

Abstract
Significant variation exists for maternal nurturing ability in inbred mice. Although classical mapping approaches have identified quantitative trait loci (QTL) that may account for this variation, the underlying genes are unknown. In this study, lactation performance data among the mouse diversity panel were used to map genomic regions associated with this variation. Females from each of 32 inbred strains (n = 8–19 dams/strain) were studied during the first 8 days of lactation by allowing them to raise weight- and size-normalized cross-foster litters (10 pups/litter). Average daily weight gain (ADG) of litters served as the primary indicator of milk production. The number of pups successfully reared to 8 days (PNUM8) also served as a related indicator of maternal performance. Initial haplotype association analysis using a Bonferroni-corrected, genome-wide threshold revealed 10 and 15 associations encompassing 11 and 13 genes for ADG and PNUM8, respectively. The most significant of these associated haplotype blocks were found on MMU 8, 11, and 19 and contained the genes Nr3c2, Egfr, Sec61g, and Gnaq. Lastly, two haplotype blocks on MMU9 were detected in association with PNUM8. These overlapped with the previously described maternal performance QTL, Neogq1. These results suggest that the application of in silico QTL mapping is a useful tool in discovering the presence of novel candidate genes involved in determining lactation capacity in mice.
Keywords: genome-wide association study, lactation, mouse, haplotype
lactation is the most energetically demanding stage of a female's lifetime (42). In rodents, the demands of lactation on the mother are twofold: 1) production of milk for the suckling pups, and 2) increased heat dissipation in response to higher food intake. Maternal food intake doubles with the onset of lactation, and there is significant growth in both the mammary gland and the maternal gut to support these demands. The identification of genes underlying these complex changes has been challenging, not only in rodents but also in dairy animals, which are more amenable to direct measurements of milk production.
The contribution of genetic factors to lactation has been extensively studied in agricultural species such as the cow and sheep (2, 10, 17, 20, 30, 34, 37, 47, 54, 60, 62, 68, 75). In the cow, heredity accounts for 25–50% of the variation in milk production traits (21). As a consequence genotype-phenotype associations mapped as quantitative trait loci (QTL) for lactation-associated traits have been found on all 29 autosomes (10). However, only a few of the causal genes for these QTL have been identified. The most noteworthy of these are Acyl-CoA:diacylglycerol acyltransferase 1 (Dgat1), growth hormone receptor (Ghr), ATP binding cassette sub-family G member 2(Abcg2), and osteopontin (Spp1) (4, 14, 23, 69). Similar limitations have been observed with the analysis of lactation or “maternal nurturing ability” in the mouse (44, 57, 71, 72, 79). To date at least eight QTL have been described in association with maternal nurturing ability in mice. In addition, there are estimated to be ∼15 epistatic interactions around 23 QTL distributed across the mouse genome (57). Despite these discoveries, the genes responsible for these QTL remain to be identified.
Recent advances in genomic sequence analysis and single nucleotide polymorphism (SNP) discovery now provide new and potentially more efficient resources for the discovery of QTL candidate genes using inbred mouse strains such as the mouse diversity panel (MDP) and the inbred laboratory mouse haplotype map (39, 50). These publicly available databases, have allowed for the development of a novel gene mapping approach known as in silico genome-wide association (GWA) mapping (24). Although not without controversy (12, 16), in silico GWA has now successfully been used in several instances to either refine previously described QTL intervals or identify completely new QTL genes (26, 45, 51, 52, 61, 73). The key advantage to this approach is the speed inherent in simply phenotyping existing individuals from defined panels of inbred strains (24). A major criticism of the approach is a tendency to produce false positive associations because of inherent “population structure” within the genomes of the classical inbred strains (6–8, 35). However, effective computation strategies have been developed to overcome this limitation (35).
Our laboratory has used litter cross-fostering approaches to measure relative milk production in the mouse (27–29). These approaches have allowed for indirect measurement of milk production capacity during all stages of the cycle with a degree of increased sensitivity and precision that has not been present in previous studies. The overall goal of this study was to collect quantitative data on maternal nurturing capacity from mouse strains within the MDP and use these data with in silico GWA mapping to discover novel genes that may contribute to the variation in milk production. In our analysis, we employed a correction for kinship that has been shown by others to provide an effective means of removing effects due to population structure (35, 39). On the basis of this work we confirmed the existence of one previously discovered litter gain QTL and we describe three potentially new ones.
