

Abstract
Lasp1 is an actin-binding, signaling pathway-regulated phosphoprotein that is overexpressed in several cancers. siRNA knockdown in cell lines retards cell migration, suggesting the possibility that Lasp1 upregulation influences cancer metastasis. Herein, we utilized a recently developed gene knockout model to assess the role of Lasp1 in modulating nontransformed cell functions. Wound healing and tumor initiation progressed more rapidly in Lasp1−/− mice compared with Lasp1+/+ controls. Embryonic fibroblasts (MEFs) derived from Lasp1−/− mice also migrated more rapidly in vitro. These MEFs characteristically possessed increased focal adhesion numbers and displayed more rapid attachment compared with wild-type MEFs. Differential microarray analyses revealed alterations in message expression for proteins implicated in cell migration, adhesion, and cytoskeletal organization. Notably, the focal adhesion protein, lipoma preferred partner (LPP), a zyxin family member and putative Lasp1 binding protein, was increased about twofold. Because LPP gene disruption reduces cell migration, we hypothesize that LPP plays a role in enhancing the migratory capacity of Lasp1−/− MEFs, perhaps by modifying the subcellular localization of other motility-associated proteins. The striking contrast in the functional effects of loss of Lasp1 in innate cells compared with cell lines reveals distinct differences in mechanisms of motility and attachment in these models.
Keywords: LIM and SH3 domain protein, mouse embryonic fibroblasts, tumor formation, zyxin, lipoma preferred partner
lasp1 (lim and sh3 domain protein 1) is a widely but differentially expressed actin-binding protein that is localized within multiple sites of dynamic actin assembly (7, 30, 50). Lasp1 was initially identified as a 40-kDa phosphoprotein, pp40, in gastric parietal cells (5). pp40 was later found to be Lasp1 (11), the gene for which was initially identified as an overexpressed product in some breast cancers (53, 54).
Several lines of evidence support a role for Lasp1 in regulating cellular functions associated with actin-cytoskeleton-associated membrane rearrangements. In parietal cells within the gastric mucosa, histamine-stimulated HCl secretion results in the translocation of the H+,K+-ATPase (proton pump) from a pool of cytoplasmic tubulovesicles to the apically directed, F-actin-rich intracellular canalicular membrane (19, 39). This process is mediated by the cAMP signaling pathway, and cAMP-dependent phosphorylation of Lasp1 on serine residues is correlated with the activation of HCl secretion and the partial translocation of Lasp1 to the intracellular canalicular membrane region (10, 11). Lasp1 also associates with F-actin in a phosphorylation-dependent manner (4, 7, 50), binds to several endocytosis-associated proteins including Huntington interacting protein 1 related (Hip1r), clathrin, and dynamin2, and colocalizes with Hip1r and dynamin2 within parietal cell intracellular canalicular region (29, 38, 40). Because disruption of the Lasp1 gene in mice leads to a more robust acid secretory response to histamine, and histamine H2 receptor antagonist-dependent blockade is delayed in gastric glands isolated from these mice, it has been postulated that Lasp1 serves as a negative regulator in this process (6).
In cultured cells, Lasp1 is present within focal adhesions (7, 10, 30, 35, 50), and there is evidence that this protein can interact with a number of motility-associated, focal adhesion proteins including zyxin, the zyxin family member, LPP, the cAMP-dependent phosphoprotein, vasodilator-stimulated phosphoprotein (VASP), and Krp1 (sarcosin), which is expressed mainly in skeletal and cardiac muscle but has also been found to be upregulated in transformed rat fibroblasts (30, 32, 51). Because Lasp1 is overexpressed in a range of cancers, including breast, prostate, liver, and ovarian, for example (22–24, 34, 53, 54, 57), it has been proposed to play a role in initiating metastasis (22, 54). Several recent studies have sought to clarify the role of Lasp1 in the regulation of cell migration using transient overexpression/knockdown approaches with conflicting results. Thus, siRNA-dependent reduction in Lasp1 expression, decreased cell migration in the BT-20, SKOV-3, COS-7, and NIH3T3 cell lines and several hepatoma cancer cell lines (HepG2, Hep3B, and Huh-3) (23, 24, 35, 57), but enhanced migration in the SKHep1C3 carcinoma cell line (47). Lasp1 overexpression also reduced migration in several breast cancer cell lines (23, 35, 55) and in two transformed cell lines, COS-7 and HEK293 (35), but enhanced the migratory activity of nontransformed PTK2 cells (23) and had no significant effect on primary human umbilical vein endothelial cell migration (23).
In this study, we used a recently generated Lasp1-null mouse model (6) to analyze the effects of chronic, physiological loss of Lasp1 on cell migration. Our findings provide the first evidence that this protein can serve as a negative, rather than a positive modulator not only of cell migration but also tumor progression.
MATERIAL AND METHODS
Animals.
Lasp1-null mice and wild-type littermates were produced and backcrossed to wild-type C57Bl/6NCr mice for 10 generations to ensure genetic homogeneity (41) as previously described (6). All procedures and treatment of mice were reviewed and approved by the Medical College of Georgia (MCG) Animal Care and Use Committee.
Mouse embryonic fibroblast isolation.
Embryonic fibroblasts were derived from day 12.5 embryos of Lasp1+/+ (n = 3, 8 embryos/mouse) and Lasp1−/− (n = 4, 9 embryos/mouse) mice as previously described (28, 59). Primary mouse embryonic fibroblast (MEFs) were plated and frozen in liquid nitrogen as passage 0. Cells were grown at 37°C in a 5% CO2 atmosphere in high glucose DMEM (Cellgro) supplemented with 10% FBS (HyClone), 2 mM l-glutamine (Cellgro), and 1% penicillin/streptomycin (Cellgro). MEFs of passages 2–6 were used in all experiments. Unless otherwise stated, cells were seeded directly onto glass coverslips or culture dishes.
Molecular cloning, plasmid constructs, and transfection.
Total RNA from wild-type and Lasp1-null MEFs was isolated with a Perfect RNA Tissue kit (5 Prime, Gaithersburg, MD) following manufacturer's instructions. Mouse Lasp1 cDNA (GenBank accession no. NM_010688) was cloned from MEF mRNA using a RT-PCR-based strategy as previously described (9). Total RNA from wild-type MEFs was converted to single-stranded DNA (Superscript First-Strand cDNA synthesis kit; Invitrogen, Carlsbad, CA) and used as a PCR template to generate the complete open reading frame for murine Lasp1 using the following primers (5′→3′): sense with HindIII cut site (5′-AAG CTT CAT GAA CCC TAA CTG TGC CC-3′); antisense with BamHI cut site (5′-GGA TCC CGA TGG CCT CCA CGG TAG TT-3′) that were synthesized by Integrated DNA Technologies (Coralville, IA). RT-PCR was performed using AccuPrime Pfx taq (Invitrogen) as follows: 94°C, 5 min followed by 30 cycles (94°C, 30 s, 55°C, 1 min, 72°C, 1 min) with a final 72°C, 7 min extension. The resulting DNA product was gel purified (Qiagen Gel Extraction Kit, Valencia, CA), ligated into pGEM-T Easy vector (Promega, System II), and sequenced using T7 and SP6 primers (MCLAB, San Francisco, CA) to ensure there were no PCR-introduced errors. The mouse Lasp1 insert was then subcloned into the pAcGFP1-C2 expression vector (Clontech), purified with a Qiagen Endofree Maxiprep kit as previously described (42), and sequenced (MCLAB) using a pAcGFP1-C2 vector-specific primer (5′-AAC CTC CCA CAC CTC CCC-3′). Lasp1−/− MEFs were transiently transfected with pAcGFP1-C2/Lasp1 with Effectene (Qiagen) using a manufacturer-supplied enhancer at a ratio of 1:6.4 (DNA to enhancer) and 1:20 (DNA to Effectene) (42).
Real-time RT-PCR.
Total RNA (5 μg) from Lasp1+/+ and Lasp1−/− MEFs and RNA (5 μg) isolated from mouse brain (C57BL/6 strain) were reverse-transcribed using a Superscript First-Strand cDNA synthesis kit (Invitrogen) as described in the previous sections. The resulting cDNA was then subjected to real-time PCR (ABI 7500 Fast Real Time PCR System; Applied Biosystems, Foster City, CA) to quantify gene expression using TaqMan Fast Universal PCR Master mix, No AmpErase UNG. Predesigned primers and probes [β-actin (Mm00607939_s1), Lasp2 (Mm00503886_m1)] were obtained from Applied Biosystems. β-Actin was used as an internal control. Thermal cycling conditions were as follows: 95°C, 20 s; 50 × 3 s cycles, 95°C; 60°C, 30 s. Each sample was analyzed in triplicate. Fold changes were determined using the 2−ΔΔCt method (36) with Sequence Detection Software 1.3 (Applied Biosystems).
Cell migration analyses.
In vivo wound healing experiments were performed on Lasp1−/− and Lasp1+/+ mice beginning at 58 days of age. Prior to being wounded, mice were deeply anesthetized (Nembutal, 50 mg/kg ip) and shaved dorsally. Full-thickness wounds were generated on backs with a biopsy punch (4-mm diameter, Miltex, York, PA) (48). Wound healing progress was recorded daily for 7 days with a digital camera (Canon IXUS60). Images were analyzed with NIH Image J software (version 1.38x). The unhealed area was defined as the “wound” (mm2), followed by normalization to the percentage of unhealed area to the original wound area [% of wound = unhealed area (mm2) on day n × 100/original wound area (mm2) on day 0 (n = days postwounding)].
