to the mitochondria after mechanical fractionation (Fig. 1) (19), it is plausible to speculate that the SR terminal cisternae (jSR) could be tethered to OMM-IMM contact points. To clarify this point, we next carried out TEM analysis of jSR-mitochondria junctions in rat cardiac muscle.
Colocalization of jSR-mitochondria junctions and SR-mitochondrial tethers with OMM-IMM contact points.
The prevalence of OMM-IMM contact points at the junctions of jSR with the mitochondria was determined in TEM images of left ventricular walls. The segment of OMM interfacing jSR ranged from 91 to 428 nm (mean: 250 ± 23 nm) that was 3.5% to 18% (mean 8.8 ± 1%) of the perimeter of the whole mitochondrial cross section (3 ± 0.2 μm in average). At these regions, the presence of OMM-IMM contacts was evaluated. For reference, we also assessed the presence of OMM-IMM contacts at 1–3 size-matched segments of SR-free OMM (see drawing in Fig. 5B). At the SR-free regions, OMM-IMM contact points were observed in approximately half of the cases (0.56 ± 0.07, 19 mitochondrial cross sections obtained from 3 different animals; Fig. 5B). By contrast, at the junctional area, at least one OMM-IMM contact point coincided in all the cases.
Fig. 5.

Correlating junctional SR(jSR)-mitochondria junctions and SR-mitochondrial tethers with OMM-IMM contact points using transmission electron microscopy (TEM). Cardiac muscle samples from rat left ventricle were fixed and processed for TEM. High-magnification (76,000× at 7 inches print size) images were taken from longitudinal as well as transversal sections of regions containing well recognizable dyads and mitochondria. A: representative high-magnification (left) TEM image of a jSR-OMM junction showing the cross section of a T-tubule (T) and a jSR sack (SR) in association with a mitochondrion (M). Note the contact points (cp) between the OMM and IMM and the discrete electron densities interconnecting the SR and OMM corresponding to tethers (white arrows: the bottom one attaching to a cp, whereas the top one attaches to an OMM segment free of IMM). The high-magnification image at left is zoomed out by a factor of 10× at right to show the organellar context. B: bar graph analyzing the likelihood of an OMM-IMM contact point being located at a jSR-OMM interface or at size-matched SR-free OMM segments selected as shown in the scheme. For each segment the presence and absence of a contact point scores 1 and 0, respectively. For each mitochondrion there is one OMM-jSR junction, and 1–3 SR-free OMM segments are averaged (in some cases it was not possible to have 3 SR-free OMM segments equal in size to the jSR-OMM junction and having well recognizable membranes). Nineteen mitochondria from 3 animals were analyzed. The difference between the 2 groups is significant (P < 0.005, using Wilcoxon signed rank test). C: bar graph summarizing the number of SR-OMM tethers observed in each jSR-mitochondria junction that ended at an OMM-IMM contact point or at an IMM-free OMM region. Data are means ± SE (n = 19; P > 0.05 using Wilcoxon signed rank test).
These results, along with the fractionation data, support the idea that the jSR is preferentially anchored to mitochondrial surface segments accommodating OMM-IMM contact points. Moreover, as Fig. 5A illustrates, at high magnification discrete electron densities traversing the jSR-OMM cleft (white arrowheads) could be resolved that likely corresponded to SR-OMM tethers (as in Ref. 8). The number of tethers counted per SR-OMM junctional section was in average 3.7 ± 0.3 with no significant difference between those ending on contact points and IMM-free OMM (Fig. 5C). It is noteworthy that in almost all (except 1 out of 19) jSR-OMM junctions evaluated, there was at least one tether ending at an OMM-IMM contact point, whereas multiple junctions (5 out of 19) were lacking tethers ending outside the contact points. Collectively, these morphological data suggest that jSR-mitochondrial junctions are aligned with mitochondrial contact points via physical tethers.
Selective enrichment of a low-molecular weight MFN2 immunoreactive protein in mitochondrial contact points.