MATERIALS AND METHODS
Animals.
Males and females (6–10 wk old) from each of 31 inbred mouse strains were purchased from the Jackson Laboratories (Bar Harbor, ME). The strains used in this study were CZECHII/EiJ, PL/J, A/J, MA/MyJ, RIIIS/J, SJL/J, DBA/1J, C58/J, LP/J, BUB/BnJ, CBA/J, DBA/2J, SM/J, CAST/EiJ, AKR/J, SWR/J, I/LnJ, C57BL/6J, NZW/LacJ, CE/J, C57L/J, LG/J, KK/HlJ, 129S1/SvImJ, BALB/cByJ, C3H/HeJ, NON/LtJ, NOD/LtJ, FVB/NJ, SEA/GnJ, and BTBR T+ tf/J. The QSi5 strain was also included in this study as these mice have previously been described as being highly fecund and capable of rearing much larger litters than other inbred strains (33, 64, 65). This strain was obtained from Dr. Ian Martin of The University of Sydney, rederived into the Children's Nutrition Research Center mouse facility, and then maintained as an inbred line. To provide cross-foster litters, CD-1 females (Charles River Laboratories) were mated alongside the experimental females. To minimize variation, all CD-1 cross-foster donor dams used in this study originated from the Charles River's Raleigh, NC, production facility.
Study design.
The experiments described in this paper were conducted in accordance with procedures outlined in the NIH Guide to Care and Use of Experimental Animals, and they were approved by the Baylor College of Medicine Animal Care and Use Committee. All animals were maintained at an ambient temperature of 21°C, and a light-dark cycle of 14 h:10 h. To allow all females used in the study to complete one lactation, and to provide females for subsequent study, individual matings were set up between males and females within each strain. The animals were fed the 2020X pelleted diet (Harlan Teklad, Indianapolis, IN) through day 15 of their second pregnancy. After the females were allowed to complete their first lactation they were mated a second time to CD-1 males (Charles River Laboratories, Wilmington, MA) and then studied during the second lactation. At 15 days of pregnancy, study females were switched to a powdered form of the diet AIN-93G (Harlan Teklad), which was dispensed in funnel feeders (Research Products International, Mt. Prospect, IL). Each female was also given a single nestlet pad (Ancare, Bellmore, NY) on day 15 of pregnancy to provide a standardized material for nest building. On day 1 postpartum, the number of pups born to each female was counted and litters were weighed. Each female then had her own litter replaced with a litter made up of 10 1-day-old CD-1 pups. The cross-foster litters were adjusted so that all had similar weight. Litter weight and pup number were then recorded for each of the females daily through day 8 postpartum. On day 10 postpartum, all study animals were euthanized for tissue collection. The phenotypes for this study were cross-foster litter weight gain and cross-foster litter pup number during the first 8 days postpartum. Additional phenotypes were measured, but will be the subject of future manuscripts.
Data processing, quality control, and descriptive statistics.
Data were summarized using the explore function of IBM SPSS version 19 (IBM, Armonk, NY). Outliers were identified and removed by using extreme studentized deviate method (66). Strain effects were then tested for using the univariate GLM function in SPSS. Covariates were not used in the analysis. To identify strains with unusually high or low phenotypic values, the 95% confidence interval (CI) about the mean for the entire study group was used compared with the 95% confidence intervals for individual strain means. All phenotypic data are presented as the mean ± 95% CI.
Heritability estimates.
Broad sense heritability estimates for pups born, litter gain, and pup survival were calculated from the sums of squares in the ANOVA analysis as previously described (52).
SNP data source for the in silico haplotype mapping.
The Primary SNP data used in the association mapping for these studies was obtained from the Inbred Laboratory Mouse Haplotype Map resource(http://www.broadinstitute.org/mouse/hapmap/), and consisted of 132,285 SNPs mapped (132k set) across 94 strains according to National Center for Biotechnology Information build 37 of the mouse genome.
Construction of haplotype blocks.