In vitro monolayer wound healing assays were performed as previously described (59) using confluent Lasp1+/+ and Lasp1−/− MEFs. Migration progress was recorded at 0, 1, 2, 4, 6, 8, 10, and 12 h using an inverted microscope (Zeiss Axiovert H25) equipped with an Axio HRm camera, a ×10 phase-contrast objective, and Axiovision AC (release 4.5) acquisition software. Images were analyzed with NIH Image software. The average of 10 measurements across the “wounded” area was defined as the width of the “wound.” The results were converted to migration rates in which cell migration was expressed as μm migrated/h (migration rate = distance migrated/temporal duration).
In vitro Transwell cell migration assays were performed using modified Boyden chambers [Innocyte Cell Migration Assay (Calbiochem)] according to manufacturer's instructions. In brief, cells were serum starved overnight then trypsinized, washed, and resuspended in serum-free medium. We added 2.5 × 105 cells to culture inserts (8-μm pore size) and allowed them to migrate through the membrane for 6 h at 37°C in a 5% CO2 atmosphere. Migratory cells that attached to the lower side of the membrane were resuspended in 200 μl of labeling/cell detachment buffer containing Calcein-AM fluorescent dye. Fluorescence signals were quantified with a Synergy fluorescent plate reader [485 nm (excitation) 520 nm (emission) wavelengths]. Three independent experiments with three replicates were performed.
Tumor induction.
A two-stage “high-dose” chemical regimen was used to induce papilloma formation (18) in wild-type and Lasp1−/− mice (n = 7–9, 7–9 mice/group, each of which contained approximately equal numbers of males and females). Areas (∼15 cm2) on the backs of 6-wk-old mice were shaved. One week later, a single application of tumor initiator, 7,12-dimethylbenz[a]anthracene [DMBA (Sigma-Aldrich), 400 nmol dissolved in 100 μl acetone] was applied to the area followed by biweekly applications of the tumor promoter, 12-O-tetradecanoylphorbol-13-acetate [TPA (Sigma), 20 nmol dissolved in 50 μl 100% ethanol]. Animals were assessed on a weekly basis for tumor appearance and progression. Tumors with a diameter of 1 mm or greater that persisted for longer than 2 wk were counted. For histological analyses, tumors were excised and fixed overnight in 10% buffered formalin. After fixation, tissue was paraffin embedded, sectioned, and stained with hemotoxylin and eosin in the Electron Microscopy & Histology Core Laboratory at MCG.
Cell proliferation and adhesion analyses.
Cell proliferation rates were assessed using Vybrant MTT Cell Proliferation Assay Kits (Invitrogen) at 24 and 48 h after cell seeding according to manufacturer's instructions. Cell adhesion assays were performed as previously described (59), using cells seeded in 96-well plates (Corning) coated with collagen type I (Sigma) at different concentrations (1, 2, 5, and 10 μg/ml). Cells were allowed to attach for 25 min at 37°C before assay. Nonadherent cells were removed by gentle washing with PBS. Attached cells were fixed in 10% formalin for 30 min and then stained with 1% toluidine blue for 60 min. The blue dye was eluted in 2% SDS solution, and the absorbance was measured at OD620. Nonspecific cell adhesion was measured on BSA-coated wells and was subtracted from each reaction value.
Immunofluorescent localization.
MEFs were fixed with 4% paraformaldehyde, permeabilized with 0.25% Triton X-100, and stained with various antibodies as previously described (9) at the following dilutions: monoclonal mouse anti-Lasp1 (clone 8C6), 1:50; monoclonal mouse anti-vinculin (Sigma, clone hVIN-1), 1:100; monoclonal rabbit anti-VASP (Cell Signaling, clone 9A2), 1:100; monoclonal mouse IgA anti-Mena (BD Transduction Laboratories, clone 21), 1:10; polyclonal rabbit anti-zyxin (Protein Tech Group), 1:10; polyclonal goat anti-LPP, 1:100 (Santa Cruz), followed by incubation with Cy3/Cy5-tagged secondary antibodies (1:100 dilution, Jackson ImmunoResearch Laboratories), Sytox Green (0.5 μM, Molecular Probes) for nuclear staining, and/or Alexa Fluor 647/568 phalloidin/Oregon Green 488 (1:40 dilution, Invitrogen/Molecular Probes) for F-actin detection. Stained cells were mounted on slides with Prolong Gold anti-fade reagent (Molecular Probes). Images were acquired with a Zeiss LSM 510 confocal microscope equipped with a krypton/argon laser and Meta software (version 3.2). For dual/triple labeling experiments, emission filter band passes, gains, and photomultiplier tube settings were adjusted to avoid signal cross-over (9, 10, 42). Images were processed using Adobe Photoshop (version 7.0). Ten nonoverlapping cells from Lasp1+/+ and Lasp1−/− MEFs, respectively, were selected randomly for quantitation of focal adhesion numbers.
Focal adhesion turnover analyses.
Focal adhesion disassembly assays were performed as previously described (17). In brief, cells were grown on glass coverslips, serum-starved overnight, and then treated with 10 μM nocodazole (Sigma) for 4 h. MEFs were fixed, permeabilized, and stained with monoclonal mouse anti-vinculin for focal adhesion visualization and Oregon Green 488 for F-actin detection, as described in the preceding section, before and after nocodazole addition and 30 min–3 h after removal.
Western blotting.
Lasp1+/+ and Lasp1−/− MEFs were rinsed with cold PBS and protein extracted by boiling (5 min) in SDS-βME [0.3% (wt/vol) SDS, 1% (vol/vol) 2-mercaptoethanol]. Proteins were quantified using a Protein Quant-iT Assay kit (Invitrogen). Western blots were analyzed with enhanced chemiluminescence (ECL) detection (Amersham Biosciences) and signals quantified using a Syngene GeneGnome System and the Gene Tools software (Synoptics) as previously described (7–9). Antibody dilutions were as follows: Monoclonal mouse anti-Lasp1, 1:2,000 (clone 8C6) (6, 10, 29); monoclonal mouse anti-β-actin, 1:25,000 (Sigma, clone AC-74); monoclonal rabbit anti-VASP (Cell Signaling, clone 9A2), 1:1,000; monoclonal mouse IgA anti-Mena (BD Transduction Laboratories, clone 21), 1:250; polyclonal rabbit anti-zyxin, 1:1,000 (Protein Tech Group); polyclonal goat anti-LPP, 1:1,000 (Santa Cruz), and polyclonal rabbit anti-Lasp2, 1:50–1:200 [a generous gift from Asako Terasaki, Chiba, Japan (52)]. Incubations with primary antibodies were followed by incubation with appropriate secondary horseradish peroxidase-tagged IgG: sheep anti-mouse IgG, donkey anti-rabbit IgG, mouse anti-goat/sheep IgG (clone GT-34) (1:5,000 dilution, Amersham Biosciences), or goat anti-mouse IgG (H+L) (1:5,000 dilution, Jackson ImmunoResearch Laboratories).
Microarray analyses.
Total RNA (200 ng) was hybridized to mouse WG-6v2.0 expression BeadChips. Arrays were scanned using the Illumina BeadArray Reader (Illumina) in the MCG Proteomics & Mass Spectrometry Core Facility. Data were processed by Mean/median scaling normalization and Welch t-test analysis using Array Track software (version 3.3, National Center for Toxicological Research, Jefferson, AR). Gene Ontology For Functional Analysis (GOFFA) was used as a comprehensive tool to functionally profile genes that were significantly affected by changes in Lasp1 gene expression. A differential expression cut-off of twofold and a P value of <0.05 were the basic criteria for further analysis. The complete data set has been deposited in the NCBI Gene Expression Omnibus (GEO) (16) and is accessible through GEO Series accession number GSE14800 at the following web address: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE14800.
Statistical analyses.
Data were analyzed and curves fitted with GraphPad Prism (version 5.0). Student's t-tests were used for paired comparisons. Multiple comparisons were analyzed by ANOVA with post hoc tests (Bonferroni and Dunnett's multiple comparison). Log-rank tests of the survival curves were used for tumor incidence analyses. Values in figures are expressed as means ± SE with n = the number of independent experiments.
RESULTS
Effects of Lasp1 gene disruption on wound healing and tumor promotion.
Findings in several cell lines that decreased expression of Lasp1 suppresses cell migration (23, 24, 35, 57), and, in some cases, proliferation (23, 24) predicted that chronic loss of Lasp1 would have a more profound effect on these processes. Because cell migration and proliferation are prominent components in wound healing (33) as well as in cancer progression (26), we used two different, well-established in vivo approaches, skin wounding and chemically induced tumor formation, to determine if chronic loss of Lasp1 influenced the outcome of these perturbations. For wounding experiments, full-thickness wounds were produced in anesthetized mice with a biopsy punch, and healing progress was analyzed as described in material and methods. A two-stage skin carcinogenesis model with a high-dose regimen was used to induce tumor formation because the Lasp1-null mouse strain was backcrossed to the C57BL/6 strain, which is highly resistant to tumor promotion protocols (14, 15, 27). Unexpectedly, we found that wound healing progressed more rapidly in Lasp1−/− mice compared with their wild-type littermates (Fig. 1, A and B). As shown in Fig. 2, tumor formation (Fig. 2A) and incidence (Fig. 2B) were also significantly greater in Lasp1-null mice compared with wild-type littermates. Representative examples of tumor formation and histological analyses are shown in Fig. 2, C and D.