Because MFN2 is an SR-mitochondrial tether candidate and is abundantly expressed in the cardiac muscle (16), we decided to test its distribution in the submitochondrial fractions. Two fractions (out of the total of 22 to 23) were selected for comparison: light fraction No. 5 that was lacking IMM and SR but was rich in OMM markers/enzymes and a heavy fraction (No. 16–17) enriched in OMM, IMM, and SR markers (see Figs. 4 and 6A). A crude rat heart mitochondrial fraction was used as reference. Three different antibodies were used to detect MFN2: one raised against the NH2 terminus (rabbit polyclonal against aa38-55 of human MFN2; Sigma M6319), another raised against the COOH terminus (mouse monoclonal against aa661-758 of human MFN2; Abcam ab56889), and one raised against both termini (mouse polyclonal against a mixture containing both recombinant Mfn2(710–757)-GST and a synthetic Mfn2 NH2-terminal peptide CNSIVTVKKNKRIIM-OH, a kind gift from Heidi McBride) (26). As shown in Fig. 6B, all antibodies detected a band at the 75–85 kDa region corresponding to the full length MFN2 monomer (30). Surprisingly, these bands came out stronger in the light fraction (No. 5) and were relatively weak in the heavier fraction (Fig. 6B), suggesting a concentration in the IMM-free OMM. Notably, the difference in the ∼75 kDa bands between F5 and F16/17 was only approximately twofold as detected by the anti-COOH terminus antibody (Fig. 6B, bottom) in contrast with 25- to 50-fold differences reported by the antibodies against the NH2 terminus (Fig. 6B, middle) or the NH2- and COOH termini (Fig. 6B, top). This discrepancy was at least in part due to the fact that the COOH terminus-only antibody appeared to be overall weaker-reacting than the other two and more prone to nonspecific binding (see the pale ladder-like banding in the rat heart mitochondrial fraction). On the other hand, a lighter (∼50 kDa) band detected only by the antibodies that could recognize the NH2 terminus (NH2 terminus only or NH2 and COOH termini) but not the one raised against the COOH terminus displayed a distribution pattern practically reciprocal of the full-length MFN2 (Fig. 6B, top 2 blots). The specificity of the ∼75-kDa bands could be confirmed using whole cell lysates of embryonic fibroblasts (MEF) derived from wild-type and MFN2 knockout mice (from which the ∼75 kDa band was missing). However, none the WT or MFN2 knockout MEFs expressed detectable levels of the protein corresponding to the 50-kDa band, probably owing to the tissue and/or species difference (not shown).
Thus, full-length MFN2 appears to localize primarily to IMM-free OMM segments in the cardiac muscle. The finding that a shorter protein detected by antibodies against the N- but not the COOH terminus of MFN2 displayed reciprocal distribution with the full-length MFN2 raises the interesting possibility of an MFN2 variant being confined to the contact points and perhaps working as an SR-contact point tether. This variant could have possibly derived from alternative transcription/splicing or from proteolytic cleavage (a similarly sized MFN2 N-terminal cleavage product of proteinase K treatment has been described before; Ref. 30).
Taken together, the data demonstrate that the OMM-IMM contact points are preferentially aligned with jSR-mitochondrial associations and also serve as anchorage points for a subset of jSR-OMM tethers. These conclusions are supported by 1) mitochondrial fractionation showing that mitochondria-associated SR is purified in the membrane fractions containing both OMM and IMM markers and not in those enriched in OMM only and 2) TEM analysis of rat ventricular cardiac muscle revealing mitochondrial contact points being practically invariably present at and tethered to the OMM interfaces with jSR. Finally, this work uncovers a ∼50-kDa MFN2 immunoreactive protein (possibly a C-terminal truncated variant of MFN2) as a possible SR-contact point tether candidate in the cardiac muscle based on its enrichment in the membrane fractions containing the SR-associated OMM and mitochondrial contact points.
Out of the primary Ca2+ transport mechanisms across the OMM and IMM the Ca2+ uniporter of the IMM has just been very recently identified (6, 14), but its intramembrane distribution pattern is yet to be established, for which our gradients lacking pure IMM were not suitable. On the other hand, OMM-IMM contact points have a unique lipid composition (nonlamellar lipids, phosphatidylethanolamine, and cardiolipin) that favors enrichment of VDAC, the main gate for Ca2+ crossing the OMM (3, 31). This notion was also supported by our Western blot data. The availability of VDAC has been shown as a limiting factor in the rapid local Ca2+ transfer from the ER to the mitochondria (29).
Taken together, alignment of jSR anchorage to the OMM with the mitochondrial contact points provides an arrangement of the three membranes, which could effectively support the local Ca2+ transfer and the consequent Ca2+-regulated mitochondrial responses (see scheme in Fig. 7).
GRANTS
These studies were supported by an American Heart Association Scientist Development Grant (SDG 0435236N) and a Pilot Research Award from Thomas Jefferson University (to G.C.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
ACKNOWLEDGMENTS
We thank Dr. Shey-Shing Sheu for helpful discussions and critical reading of the manuscript.