With the 132k set, data were first filtered to exclude nonpolymorphic SNPs or those with minor-allele frequency of < 0.1, then linkage disequilibrium (LD) blocks were constructed in Haploview (3) using the four-Gamete Rule option, which generated a greater number of LD blocks compared with the other options. The tag SNPs in each LD block were also identified and marked and the LD block information was exported as text files, separately for each chromosome. These tag SNP files were then used to extract the haplotype for each strain, using R codes (74). The code is available on request from Peter C. Thomson (University of Sydney). One haplotype file was generated for each chromosome.
Construction of the similarity matrix.
Unless the genetic similarity across mouse strains is taken into account, an evaluation of association between haplotypes (or individual SNPs) and phenotypic strain means may result in spurious associations. To accommodate this, a genetic similarity matrix (S) was derived across all mouse strains in the database. The correction is essentially the same as the previously described kinship matrix approach (35). This was constructed by considering for each pair of mouse strains, the proportion of the genome they have in common, as assessed by the haplotypes. A weighting is included in this calculation for the number of tag SNPs. Formally, the calculation method for the strain similarity, sij between mouse strains i and j is
where nijk = number of tag SNPs in haplotype block k; H = total number of haplotype blocks over the genome; and where
Association analysis.
To evaluate the association between the phenotype (strain mean) and the haplotype, adjusting for strain similarity, a mixed-model framework was used, namely
where y = vector of phenotypic values, X = incidence matrix incorporating terms for the observed haplotype and any other fixed effects; β = vector of effects corresponding to these terms, Z = incidence matrix linking phenotype records to genetic groups, u = vector of polygenic genetic effects, and ε = vector of residual random errors.
The random errors are assumed to be independent with constant variance σε2, i.e., var(ε) = σε2 I, whereas the values of u are assumed to be correlated, according to the strain similarity, i.e., var(u) = σu2S. (Since only a subset of the strains were used here because of phenotypic information availability, a subset of the matrix S was extracted.) Model fitting is conducted iteratively using Henderson's mixed model equations (32), with a REML procedure used to estimate σε2 and σu2 at each iteration, as well as obtain estimates of β (and predictions of u). The effect of the haplotype is obtained by extracting the required elements of β̂, say β̂k. The significance of the haplotype association was assessed using a likelihood ratio χ2 test, comparing the fit of the model with (full) and without (reduced) the haplotype. The degrees of freedom for each test were the number of different haplotypes in the block less one. The mixed model analysis was conducted with user-written code in R (74). The code is available on request to Peter C. Thomson (University of Sydney). Haplotype associations were considered significant at a genome-wide P value threshold of 2.75 × 10−6 based on the Bonferroni correction of α/n, where n was the number of blocks tested (18,124) and α was 0.05.
Individual SNP association.
To confirm the results from the haplotype analysis and to focus on individual SNPs within the genome, an efficient mixed-model analysis was conducted as previously described (35) using the UCLA EMMA webserver (http://mouse.cs.ucla.edu/emmaserver/). The genome-wide significance threshold for this analysis was P = 10−5.
Real-time PCR analysis of gene expression.
Real-time PCR was used to measure the mRNA levels for genes identified from the genome-wide association study (GWAS) analysis as potential candidates. The genes in this analysis included epidermal growth factor receptor (Egfr), guanine nucleotide binding protein Q polypeptide (Gnaq), nuclear receptor subfamily 3, group C, member 2 (Nr3c2), and Sec61γ (Sec61g). The primer sequences for these genes are as follows: ATGCGAAGACGTCACATTGTT and CCATCACATAGGCTTCGTCA for Egfr, TGAACACAATAAGGC TCATGC and TGTAGGCAGAT AGGAAGGGTC for Gnaq, ACTTGCCTCTTGAGGACCAA and TTTGGAACTGTGCTCAGTAGC for Nr3cs, and GCAGTTTGTGGAGCCCAGCC and TGAGAAGGACTCAGCCACCCACAA for Sec61g. Total RNA from mammary tissue collected from lactating dams (five per strain) at day 10 postpartum was isolated with TRIzol reagent (Invitrogen) as per manufacturer's instruction. Reverse transcription PCR was performed using Iscript Reverse Transcription (Bio-Rad catalog 170-8841) according to the manufacturer's manual. In brief, 1,000 ng total RNA was used for each 20 μl reaction. The reverse transcription was incubated at 25°C for 5 min, followed by 42°C for 30 min and 85°C for 5 min. Quantitative (q) PCR was performed in the Bio-Rad CFX 96 Real Time System under the following conditions: 20 μl reaction contained 50 ng of cDNA template, 125 nM of each primer, and 1× SsoAdvanced SYBR Green Supermix (Bio-Rad, catalog 1725261). Samples were denatured at 95°C for 30 s, followed by a 40 cycle-PCR or 95°C for 5 s, 65°C for 30 s. Upon completion of 40 cycles the samples were heated to 72°C for 5 min and followed by dissociation analysis to determine amplicon size. Reference genes were chosen based on the presence of stable expression and a high pair-wise correlation using the Bestkeeper software (59). Relative mRNA levels were normalized and expressed as 2−ΔCT using a Bestkeeper index based on the CT of the two reference genes nucleolin (Ncl) and peptidylprolyl isomerase A (Ppia), which are both highly stable and displayed a Pearson's correlation of 0.7. The primer pairs for Ncl and Ppia were CATGGTGAAGCTCGCAAAG and TCACTATCCT CTTCCACCTCCT, and GAGCTGTTTG CAGACAAAGTTC and CCCTGGCACATGAATCCTGG, respectively.