Fig. 1.
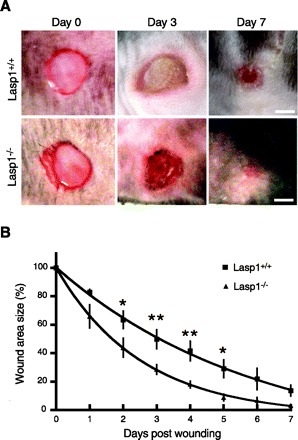
Effect of Lasp1 gene disruption on wound healing. Wounds were generated on the backs of Lasp1+/+ and Lasp1−/− mice with a biopsy punch. Healing progress was recorded with a digital camera on a daily basis for 1 wk then quantitated as described in material and methods. A: representative images from selected time points (0, 3, and 7 days postwounding). Bars, 20 mm. B: quantitation of data from wound healing experiments. The rate of wound healing was significantly faster in Lasp1−/− mice compared with Lasp1+/+ mice. *P < 0.05, **P < 0.01; n = 4–6.
Fig. 2.
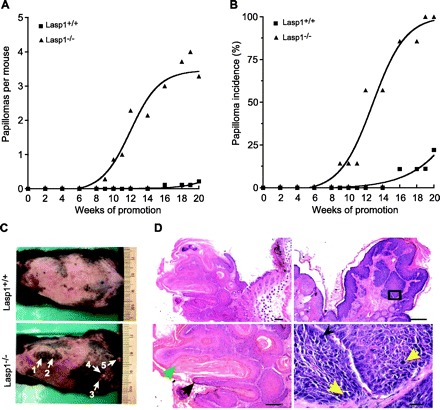
Effects of Lasp1 gene disruption on skin tumor initiation and promotion. Tumors were initiated with DMBA then promoted with TPA for 20 wk as described in material and methods. A: tumor multiplicity expressed as the average number of tumors formed during the promotion period. Tumor multiplicities were significantly increased in Lasp1−/− mice compared with Lasp1+/+ mice (P < 0.05, n = 7–9). Error bars were omitted for clarity. B: skin tumor incidence expressed as the percentage of mice developing tumors within each group during the promotion period. The rate of tumor formation was significantly increased in Lasp1−/− mice compared with Lasp1+/+ mice (P < 0.05; n = 7–9). C: representative dorsal photographs of Lasp1+/+ and Lasp1−/− littermates at the end of promotion. Arrows indicate location of tumors. D: histological analyses of the 2 largest tumors (#4 and 5) from the Lasp1-null mouse in C. Left: sections of tumor #4 in C, which depict papillomatosis (black arrow), hypergranulosis (black arrow), and hyperkeratosis (green arrow). Magnifications: top, ×2.5; bottom, ×5. Right: sections of tumor #5 in C, which also show papillomatosis and hyperkeratosis, but with focal areas suggestive of carcinoma in situ arising in a verruca. Bottom image is a higher magnification view of the region within the box above, showing marked atypical epithelium. Yellow arrows indicate mitoses. Black arrowhead: large atypical nucleus. Magnifications: top, ×5; bottom, ×40. Bars, 200 μm (×2.5 and ×5); 20 μm (×40).
Effects of chronic loss of Lasp1 on MEF migration, proliferation, and attachment.
The observations that both wound healing and tumor progression occurred more rapidly in Lasp1-null mice suggested the possibility that Lasp1 plays a negative, rather than a positive, role in regulating cell motility and proliferation. To test this hypothesis, we analyzed cell migration in vitro using MEFs isolated from Lasp1-null and wild-type mice. As shown in Fig. 3, two different approaches, including a cell wounding assay (Fig. 3, A and B) and a modified Boyden chamber cell migration assay (Fig. 3C), provided independent confirmation that Lasp1-null MEFs migrate more rapidly than do Lasp1+/+ MEFs. Furthermore, although chronic loss of Lasp1 did not alter the rate of proliferation as assessed under standard culture conditions (Fig. 4A), the rate of MEF attachment to collagen type I was significantly faster in Lasp1-null MEFs (Fig. 4B). The lack of effect of chronic loss of Lasp1 on normal cell proliferation suggests that some type of perturbation, such as chemically-induced tumor formation (Fig. 2), is necessary to unveil the enhancement in proliferation that is conveyed by disruption of the Lasp1 gene.
Fig. 3.
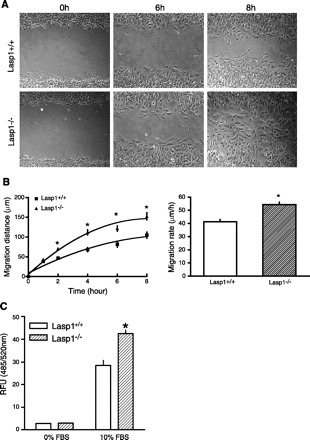
Migration rates of embryonic fibroblasts isolated from wild-type and Lasp1-null littermates assessed using monolayer wound healing and Transwell cell migration assays. A: monolayer wound healing assay: mouse embryonic fibroblasts (MEFs) were plated in 35 mm culture dishes and grown to confluence. A “wound” was generated in the center of each dish with a Gilson P-200 pipette tip, and migratory progress was recorded by phase-contrast microscopy. Representative images from selected time points postwounding are shown in the figure. B: quantitation of data from wound healing assays. Left: time course of MEF migration. Right: rate of migration (changes in the width of the wound over time). There was a significant difference in the distances covered by Lasp1−/− MEFs compared with Lasp1+/+ MEFs. *P < 0.01; n = 4. C: modified Boyden chamber assays: Innocyte Cell Migration chambers were used to measure cell migration over a 6 h period as described in material and methods. Lower chambers containing serum-free media were used as negative controls. The number of MEFs that migrated through the membranes of the chamber inserts during this time period was significantly greater for Lasp1−/− MEFs compared with Lasp1+/+ MEFs. *P < 0.05; n = 3.
Fig. 4.
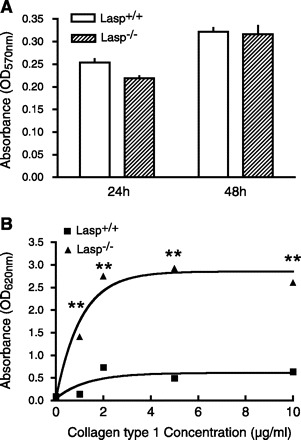
Cell proliferation and adhesion in Lasp1+/+ and Lasp1−/− MEFs. A: cell proliferation was analyzed using a Vybrant MTT Cell Proliferation Assay (Invitrogen). There was no significant difference in proliferation between Lasp1+/+ and Lasp1−/− MEFs at either 24 or 48 h after cell plating. P > 0.05; n = 4. B: the attachment of both Lasp1+/+ and wild-type MEFs was positively correlated with increases in collagen type I concentrations; however, the attachment of Lasp1−/− MEFs was significantly greater than Lasp1+/+ MEFs at all collagen concentrations tested. **P < 0.01; n = 4.
Focal adhesion numbers and dynamics in Lasp1 null MEFs.
Focal adhesions play a central role in mediating attachment of cells to the substratum and coordinating interrelationships between attachment and cytoskeletal assembly and disassembly during cell migration (49). Lasp1 has been localized within focal adhesions in several cell lines and in gastric mucosal fibroblasts (7, 10, 30, 35, 50) along with the focal adhesion protein, zyxin (32). Lasp1 also binds to zyxin in vitro (30, 32), and transient knockdown of Lasp1 has been linked to the loss of zyxin from focal adhesions in breast and ovarian cancer cell lines (23, 24). To determine if zyxin localization is similarly altered when Lasp1 is chronically absent, we used confocal microscopy to immunolocalize zyxin in wild-type and Lasp1-null MEFs. As shown in Fig. 5A, both Lasp1 and zyxin were found to be present within focal adhesions in wild-type MEFs, as expected. However, there was no evidence that zyxin localization within focal adhesions was altered in Lasp1-null MEFs (Fig. 5A). In addition, the morphologies of zyxin-containing focal adhesions and actin stress fibers were similar in wild-type and Lasp1-null MEFs (Fig. 5A). These latter findings are in agreement with a previous report in which transient knockdown of Lasp1 was not found to alter the subcellular localizations of F-actin or another focal adhesion protein, vinculin (23). Interestingly, we further observed that the number of zyxin-containing focal adhesions was significantly increased in Lasp1−/− MEFs (Fig. 5B). Similar results were obtained when vinculin-containing focal adhesions were quantified (not shown). Reconstitution of Lasp1 in Lasp1−/− MEFs reduced focal adhesion numbers (Fig. 5B), providing additional support for a reciprocal relationship between Lasp1 expression and the number of focal adhesions.
Fig. 5.