REFERENCES
- 1. Adams V, Bosch W, Schlegel J, Wallimann T, Brdiczka D. Further characterization of contact sites from mitochondria of different tissues: topology of peripheral kinases. Biochim Biophys Acta 981: 213–225, 1989 [DOI] [PubMed] [Google Scholar]
- 2. Ardail D, Gasnier F, Lerme F, Simonot C, Louisot P, Gateau-Roesch O. Involvement of mitochondrial contact sites in the subcellular compartmentalization of phospholipid biosynthetic enzymes. J Biol Chem 268: 25985–25992, 1993 [PubMed] [Google Scholar]
- 3. Ardail D, Privat JP, Egret-Charlier M, Levrat C, Lerme F, Louisot P. Mitochondrial contact sites. Lipid composition and dynamics. J Biol Chem 265: 18797–18802, 1990 [PubMed] [Google Scholar]
- 4. Balaban RS. Domestication of the cardiac mitochondrion for energy conversion. J Mol Cell Cardiol 46: 832–841, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bathori G, Csordas G, Garcia-Perez C, Davies E, Hajnoczky G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J Biol Chem 281: 17347–17358, 2006 [DOI] [PubMed] [Google Scholar]
- 6. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341–345, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beutner G, Sharma VK, Giovannucci DR, Yule DI, Sheu SS. Identification of a ryanodine receptor in rat heart mitochondria. J Biol Chem 276: 21482–21488, 2001 [DOI] [PubMed] [Google Scholar]
- 8. Boncompagni S, Rossi AE, Micaroni M, Beznoussenko GV, Polishchuk RS, Dirksen RT, Protasi F. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol Biol Cell 20: 1058–1067, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brdiczka D. Contact sites between mitochondrial envelope membranes. Structure and function in energy- and protein-transfer. Biochim Biophys Acta 1071: 291–312, 1991 [DOI] [PubMed] [Google Scholar]
- 10. Brdiczka DG, Zorov DB, Sheu SS. Mitochondrial contact sites: their role in energy metabolism and apoptosis. Biochim Biophys Acta 1762: 148–163, 2006 [DOI] [PubMed] [Google Scholar]
- 11. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160: 189–200, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174: 915–921, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610, 2008 [DOI] [PubMed] [Google Scholar]
- 14. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336–340, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dedkova EN, Blatter LA. Mitochondrial Ca2+ and the heart. Cell Calcium 44: 77–91, 2008 [DOI] [PubMed] [Google Scholar]
- 16. Dorn GW, 2nd, Scorrano L. Two close, too close: sarcoplasmic reticulum-mitochondrial crosstalk and cardiomyocyte fate. Circ Res 107: 689–699, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci 14: 1197–1218, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Franzini-Armstrong C. ER-mitochondria communication. How privileged? Physiology (Bethesda) 22: 261–268, 2007 [DOI] [PubMed] [Google Scholar]
- 19. Garcia-Perez C, Hajnoczky G, Csordas G. Physical coupling supports the local Ca2+ transfer between sarcoplasmic reticulum subdomains and the mitochondria in heart muscle. J Biol Chem 283: 32771–32780, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hayashi T, Martone ME, Yu Z, Thor A, Doi M, Holst MJ, Ellisman MH, Hoshijima M. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J Cell Sci 122: 1005–1013, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325: 477–481, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kornmann B, Walter P. ERMES-mediated ER-mitochondria contacts: molecular hubs for the regulation of mitochondrial biology. J Cell Sci 123: 1389–1393, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maack C, O′Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol 102: 369–392, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Massey JB, Churchich JE. Kynuramine, a fluorescent substrate and probe of plasma amine oxidase. J Biol Chem 252: 8081–8084, 1977 [PubMed] [Google Scholar]
- 25. Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem 76: 723–749, 2007 [DOI] [PubMed] [Google Scholar]
- 26. Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H. Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem 280: 25060–25070, 2005 [DOI] [PubMed] [Google Scholar]
- 27. Pacher P, Csordas G, Hajnoczky G. Mitochondrial Ca2+ signaling and cardiac apoptosis. Biol Signals Recept 10: 200–223, 2001 [DOI] [PubMed] [Google Scholar]
- 28. Pacher P, Hajnoczky G. Propagation of the apoptotic signal by mitochondrial waves. EMBO J 20: 4107–4121, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, Tuft RA, Fogarty KE, Rizzuto R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol 159: 613–624, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rojo M, Legros F, Chateau D, Lombes A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci 115: 1663–1674, 2002 [DOI] [PubMed] [Google Scholar]
- 31. Rostovtseva TK, Kazemi N, Weinrich M, Bezrukov SM. Voltage gating of VDAC is regulated by nonlamellar lipids of mitochondrial membranes. J Biol Chem 281: 37496–37506, 2006 [DOI] [PubMed] [Google Scholar]
- 32. Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175: 901–911, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]