RESULTS
Analysis of litter gain and pup survival.
A total of 419 dams were placed on the study at the beginning of their second parity by giving them weight-normalized litters of 10 CD-1 pups at day 1 postpartum. Of these, 379 completed the study without complications. Of these, 358 completed the study with at least 1 pup remaining by day 8 postpartum. The traits analyzed were number of pups reared to day 8 postpartum (PNUM8), and average daily litter gain (ADG) during the first 8 days postpartum. The broad-sense heritability estimate for PNUM8 was 0.32. For average daily gain, the heritability estimate was 0.47. For both phenotypes, strain accounted for a highly significant proportion of the observed variation (all P values <0.0001). In addition both PNUM8 and ADG were highly correlated (P < 0.0001) with a Pearson's r value of 0.85. The distribution strain means for ADG (Fig. 1A) ranged from −0.71 ± 1.37 to 4.59 ± 0.53 g/day (mean ± 95% CI) and an overall mean of 2.57 ± 0.19 g/day (95% CI, 2.37–2.76 g/day). Extreme strains at the upper end of the distribution included QSi5, BTBRT + tf/J, and SEA/GnJ. Strains that were extremes at the lower end of the distribution included RIIIS/J, MA/MyJ, and A/J. Overall, the average PNUM8 was 8.98 ± 0.24 (95% CI: 8.74–9.22). This trait showed a skewed distribution that was the result of limiting the cross-foster litter size to 10. When compared across strains, means for this trait ranged from 6.46 ± 2.38 to 10.0 ± 0.00 (Fig. 1B). Of the 32 strains studied, eight maintained all 10 pups out through day 8 postpartum. Extreme strains at the lower end of the distribution included A/J, MA/MyJ, and CZECHII/EiJ.
Fig. 1.
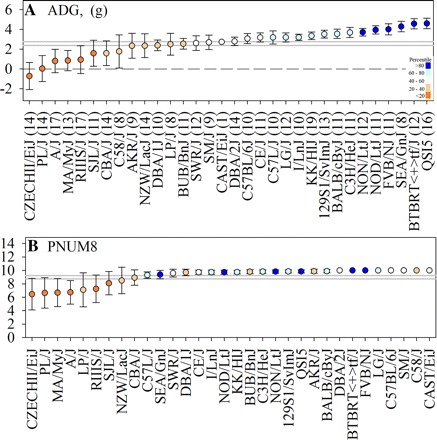
Average daily litter gain (ADG) and pup survival among 32 inbred mouse strains. Females from each strain were studied in their 2nd lactation. Females were induced to ovulation by treatment with PMS and HCG and then mated to CD-1 males. On day 1 postpartum, number of pups born per litter was recorded and independent cross-foster litters of 10 1-day-old CD-1 pups were then given to each dam. ADG weight gain (A) and maternal litter rearing capacity (B) were recorded daily through day 8 postpartum (PNUM8). Each symbol represents the mean ± 95% confidence interval (CI) for 8–16 dams with the exception of CAST/EiJ for which only a single dam was obtained. Numbers in parentheses indicate the number of animals studied. The solid gray lines indicate the 95% CIs about the overall mean for each trait. Symbols are color coded by the percentile ranking of the strain mean for ADG.
Phenotypic distributions in relation to strain relatedness.