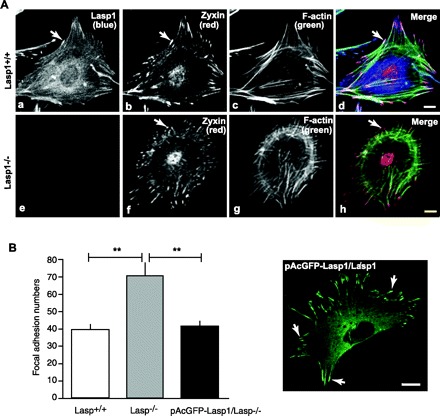
Analysis of focal adhesion numbers and morphologies in Lasp1+/+ and Lasp1−/− MEFs. A: Lasp1+/+ and Lasp1−/− MEFs were fixed and stained with a zyxin polyclonal antibody and Cy3-tagged donkey anti-rabbit secondary antibody to localize zyxin, a Lasp1 monoclonal antibody, and Cy5-tagged donkey anti-mouse antibody to localize Lasp1 and with Oregon green 488 phalloidin to localize F-actin as described in material and methods. In wild-type MEFs, Lasp1 was localized within focal adhesions and in the perinuclear region (a). Zyxin was similarly localized within focal adhesions but was also prominently localized within nuclei (b). In Lasp1−/− MEFs (e), the subcellular localization of zyxin (f) was not affected. Focal adhesion morphologies and F-actin localization within stress fibers were also similar in Lasp1−/− and Lasp1+/+ MEFs (c, g). Composite images (d, h). B: focal adhesion numbers were quantified as described in material and methods. Where indicated, Lasp1−/− MEFs were transfected with Effectene using a pAcGFP1-C2-Lasp1 construct containing the complete open reading frame of murine Lasp1. Left: the number of zyxin-containing focal adhesions was increased in Lasp1−/− MEFs compared with wild-type MEFs, and transient expression of pAcGFP1-C2-Lasp1 in Lasp1−/− MEFs led to a decrease in focal adhesion numbers. **P < 0.01; n = 4. Right: representative image of transfected Lasp1−/− MEFs demonstrating appropriate localization of pAcGFP1-C2-Lasp1 within focal adhesions and the perinuclear region. Arrows, focal adhesions. Bars 20 μm.
Although focal adhesions are required to support cellular attachment and migration, an increased level of focal adhesion formation could lead to decreased cell migration, depending upon the strength of attachment as well as the interplay between focal adhesion dynamics and actomyosin-dependent contraction (25). To determine if the increase in focal adhesion numbers in Lasp1-null MEFs is linked to an increase in the turnover of these structures, which could serve to enhance migratory capacity, we used a recently described protocol in which effects of nocodazole-induced formation of focal adhesions and stress fibers and loss of these structures after nocodazole removal are followed over time under serum-starved conditions (17). Focal adhesions were localized using an anti-vinculin antibody, and actin cytoskeletal structure was assessed using Oregon green 488 phalloidin. As expected (3, 17), serum starvation disrupted focal adhesions and stress fibers in Lasp1-null and wild-type MEFs (Fig. 6, A, A′, B, and B′), and nocodazole induced focal adhesion and stress fiber formation in both models (Fig. 6, C, C′, D, and D′). Thirty minutes after nocodazole removal, focal adhesions and stress fibers remained prominent in wild-type MEFs (Fig. 6, E and E′) but were almost completely disrupted in Lasp1-null MEFs (Fig. 6, F and F′). One hour later, stress fibers and focal adhesions were obviously disrupted in both models (Fig. 6, G, G′, H, and H′); however, within 2 h, focal adhesions and stress fibers were beginning to reform in Lasp1−/− MEFs (Fig. 6, J and J′) but not Lasp1+/+ MEFs (Fig. 6, I and I′). After 3 h, focal adhesion formation appeared to have stabilized in both cellular models (Fig. 6, K, K′, L, and L′). Thus, this experimental approach provided strong evidence that focal adhesion and actin stress fiber turnover occurs more rapidly in MEFs lacking Lasp1.
Fig. 6.
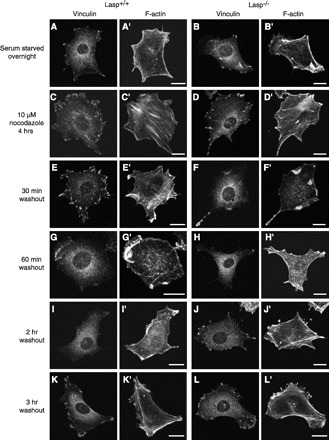
Focal adhesion turnover. Serum-starved cells were treated with 10 μM nocodazole for 4 h and then fixed and stained with anti-vinculin antibody, Cy3-tagged donkey anti-mouse secondary antibody along with Oregon green 488 phalloidin to localize F-actin at different time points after nocodazole removal as described in material and methods. Bars, 20 μm. When MEFs were serum starved overnight, few focal adhesions (A, B) were observed and stress fibers were disrupted in both Lasp1+/+ (A′) and Lasp1−/− (B′) MEFs. Four hours after nocodazole treatment, focal adhesions (C, D) and stress fibers (C′, D′) had reassembled in both models. Thirty minutes after removal of nocodazole, Lasp1+/+ MEFs still contained prominent focal adhesions and stress fibers (E, E′), whereas focal adhesions were disassembled and stress fibers were disrupted in Lasp1−/− MEFs (F, F′). Sixty minutes after nocodazole removal, focal adhesions were disassembled, and stress fibers were disrupted in both Lasp1+/+ (G, G′) and Lasp1−/− (H, H′) MEFs. Two hours after washout, focal adhesions and stress fibers were beginning to reform in Lasp1−/− MEFs but not Lasp1+/+ MEFs (I, J, I′, J′). Within 3 h after washout, focal adhesion (K, L) and stress fibers (K′, L′) had reformed in both models. Images representative of 10 cells per time point analyzed for each model.
Effects of chronic loss of Lasp1 on the expression and localization of putative Lasp1 binding partners.
The lack of effect of loss of Lasp1 on zyxin localization led us to explore possible effects on other putative Lasp1 binding partners. Lipoma preferred partner (LPP) is a zyxin family member that also binds to Lasp1 in vitro (30). LPP is also present within focal adhesions (20, 21, 44, 46) and can shuttle between cellular adhesion sites and the nucleus, at least in response to specific drug challenges (21, 44). Confocal microscopic analyses immunolocalized LPP, along with zyxin and Lasp1, within focal adhesions in wild-type MEFs (Fig. 7, A, B, E, and F). However, the subcellular patterns of distribution of zyxin and LPP were not identical in that zyxin, but not LPP, was localized within the nuclei of both Lasp1-null and wild-type MEFs (Fig. 7, B, E, F, I, and J). In contrast, both LPP and Lasp1 were localized within the perinuclear region in wild-type MEFs (Fig. 7, A, B, and E). Although the subcellular localization of LPP was not affected by loss of Lasp1 (Fig. 7I), Western blot analyses detected a significant increase in LPP expression in Lasp1−/− MEFs, whereas the expression levels of zyxin and β-actin were unchanged (Fig. 8).
Fig. 7.
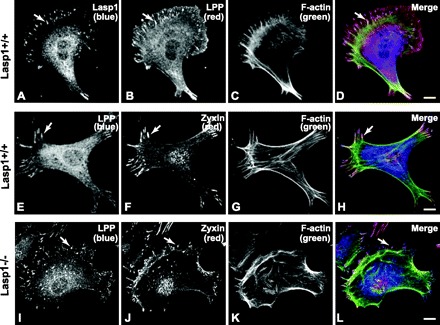
Subcellular immunolocalization of candidate Lasp1 binding proteins in MEFs. Cells were cultured in serum-containing medium for 1 day after seeding on coverslips and then fixed and stained with Lasp1 and zyxin antibodies and Oregon green 488 phalloidin as described in Fig. 5. Lipoma preferred partner (LPP) was immunolocalized with a polyclonal antibody and a Cy3-tagged (B) or Cy5-tagged (E, I) donkey anti-goat secondary antibody. In agreement with data shown in Fig. 5, Lasp1 was localized within focal adhesions and in the perinuclear region in wild-type MEFs (A). In wild-type MEFs, LPP was localized within focal adhesions along with Lasp1 and zyxin (A, B, E, F), but had a subcellular distribution that more closely resembled that of Lasp1 than zyxin (A, B, E, F). In contrast to zyxin, both Lasp1 and LPP were localized above and below nuclei, but not within nuclei based on the analysis of multiple, contingent image sections (not shown). F-actin was similarly localized within stress fibers of Lasp1−/− and Lasp1+/+ MEFs (C, G, K). Chronic loss of Lasp1 did not alter the subcellular localizations of LPP (I) or zyxin (J). Composite images (D, H, L). Arrows, focal adhesions. Bars, 20 μm.
Fig. 8.