To visualize variations in phenotypic performance in relation to genetic relatedness, a heat map illustrating phenotypic performance quintiles for ADG and PNUM8 aligned to a dendrogram based on genetic relatedness (Pearson's distance, d) was constructed (Fig. 2). Inspection of the figure suggested that low performance was not confined to any particular cluster of strains but could be found in at least four different branches and in some cases closely related to strains with high performance. There did appear to be a high-performance cluster (average d = 0.41) that contained NON/LtJ, FVB/NJ, NOD/LtJ, and QSi5. Interestingly, within this cluster there were also strains, BUB/BnJ, SJL/J, and SWR/J, that displayed average to below-average performance on the two traits. In addition, SEA/GnJ and BALB/cByJ are both somewhat closely (average d = 0.39) related to A/J, which is a strain that displayed below-average performance. The two strains BTBRT + tf/J and 129S1/SvImJ, though both above average performance and somewhat closely related (d = 0.42), are distinct (d = 0.6) from the other high performance strains in the set. Although the CZECHII/EiJ and CAST/EiJ strains are quite distinct from the rest of the strains it appears that they may differ in their capacity to rear litters and they are also quite different from each other genetically (d = 0.65). Unfortunately, we were unable to obtain more than a single second-parity dam from the CAST/EiJ strain during the course of the experiment. However, the fact that pup survival was so high suggested that further work with this strain may be informative.
Fig. 2.
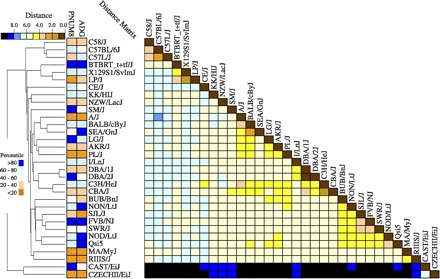
Comparison of genetic relatedness with ADG and maternal pup survival among 32 inbred mouse strains. A dendrogram (left) illustrating the genetic relatedness among the strains used in this study is presented alongside a heat map (middle) illustrating the relative ranking for ADG and PNUM8. An additional heat map (right) illustrates the pairwise relatedness among the strains expressed as a Pearson's distance (d = 1 − r, where r is Pearson's correlation). The dendrogram is a UPGMA tree based on average haplotype block identity across the entire genome. Pair-wise genetic distances between strains were computed as described under genetic similarity in materials and methods.
Identification of regions associated with ADG and PNUM8.
The application of Haploview generated a total of 18,124 haplotype blocks with block sizes ranging from 1 bp to 499 kbp with an average of 50.75 ± 0.51 kbp (Supplemental Table S1).1 The distance between haplotype blocks ranged from 0.02 to 20,798.75 with an average of 89.79 ± 1.99 kbp. The density of haplotype blocks across the genome ranged from 0 to 141 with an average of 69 ± 2 blocks per 10 Mbp. The highest average haplotype density was found for chromosome 9 (83 ± 10), while the X chromosome had a density of 8 ± 1 blocks per 10 Mbp.
To focus on only the strongest haplotype associations, a genome-wide Bonferroni-corrected P value of 2.75 × 10−6 was used. This threshold allowed for the detection of 10, and 15 associations for ADG (Fig. 3A), and PNUM8 (Fig. 3B), respectively (Table 1). Although calculating the effect size for each of the blocks in this analysis was not practical because many of the blocks had more than two haplotypes, phenotypic means for each haplotype in the associated blocks (Supplemental Table S2) were used to calculate a percent change as an estimate of relative effect size (Table 1). This allowed for prioritizing the associations on either P value or relative effect size. The initial work described here is based on using P values to prioritize the blocks. Overall, the most significant (P = 10−10) associations were found for PNUM8 on MMU11 (Fig. 4A). On this chromosome there were six blocks exhibiting strong association (Table 1). Of these there were three that contained genes. A block at MMU11:16392211 (P = 2 × 10−6) was 53 kbp and contained the gene Sec61g (Fig. 4B). A single SNP at MMU11:16509601 (P = 2 × 10−9) was located between the Sec61g and Egfr genes. There was also a block at MMU11:1000111136 that was 18 kbp and overlapped with Krt17 and Krt42. For the region adjacent to Egfr, an independent association analysis based on individual SNPs confirmed the significance of the haplotype associations by detecting a cluster of highly significant single SNP beginning at MMU11:156483344 and ending at 15606840 (Fig. 4B). Although none of these SNPs were located within any known genes, a few were present in an 18 kbp region that was 145 kbp upstream of Egfr and 79 kbp downstream of Sec61g. Further analysis using the software package Haploview indicated that although the region between these two genes had a generally high LD (Fig. 4B), there could be as many as five independent blocks across the region. Furthermore, follow-up analysis using the UCSC Genome Browser (http://genome-test.cse.ucsc.edu/index.html) failed to indicate the presence of significant regulatory elements in this region. Analysis of individual SNPs in the block containing the Krt17 and Krt42 genes failed to demonstrate significant associations. Therefore these genes were not studied further.