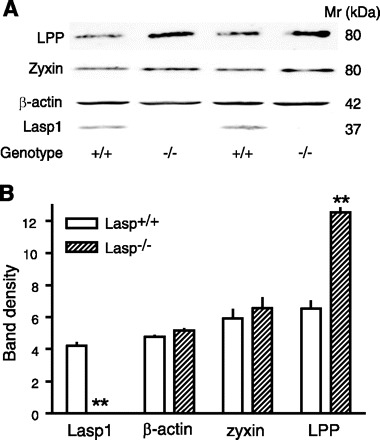
Effects of chronic loss of Lasp1 on zyxin and LPP expression as assessed by Western blotting. Lasp1+/+ and Lasp1−/− MEFs were seeded in culture dishes and grown to confluence. Cellular protein was extracted and subjected to Western blotting as described in material and methods (25 μg protein was loaded/lane). A: representative images showing strong Lasp1 immunoreactivity in Lasp1+/+ extracts but no detectable signal in Lasp1−/− MEFs. LPP and zyxin, which migrate on SDS PAGE gels with an apparent molecular ratio (Mr) of ∼80 kDa, were detected in both Lasp1+/+ and Lasp1−/− MEFs. β-Actin was used as a loading control. B: quantitation of data from Western blots showing a significant increase in LPP, but not zyxin or β-actin expression in Lasp1−/− MEFs. **P < 0.01; n = 3.
Zyxin also binds to the focal adhesion, motility-associated protein, VASP, and zyxin gene disruption alters the subcellular localization of VASP as well as mammalian enabled (Ena, also known as Mena/ENAH) (28), a closely related VASP family member (31). Because there is some evidence that Lasp1 can interact with VASP (30), we analyzed effects of Lasp1 gene disruption on the subcellular localization of Ena/VASP proteins. As assessed by immunofluorescent localization analyses, the loss of Lasp1 had no effect on the subcellular localization of either VASP or Mena (not shown).
Microarray and quantitative RT-PCR analyses.
To determine if the chronic absence of Lasp1 alters the expression of other genes associated with cell motility/attachment and/or other cellular functions, we performed global gene expression analyses using RNA isolated from Lasp1+/+ and Lasp1−/− MEFs (n = 3, 3 repetitions/group). We found 154 genes that displayed more than a twofold difference in expression with a P < 0.05 (Supplemental Table S1).1 Of these, 73 were upregulated and 81 were downregulated. Approximately one-third of the affected genes have recognized functions related to cell motility, adhesion, cytoskeleton organization, cell proliferation, and transcriptional regulation (Table 1). In agreement with protein expression analyses (Fig. 8), LPP gene expression was significantly increased in Lasp1−/− MEFs, whereas expression levels of zyxin and the other known zyxin family members (Trip6, LimD1, and ajuba) and VASP were unchanged. Similar levels of transcript expression were also detected for the VASP family member, Mena, with two Illumina probe sets. A third set, which recognizes a truncated form of Mena (accession no. AK020248), predicted an approximately twofold upregulation (Supplemental Table S1). However, we were unable to detect any change in protein expression levels by Western blotting of Mena using an antibody predicted to cross react with the known Mena isoforms (not shown). The findings that chronic loss of Lasp1 does not influence the expression of Mena/VASP are similar to those previously reported by Grunewald and colleagues (23) in which siRNA knockdown of Lasp1 in the BT-20 cell line had no effect on the expression of VASP or ENAH as assessed by Western blotting.
Table 1.
Differentially expressed genes in Lasp1−/− mouse embryonic fibroblasts categorized by recognized functions
| Gene Symbol | Description | Accession Number | Fold Change(s) | P Value(s) | ||||
|---|---|---|---|---|---|---|---|---|
| Cell motility/migration | ||||||||
| Acta1* | actin, α1, skeletal muscle | NM_009606 | 2.870 | 0.015 | ||||
| Lpp† | LIM domain containing preferred translocation partner in lipoma | AK045341 | 2.24; 2.262; 2.298 | 0.039; 0.038; 0.002 | ||||
| Slit3‡ | slit homolog 3 (Drosophila) | XM_203363 | −2.985 | 0.002 | ||||
| Tnn‡ | tenascin N (tenascin-W) | NM_177839 | −2.316 | 0.000 | ||||
| Flt1‡ | FMS-like tyrosine kinase 1 | NM_010228 | −2.128 | 0.035 | ||||
| Cell adhesion/extracellular matrix organization and biogenesis | ||||||||
| Col2a1† | procollagen, type II, α1 | NM_031163 | 6.674; 3.020; 5.171 | 0.000; 0.000; 0.002 | ||||
| Nrp | neuropilin 1 | AK030358 | 2.454 | 0.039 | ||||
| Nid1 | nidogen 1 | AK049585 | 2.435 | 0.026 | ||||
| Klra4 | killer cell lectin-like receptor, subfamily A, member 4 | NM_010649 | 2.228 | 0.002 | ||||
| Col10a1 | procollagen, type X, α1 | NM_009925 | 2.181 | 0.005 | ||||
| Matn4 | matrilin 4 | NM_013592 | −3.356 | 0.039 | ||||
| Mfap4 | microfibrillar-associated protein 4 | NM_029568 | −2.725 | 0.040 | ||||
| Cdh3 | cadherin 3 | NM_007665 | −2.231 | 0.007 | ||||
| Dpt | dermatopontin | NM_019759 | −2.179 | 0.002 | ||||
| Comp | cartilage oligomeric matrix protein | NM_016685 | −2.075 | 0.044 | ||||
| Cytoskeleton organization and biogenesis | ||||||||
| Msn‡ | moesin | AK031171 | 2.026 | 0.022 | ||||
| Krt1-14 | keratin 14 | NM_016958 | −6.024 | 0.003 | ||||
| Fgf21 | fibroblast growth factor 21 | NM_020013 | −2.519 | 0.008 | ||||
| Regulation of cell proliferation through cell cycle | ||||||||
| Cxcl1 | chemokine (C-X-C motif) ligand 1 | NM_008176 | 3.199 | 0.001 | ||||
| Fgf7 | fibroblast growth factor 7 | NM_008008 | 2.430 | 0.001 | ||||
| Bcl11b | B-cell leukemia/lymphoma 11B | NM_021399 | −3.745 | 0.009 | ||||
| Osr2† | odd-skipped related 2 (Drosophila) | NM_054049 | −2.519; −2.331 | 0.013; 0.005 | ||||
| Cell growth | ||||||||
| Socs2 | suppressor of cytokine signaling 2 | NM_007706 | −2.346 | 0.007 | ||||
| Igfbp3‡ | insulin-like growth factor binding protein 3 | NM_008343 | −2.124 | 0.015 | ||||
| Htra1† | HtrA serine peptidase 1 | NM_019564 | −2.062; −2.016 | 0.001; 0.002 | ||||
| Apoptosis | ||||||||
| Phlda1 | pleckstrin homology-like domain, family A, member 1 | NM_009344 | 2.206 | 0.001 | ||||
| Cd40† | CD40 antigen | NM_170702 | −2.160; −2.387 | 0.001; 0.000 | ||||
| Iron ion transport | ||||||||
| Cp | ceruloplasmin | NM_007752 | 2.259 | 0.001 | ||||
| Regulation of transcription | ||||||||
| Hoxa5 | homeo box A5 | NM_010453 | 3.701 | 0.000 | ||||
| Nfkbiz | nuclear factor of κ light polypeptide gene enhancer in B-cells inhibitor, ζ | NM_030612 | 2.657 | 0.002 | ||||
| Sox5† | SRY-box containing gene 5 | AK081802 | 2.279; 2.915 | 0.005; 0.006 | ||||
| Foxc1 | forkhead box C1 | NM_008592 | 2.256 | 0.003 | ||||
| Gata2 | GATA binding protein 2 | NM_008090 | 2.250 | 0.001 | ||||
| Hoxa2† | homeo box A2 | NM_010451 | 2.083; 2.150 | 0.003; 0.001 | ||||
| Hoxa7 | homeo box A7 | NM_010455 | 2.027 | 0.021 | ||||
| Msx1 | homeo box, msh-like 1 | NM_010835 | −6.173 | 0.001 | ||||
| Tcfap2b† | transcription factor AP-2β | NM_001025305 | −5.434; −3.367; −3.289 | 0.000; 0.012; 0.008 | ||||
| Tbx4 | T-box 4 | NM_011536 | −2.264 | 0.006 | ||||
| Irf7 | interferon regulatory factor 7 | NM_016850 | −2.220 | 0.041 | ||||
| Parp14 | poly(ADP-ribose) polymerase family, member 14 | NM_001039530 | −2.206 | 0.038 | ||||
| Hoxd8 | homeo box D8 | XM_355338 | −2.192 | 0.001 | ||||
| Tcfap2a | transcription factor AP-2, α | NM_011547 | −2.150 | 0.003 | ||||
| Signal transduction | ||||||||
| Tgm2 | transglutaminase 2, C polypeptide | NM_009373 | 2.344 | 0.001 | ||||
| Pde1a | phosphodiesterase 1A, calmodulin-dependent | NM_016744 | 2.224 | 0.002 | ||||
| Ccl9 | chemokine (C-C motif) ligand 9 | NM_011338 | 2.163 | 0.000 | ||||
| P2ry14† | purinergic receptor P2Y, G protein-coupled, 14 | NM_133200 | 2.070; 2.282 | 0.000; 0.001 | ||||
| Gpr49 | leucine rich repeat containing G protein-coupled receptor 5 | NM_010195 | −6.250 | 0.006 | ||||
| Cxcl15 | chemokine (C-X-C motif) ligand 15 | NM_011339 | −3.257 | 0.014 | ||||
| Tbxa2r† | thromboxane A2 receptor | NM_009325 | −3.058; −2.890 | 0.004; 0.004 | ||||
Genes shown are those that could be functionally categorized, based on their most prominently recognized functions. Of these, 24 and 28 genes were found to be up- and downregulated, respectively.
BLAST analyses indicate that this probe can detect α-actin-like EST sequences in nonmuscle tissues, as skin, mammary gland, and bone. No significant alterations in the expression levels of other actin isoforms (Acta2, Actb, Actc1, Actg1, Actg2) were detected.
Genes identified by multiple probes. The corresponding fold changes and P values are listed for each probe.
Genes included in more than one category. For example, Slit3 is an extracellular matrix protein as well as a cell motility-related protein; Tnn is implicated not only in the regulation of cell motility but also proliferation as well as in and cell adhesion. Moesin has been identified as an upregulated gene in the motility aspect of the invasion signature in cancer cells (12).
Lasp2 (LIM-nebulette), a nebulin family member that is the closest known homolog to Lasp1 (32, 52), has recently been proposed to play a role in regulating cell motility (13). Lasp2 is highly expressed in neuronal tissues, but at much lower levels in peripheral tissues (52). Previously, we did not detect significant levels of Lasp2 expression in gastric mucosa of either Lasp1-null mice or wild-type littermates (6). With highly sensitive, quantitative real-time RT-PCR, a very low level of Lasp2 expression could be detected in MEFs (∼1,000× lower than in brain); however, there was no difference in the level of expression in Lasp1−/− compared with Lasp1+/+ MEFs (Fig. 9, A and B). In less sensitive microarray and Western blot analyses, no Lasp2 signal above background was detected [see Fig. 9 and microarray data (accession number, XM_358339) deposited at the following web site: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE14800]. In contrast, a strong Lasp2 signal was detected by Western blot in mouse brain with a fourfold lower dilution of the anti-Lasp2 antibody (Fig. 9B, inset). In the same samples, Lasp1 was readily detected in wild-type MEFs and in mouse brain with anti-Lasp1 antibody but was not detected in Lasp1-null MEFs (Fig. 9B, inset).
Fig. 9.
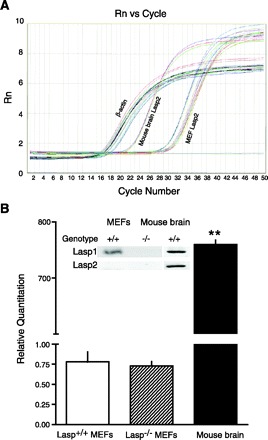
Quantitative RT-PCR and Western blot analyses of Lasp2 expression in MEFs and mouse brain. A: amplification profiles of Lasp2 message in Lasp1+/+ and Lasp1−/− MEFs and in mouse brain. β-Actin was used as an internal control for each sample. B: fold changes in Lasp2 expression relative to β-actin determined by 2−ΔΔCt method [relative expression = 2−ΔΔCt, where ΔCt = Ct (Lasp2)-Ct (β-actin)]. Very low levels of Lasp2 expression were detected in MEFs compared with mouse brain. The level of Lasp2 expression in Lasp1+/+ vs. Lasp1−/− MEFs was not significantly different. **P < 0.01; n = 3. Inset: representative images from Western blots showing Lasp2 immunoreactivity is readily detected in mouse brain, but not in MEFs, and Lasp1 is readily detected in Lasp1+/+ MEFs and mouse brain extracts but not in Lasp1−/− MEFs. Mouse brain and MEF proteins were extracted and subjected to Western blot analysis as described in material and methods (50 μg protein/lane; Lasp2 antibody dilution: MEFs, 1:50; mouse brain, 1:200; Lasp1 antibody dilution, 1:2,000).
DISCUSSION
Herein, we provide the first evidence linking chronic loss of Lasp1 to increased rates of cell migration and tumor promotion. These findings were unexpected in light of previous findings in which transient reductions in Lasp1 expression decreased cell migration and invasiveness and, in some cases, proliferation (23, 24, 35, 51, 57). It should be noted, however, that a more recent study found that reduced expression of Lasp1 in the SKHep1C3 hepatocellular carcinoma cell line enhanced migration (47). Conflicting findings have also been reported in overexpression studies in that transient overexpression of Lasp1 reduced migration in some cell lines (23, 35, 55) and enhanced, or had no effect, in others (23). One potential explanation for these conflicting observations is that both transient and chronic alterations in Lasp1 expression differentially affect cellular responses, depending on the absolute level of expression attained by the manipulation. Thus, as found in the present study, complete depletion of Lasp1 enhances cell migration. In contrast, partial and variable depletion of Lasp1, as in siRNA knockdown experiments, may differentially affect cellular responses through alterations in specific molecular interactions and, possibly, changes in the pattern of subcellular compartmentalization. Another possibility is that chronic alterations in Lasp1 expression induce different responses by altering gene expression profiles that are not similarly modified by transient alterations in Lasp1 expression. The protein expression profile of a particular cell line might also influence the outcome of these experiments by affecting different subcellular protein-protein interaction pathways. Off-target effects associated with siRNA knockdown and subcellular mistargeting resulting from protein overexpression (also considered to be a possibility by Lin and colleagues, Ref. 35) could also be factors. Whatever the mechanism, these conflicting observations raise intriguing questions regarding the role of Lasp1 in the regulation of cell motility, particularly as related to cancer development and progression. Future experimental approaches in which Lasp1 expression is chronically suppressed, as with a stable siRNA knockdown, and transiently reduced, as with an inducible siRNA knockdown, may provide further insights into the role of Lasp1 in these processes.
The Lasp1 gene, previously known as MLN50, was initially detected as an overexpressed product in some breast cancers (53, 54). Since then, Lasp1 has been found to be upregulated in a number of other cancers (22–24, 34, 57). Lasp1 upregulation has generally been interpreted as supportive of an active role for Lasp1 in the development of metastasis (22, 54). If, however, as suggested by present findings, Lasp1 plays a negative role in modulating cell migration and tumor formation, the increased expression of Lasp1 might reflect an important, positive development, serving to slow, rather than accelerate cancer progression. Recent microarray analyses of breast cancer patients provide some support to this hypothesis in that Lasp1 expression was found to be downregulated in breast cancer patients with the shortest postsurgical survival rates (1).
To begin to address the mechanisms associated with the alterations in cell migration, we analyzed known Lasp1 binding partners, focusing on those with previously established roles in the regulation of this activity. The focal adhesion protein, zyxin, was a prime candidate as zyxin associates with Lasp1 in vitro (30, 32), and transient knockdown of Lasp1 has been linked to loss of zyxin from focal adhesions (23, 24). Rather than a displacement of zyxin, however, we found that chronic loss of Lasp1 leads to a significant increase in the number of zyxin-containing focal adhesions. Since we found no evidence to suggest that zyxin expression is upregulated in Lasp1−/− MEFs, we postulate that the increase in zyxin-containing focal adhesions reflects a redistribution of zyxin from other subcellular compartment(s). Further experimentation is necessary to confirm whether this is, indeed, the case; however, the prominent presence of zyxin in focal adhesions of Lasp1-null MEFs provides a convincing counterargument to the proposal that Lasp1 is necessary and sufficient for the recruitment of zyxin to focal adhesions (24).
If zyxin levels at the cell periphery are increased at the expense of Lasp1, and the localization and/or expression of other regulatory proteins are simultaneously affected, this imbalance might lead to increased cell migration. The increased rate of cell-substrate attachment in Lasp1−/− MEFs might also be, at least partially, mediated by alterations in the number of focal adhesions. In this context, it is noteworthy that zyxin gene disruption produces an MEF phenotype that, in several respects, bears striking similarity to the Lasp1-null phenotype. Thus, compared with wild-type MEFs, zyxin-null MEFs display 1) increased rates of adherence to several extracellular matrices, including collagen type I; 2) increased migration in modified Boyden chamber assays as well as in monolayer wounding assays in the absence of changes in proliferation; and 3) similar focal adhesion and stress fiber morphologies (28). These findings suggest the possibility that both zyxin and Lasp1 are negative regulators of cell migration. Because chronic loss of zyxin, but not Lasp1, disrupted Ena/VASP localization within focal adhesions (28), the underlying mechanisms associated with altered adherence and motility are presumably different. In particular, because zyxin accumulation at focal adhesion sites has been linked to reduced membrane protrusion and traction forces (2, 60), the increased presence of zyxin in focal adhesions of Lasp1−/− MEFs may be offset by changes in the distribution and/or expression of other motility-associated proteins. Zyxin has the capacity to shuttle between focal adhesions and the nucleus, possibly transferring information from the cell periphery to nuclear transcription machinery (43–45). If a similar phenomenon occurs in MEFs, the increased recruitment of zyxin to focal adhesions in the absence of Lasp1 might also impact this process, perhaps by altering the expression of at least some the genes that we have identified that are linked to cell motility, adhesion, and/or cytoskeletal organization. Along these same lines, the finding that the zyxin family member LPP is upregulated at both the mRNA and protein levels is highly relevant. Like zyxin, LPP binds to Lasp1 in vitro (30, 32). LPP is highly expressed in smooth muscle but is also present in MEFs (Figs. 5, 7), as well as in a range of other tissues (21, 44). Furthermore, a recent proteomic analysis of the “leading pseudopodium” of chemotactic cells (the formation of which is necessary for cell migration) localized LPP and Lasp1, but not zyxin, within these structures (58). Although LPP functions are not as well established as zyxin, there is increasing evidence that this protein plays a positive role in regulating cell migration (20, 21, 37, 56), including recent observations that MEF-derived LPP-null mice exhibit reduced migratory capacity (56). Because the subcellular distribution pattern of LPP in MEFs more closely resembles that for Lasp1 than zyxin, it is possible that, at least in MEFs, the regulatory functions of Lasp1 are linked to interactions with LPP rather than to zyxin. We do not yet understand why loss of Lasp1 increases LPP expression; however, this finding, in concert with the detection of altered expression of messages for a number of other proteins that are involved not only with regulation of cell motility and adhesion but also with cell proliferation, cytoskeletal organization, signal transduction, and nuclear transcription is, indeed, fascinating and warrants further investigation.