Fig. 3.
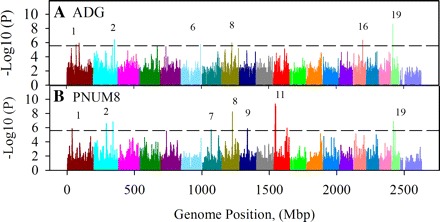
Manhattan plots illustrating significant genome-wide associations for ADG and PNUM8. The graphs present the results of haplotype association analysis for ADG (A) and PNUM8 (B). Each bar represents the location in the genome that begins a haplotype block. The significance threshold for a Bonferroni-corrected genome-wide (GW05) P value of α = 0.05 (2.75 × 10−6) is indicated by the dashed line.
Table 1.
Haplotype blocks in the murine genome associated with ADG and PNUM8
| Trait | Chr. | Location | P Value | Size, kbp | Haplotypes, n | Effect Size† | Previous QTL* | Genes* |
|---|---|---|---|---|---|---|---|---|
| ADG | 1 | 71,678,828 | 1.83E-06 | 137.7 | 3 | 28 | Apol7d, Fn1 | |
| 1 | 93,063,713 | 1.09E-06 | 6.6 | 2 | 22 | Bmd5 | Rbm44 | |
| 1 | 93,522,108 | 1.90E-06 | 21.7 | 2 | 21 | Bmd5 | ||
| 1 | 93,935,237 | 1.90E-06 | 186.5 | 2 | 21 | Bmd5 | Hdac4 | |
| 2 | 158,861,097 | 3.56E-07 | 0.1 | 2 | 37 | Bglu1, Mob5, Prdt2, Scgq3 | miR1902 | |
| 6 | 147,990,373 | 1.58E-06 | 9 | 3 | 61 | Far2 | ||
| 8 | 79,373,410 | 1.04E-06 | 64.9 | 4 | 45 | Bmd39 | Gm10649, Nr3c2 | |
| 16 | 75,030,310 | 4.11E-07 | 55.6 | 3 | 42 | |||
| 19 | 16,273,854 | 2.21E-09 | 141.9 | 2 | 33 | Gnaq | ||
| 19 | 16,323,845 | 3.54E-07 | 272.9 | 2 | 28 | Gnaq | ||
| PNUM8 | 1 | 36,965,390 | 1.33E-06 | 6.2 | 3 | 42 | Idd26 | Tmem131 |
| 2 | 94,183,967 | 2.57E-07 | 46.9 | 4 | 44 | Mob5, Mohg1, PLast2b, T2dm2 sa, Tmc1m1 | Ttc17 | |
| 2 | 143,957,762 | 1.52E-07 | 72.8 | 3 | 39 | Bglu1, Mob5, Plast2b, Prdt2, T2dm2sa | ||
| 7 | 74,436,551 | 1.98E-06 | 144.7 | 3 | 38 | Mef2a | ||
| 8 | 79,373,410 | 5.79E-09 | 64.9 | 4 | 34 | Bmd39 | Gm10649, Nr3c2 | |
| 9 | 59,194,226 | 2.67E-06 | 114 | 4 | 49 | Alpq3, El4, Neogq1 | Arih1, Bbs4 | |
| 9 | 60,328,263 | 1.35E-06 | 9.9 | 3 | 46 | Alpq3, El4, Neogq1 | Thsd4 | |
| 11 | 13,000,201 | 3.18E-07 | 163.1 | 3 | 19 | Bmch5, W6q3 | ||
| 11 | 13,526,250 | 4.45E-10 | 215.4 | 5 | 39 | Bmch5, W6q3 | ||
| 11 | 15,748,958 | 8.70E-10 | 115.7 | 3 | 27 | Bmch5, W6q3 | ||
| 11 | 16,392,211 | 2.37E-06 | 53.9 | 2 | 21 | Bmch5, W6q3 | Sec61 g | |
| 11 | 16,509,601 | 2.33E-09 | 0.001 | 2 | 23 | Bmch5, W6q3 | Between Sec61 g, Egfr | |
| 11 | 100,111,136 | 1.14E-06 | 18.2 | 2 | 41 | Cia40. Pregq2, Tmc1m2 | Krt17, Krt42 | |
| 19 | 16,323,845 | 1.24E-07 | 141.9 | 2 | 17 | Gnaq | ||
| 19 | 27,522,368 | 2.18E-06 | 28.9 | 3 | 40 |
From the Mouse Genome Database (MGD), Mouse Genome Informatics, The Jackson Laboratory (Bar Harbor, ME; http://www.informatics.jax.org).