In summary, the present findings, based on chronic depletion of Lasp1 under physiological conditions, support the hypothesis that Lasp1 serves as a negative, rather than a positive, modulator of cell migration and tumor formation in native tissues. Initial evidence suggests a potential role for the zyxin family member, LPP, in mediating these activities. Further studies are necessary to define the multitude of protein-protein interactions that are likely involved in this process and to refine the link between loss of Lasp1 and increased tumor formation in the framework of cancer development and metastasis.
GRANTS
This work was supported by National Institutes of Health Grant RO1DK-31900 to C. S. Chew.
Supplementary Material
Acknowledgments
We thank Dr. Asako Terasaki from Chiba University (Japan) for her generous gift of polyclonal rabbit anti-Lasp2 antibodies. We also thank John Nechtman and Eric Miller in the MCG Proteomics & Mass Spectrometry Core Facility for performing microarray analyses, Darren Baker in the MCG Imaging Core for assistance with imaging applications, and Penny Roon and Donna Kumiski in the MCG Electron Microscopy & Histology Core Laboratory for performing mice tumor histological analyses.
Footnotes
Address for reprint requests and other correspondence: C. S. Chew, Inst. of Molecular Medicine and Genetics, Sanders R&E Bldg., Rm. CB 2803, Medical College of Georgia, Augusta, GA 30912-3175 (e-mail: cchew@mcg.edu).
The online version of this article contains supplemental material.
REFERENCES
- 1.AsakaSAsaka S, Fujimoto T, Akaishi J, Ogawa K, Onda M. Genetic prognostic index influences patient outcome for node-positive breast cancer. Surg Today 36: 793–801, 2006. [DOI] [PubMed] [Google Scholar]
- 2.BeningoKABeningo KA, Dembo M, Kaverina I, Small JV, Wang YL. Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J Cell Biol 153: 881–888, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.BershadskyABershadsky A, Chausovsky A, Becker E, Lyubimova A, Geiger B. Involvement of microtubules in the control of adhesion-dependent signal transduction. Curr Biol 6: 1279–1289, 1996. [DOI] [PubMed] [Google Scholar]
- 4.ButtEButt E, Gambaryan S, Gèottfert N, Galler A, Marcus K, Meyer HE. Actin binding of human LIM and SH3 protein is regulated by cGMP- and cAMP-dependent protein kinase phosphorylation on serine 146. J Biol Chem 278: 15601–15607, 2003. [DOI] [PubMed] [Google Scholar]
- 5.ChewCSChew CS, Brown MR. Histamine increases phosphorylation of 27- and 40-kDa parietal cell proteins. Am J Physiol Gastrointest Liver Physiol 253: G823–G829, 1987. [DOI] [PubMed] [Google Scholar]
- 6.ChewCSChew CS, Chen X, Bollag RJ, Isales C, Ding KH, Zhang H. Targeted disruption of the Lasp-1 gene is linked to increases in histamine-stimulated gastric HCl secretion. Am J Physiol Gastrointest Liver Physiol 295: G37–G44, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ChewCSChew CS, Chen X, Parente JA Jr, Tarrer S, Okamoto C, Qin HY. Lasp-1 binds to non-muscle F-actin in vitro and is localized within multiple sites of dynamic actin assembly in vivo. J Cell Sci 115: 4787–4799, 2002. [DOI] [PubMed] [Google Scholar]
- 8.ChewCSChew CS, Okamoto CT, Chen X, Qin HY. IQGAPs are differentially expressed and regulated in polarized gastric epithelial cells. Am J Physiol Gastrointest Liver Physiol 288: G376–G387, 2005. [DOI] [PubMed] [Google Scholar]
- 9.ChewCSChew CS, Okamoto CT, Chen X, Thomas R. Drebrin E2 is differentially expressed and phosphorylated in parietal cells in the gastric mucosa. Am J Physiol Gastrointest Liver Physiol 289: G320–G331, 2005. [DOI] [PubMed] [Google Scholar]
- 10.ChewCSChew CS, Parente JA Jr, Chen X, Chaponnier C, Cameron RS. The LIM and SH3 domain-containing protein, lasp-1, may link the cAMP signaling pathway with dynamic membrane restructuring activities in ion transporting epithelia. J Cell Sci 113: 2035–2045, 2000. [DOI] [PubMed] [Google Scholar]
- 11.ChewCSChew CS, Parente JA Jr, Zhou C, Baranco E, Chen X. Lasp-1 is a regulated phosphoprotein within the cAMP signaling pathway in the gastric parietal cell. Am J Physiol Cell Physiol 275: C56–C67, 1998. [DOI] [PubMed] [Google Scholar]
- 12.CondeelisJCondeelis J, Singer RH, Segall JE. The great escape: when cancer cells hijack the genes for chemotaxis and motility. Ann Rev Cell Dev Biol 21: 695–718, 2005. [DOI] [PubMed] [Google Scholar]
- 13.DengXADeng XA, Norris A, Panaviene Z, Moncman CL. Ectopic expression of LIM-nebulette (LASP2) reveals roles in cell migration and spreading. Cell Motil Cytoskeleton 65: 827–840, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiGiovanniJDiGiovanni J. Genetic factors controlling responsiveness to skin tumor promotion in mice. Prog Clin Biol Res 391: 195–212, 1995. [PubMed] [Google Scholar]
- 15.DiGiovanniJDiGiovanni J, Imamoto A, Naito M, Walker SE, Beltrán L, Chenicek KJ, Skow L. Further genetic analyses of skin tumor promoter susceptibility using inbred and recombinant inbred mice. Carcinogenesis 13: 525–531, 1992. [DOI] [PubMed] [Google Scholar]
- 16.EdgarREdgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.EzrattyEJEzratty EJ, Partridge MA, Gundersen GG. Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat Cell Biol 7: 581–590, 2005. [DOI] [PubMed] [Google Scholar]
- 18.FillerRBFiller RB, Robert SJ, Girardi M. Cutaneous two-stage chemical carcinogenesis. CSH Protocols 10.1101/pdb.prot4837, 2007. [DOI] [PubMed]
- 19.ForteJGForte JG, Yao X. The membrane-recruitment-and-recycling hypothesis of gastric HCl secretion. Trends Cell Biol 6: 45–48, 1996. [DOI] [PubMed] [Google Scholar]
- 20.GorenneIGorenne I, Jin L, Yoshida T, Sanders JM, Sarembock IJ, Owens GK, Somlyo AP, Somlyo AV. LPP expression during in vitro smooth muscle differentiation and stent-induced vascular injury. Circ Res 98: 378–385, 2006. [DOI] [PubMed] [Google Scholar]
- 21.GorenneIGorenne I, Nakamoto RK, Phelps CP, Beckerle MC, Somlyo AV, Somlyo AP. LPP, a LIM protein highly expressed in smooth muscle. Am J Physiol Cell Physiol 285: C674–C685, 2003. [DOI] [PubMed] [Google Scholar]
- 22.GrunewaldTGGrunewald TG, Kammerer U, Kapp M, Eck M, Dietl J, Butt E, Honig A. Nuclear localization and cytosolic overexpression of LASP-1 correlates with tumor size and nodal-positivity of human breast carcinoma. BMC Cancer 7: 198, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.GrunewaldTGGrunewald TG, Kammerer U, Schulze E, Schindler D, Honig A, Zimmer M, Butt E. Silencing of LASP-1 influences zyxin localization, inhibits proliferation and reduces migration in breast cancer cells. Exp Cell Res 312: 974–982, 2006. [DOI] [PubMed] [Google Scholar]
- 24.GrunewaldTGGrunewald TG, Kammerer U, Winkler C, Schindler D, Sickmann A, Honig A, Butt E. Overexpression of LASP-1 mediates migration and proliferation of human ovarian cancer cells and influences zyxin localisation. Br J Cancer 96: 296–305, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.GuptonSLGupton SL, Waterman-Storer CM. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell 125: 1361–1374, 2006. [DOI] [PubMed] [Google Scholar]
- 26.HanahanDHanahan D, Weinberg RA. The hallmarks of cancer. Cell 100: 57–70, 2000. [DOI] [PubMed] [Google Scholar]
- 27.HenningsHHennings H, Glick AB, Lowry DT, Krsmanovic LS, Sly LM, Yuspa SH. FVB/N mice: an inbred strain sensitive to the chemical induction of squamous cell carcinomas in the skin. Carcinogenesis 14: 2353–2358, 1993. [DOI] [PubMed] [Google Scholar]
- 28.HoffmanLMHoffman LM, Jensen CC, Kloeker S, Wang CL, Yoshigi M, Beckerle MC. Genetic ablation of zyxin causes Mena/VASP mislocalization, increased motility, and deficits in actin remodeling. J Cell Biol 172: 771–782, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.JainRNJain RN, Al-Menhali AA, Keeley TM, Ren J, El-Zaatari M, Chen X, Merchant JL, Ross TS, Chew CS, Samuelson LC. Hip1r is expressed in gastric parietal cells and is required for tubulovesicle formation and cell survival in mice. J Clin Invest 118: 2459–2470, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.KeicherCKeicher C, Gambaryan S, Schulze E, Marcus K, Meyer HE, Butt E. Phosphorylation of mouse LASP-1 on threonine 156 by cAMP- and cGMP-dependent protein kinase. Biochem Biophys Res Commun 324: 308–316, 2004. [DOI] [PubMed] [Google Scholar]
- 31.KrauseMKrause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Ann Rev Cell Dev Biol 19: 541–564, 2003. [DOI] [PubMed] [Google Scholar]
- 32.LiBLi B, Zhuang L, Trueb B. Zyxin interacts with the SH3 domains of the cytoskeletal proteins LIM-nebulette and Lasp-1. J Biol Chem 279: 20401–20410, 2004. [DOI] [PubMed] [Google Scholar]
- 33.LiJLi J, Chen J, Kirsner R. Pathophysiology of acute wound healing. Clin Dermatol 25: 9–18, 2007. [DOI] [PubMed] [Google Scholar]
- 34.LiZLi Z, Szabolcs M, Terwilliger JD, Efstratiadis A. Prostatic intraepithelial neoplasia and adenocarcinoma in mice expressing a probasin-Neu oncogenic transgene. Carcinogenesis 27: 1054–1067, 2006. [DOI] [PubMed] [Google Scholar]
- 35.LinYHLin YH, Park ZY, Lin D, Brahmbhatt AA, Rio MC, Yates JR 3rd, Klemke RL. Regulation of cell migration and survival by focal adhesion targeting of Lasp-1. J Cell Biol 165: 421–432, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LivakKJLivak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-delta delta C(T)] method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 37.MajeskyMWMajesky MW. Organizing motility: LIM domains, LPP, and smooth muscle migration. Circ Res 98: 306–308, 2006. [DOI] [PubMed] [Google Scholar]
- 38.OkamotoCTOkamoto CT, Duman JG, Tyagarajan K, McDonald KL, Jeng YY, McKinney J, Forte TM, Forte JG. Clathrin in gastric acid secretory (parietal) cells: biochemical characterization and subcellular localization. Am J Physiol Cell Physiol 279: C833–C851, 2000. [DOI] [PubMed] [Google Scholar]
- 39.OkamotoCTOkamoto CT, Forte JG. Vesicular trafficking machinery, the actin cytoskeleton, and H+-K+-ATPase recycling in the gastric parietal cell. J Physiol 532: 287–296, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.OkamotoCTOkamoto CT, Li R, Zhang Z, Jeng YY, Chew CS. Regulation of protein and vesicle trafficking at the apical membrane of epithelial cells. J Control Release 78: 35–41, 2002. [DOI] [PubMed] [Google Scholar]
- 41.PapaioannouVEPapaioannou VE, Behringer RR. Maintaining your mutant. In: Mouse Phenotypes: A Handbook of Mutation Analysis. New York: CSHL, 2005, p. 54–55.
- 42.ParenteJAParente JA Jr, Chen X, Zhou C, Petropoulos AC, Chew CS. Isolation, cloning, and characterization of a new mammalian coronin family member, coroninse, which is regulated within the protein kinase C signaling pathway. J Biol Chem 274: 3017–3025, 1999. [DOI] [PubMed] [Google Scholar]
- 43.PetitMMPetit MM, Crombez KR, Vervenne HB, Weyns N, Van de Ven WJ. The tumor suppressor Scrib selectively interacts with specific members of the zyxin family of proteins. FEBS Lett 579: 5061–5068, 2005b. [DOI] [PubMed] [Google Scholar]
- 44.PetitMMPetit MM, Fradelizi J, Golsteyn RM, Ayoubi TA, Menichi B, Louvard D, Van de Ven WJ, Friederich E. LPP, an actin cytoskeleton protein related to zyxin, harbors a nuclear export signal and transcriptional activation capacity. Mol Biol Cell 11: 117–129, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.PetitMMPetit MM, Meulemans SM, Alen P, Ayoubi TA, Jansen E, Van de Ven WJ. The tumor suppressor Scrib interacts with the zyxin-related protein LPP, which shuttles between cell adhesion sites and the nucleus. BMC Cell Biol 6: 1, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.PetitMMPetit MM, Meulemans SM, Van de Ven WJ. The focal adhesion and nuclear targeting capacity of the LIM-containing lipoma-preferred partner (LPP) protein. J Biol Chem 278: 2157–2168, 2003. [DOI] [PubMed] [Google Scholar]
- 47.SalviASalvi A, Bongarzone I, Micciché F, Arici B, Barlati S, De Petro G. Proteomic identification of LASP-1 down-regulation after RNAi urokinase silencing in human hepatocellular carcinoma cells. Neoplasia 11: 207–219, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.SanoSSano S, Itami S, Takeda K, Tarutani M, Yamaguchi Y, Miura H, Yoshikawa K, Akira S, Takeda J. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J 18: 4657–4668, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.SastrySKSastry SK, Burridge K. Focal adhesions: a nexus for intracellular signaling and cytoskeletal dynamics. Exp Cell Res 261: 25–36, 2000. [DOI] [PubMed] [Google Scholar]
- 50.SchreiberVSchreiber V, Moog-Lutz C, Regnier CH, Chenard MP, Boeuf H, Vonesch JL, Tomasetto C, Rio MC. Lasp-1, a novel type of actin-binding protein accumulating in cell membrane extensions. Mol Med 4: 675–687, 1998. [PMC free article] [PubMed] [Google Scholar]
- 51.SpenceHJSpence HJ, McGarry L, Chew CS, Carragher NO, Scott-Carragher LA, Yuan Z, Croft DR, Olson MF, Frame M, Ozanne BW. AP-1 differentially expressed proteins Krp1 and fibronectin cooperatively enhance Rho-ROCK-independent mesenchymal invasion by altering the function, localization, and activity of nondifferentially expressed proteins. Mol Cell Biol 26: 1480–1495, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.TerasakiAGTerasaki AG, Suzuki H, Nishioka T, Matsuzawa E, Katsuki M, Nakagawa H, Miyamoto S, Ohashi K. A novel LIM and SH3 protein (lasp-2) highly expressing in chicken brain. Biochem Biophys Res Commun 313: 48–54, 2004. [DOI] [PubMed] [Google Scholar]
- 53.TomasettoCTomasetto C, Moog-Lutz C, Regnier CH, Schreiber V, Basset P, Rio MC. Lasp-1 (MLN 50) defines a new LIM protein subfamily characterized by the association of LIM and SH3 domains. FEBS Lett 373: 245–249, 1995. [DOI] [PubMed] [Google Scholar]
- 54.TomasettoCTomasetto C, Régnier C, Moog-Lutz C, Mattei MG, Chenard MP, Lidereau R, Basset P, Rio MC. Identification of four novel human genes amplified and overexpressed in breast carcinoma and localized to the q11-q21.3 region of chromosome 17. Genomics 28: 367–376, 1995. [DOI] [PubMed] [Google Scholar]
- 55.TurnerDPTurner DP, Findlay VJ, Kirven AD, Moussa O, Watson DK. Global gene expression analysis identifies PDEF transcriptional networks regulating cell migration during cancer progression. Mol Biol Cell 19: 3745–3757, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.VervenneHBVervenne HB, Crombez KR, Delvaux EL, Janssens V, Van de Ven WJ, Petit MM. Targeted disruption of the mouse Lipoma Preferred Partner gene. Biochem Biophys Res Commun 379: 368–373, 2009. [DOI] [PubMed] [Google Scholar]
- 57.WangBWang B, Feng P, Xiao Z, Ren EC. LIM and SH3 protein 1 (Lasp1) is a novel p53 transcriptional target involved in hepatocellular carcinoma. J Hepatol 50: 528–537, 2009. [DOI] [PubMed] [Google Scholar]
- 58.WangYWang Y, Ding SJ, Wang W, Jacobs JM, Qian WJ, Moore RJ, Yang F, Camp DG 2nd, Smith RD, Klemke RL. Profiling signaling polarity in chemotactic cells. Proc Natl Acad Sci USA 104: 8328–8333, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.XuWXu W, Baribault H, Adamson ED. Vinculin knockout results in heart and brain defects during embryonic development. Development 125: 327–337, 1998. [DOI] [PubMed] [Google Scholar]
- 60.Zaidel-BarRZaidel-Bar R, Ballestrem C, Kam Z, Geiger B. Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J Cell Sci 116: 4605–4613, 2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.