Calculated as the percent difference between the lowest and highest mean among the haplotypes.
ADG, average daily litter gain; PNUM8, day 8 postpartum.
Fig. 4.
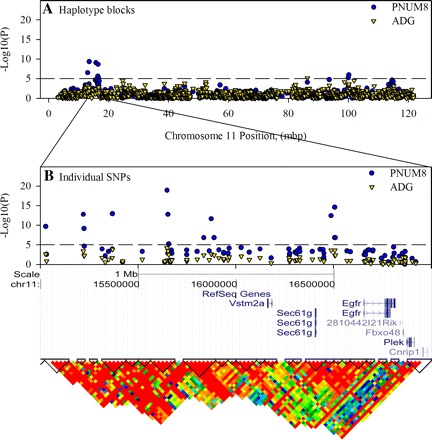
Definition of haplotype blocks for MMU11 that are in close proximity to the genes for Sec61g and Egfr. Single nucleotide polymorphism (SNP) data for MMU11 were loaded into Haploview to confirm the block structure in this region and define the major haplotypes for the region. These same data were also used in an independent single SNP analysis using the efficient mix-model analysis server (http://mouse.cs.ucla.edu/emmaserver/). Association score is plotted against the position for the start of each of 977 haplotype blocks that were defined on this chromosome (A). The region from 15 to 17 Mbp is enlarged to illustrate highly significant single SNP associations in relationship to known genes and linkage disequilibrium (LD), which is expressed as r2 in the heat map (B).
The second most highly significant (P = 2×10−9) haplotype association was between ADG and a 142 kbp block located at MMU19:16323845 (Table 1, Fig. 5A). This block was also significantly associated with PNUM8 (P = 1×10−7) and contained the gene Gnaq. Association analysis based on single SNPs (Fig. 6B) demonstrated the presence of a cluster of significantly associated SNPs that were primarily located toward the 3′-end of the Gnaq gene. The region containing these SNPs was also found to have a high LD (Fig. 5B). All of the associated SNPs were contained within introns.
Fig. 5.
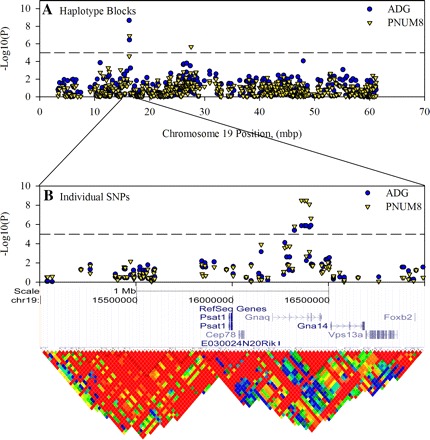
Definition of haplotype blocks on MMU19 that are in close proximity of the gene for Gnag. SNP data for MMU19 were loaded into Haploview to confirm the block structure in this region and define the major haplotypes for the region. This same data were also used in an independent single SNP analysis using the efficient mix-model analysis server (http://mouse.cs.ucla.edu/emmaserver/). Association score is plotted against the position for the start of each of 473 haplotype blocks that were defined on this chromosome (A). The region from 15 to 17 Mbp is enlarged to illustrate highly significant single SNP associations in relationship to known genes and LD, which is expressed as r2 in the heat map (B).
Fig. 6.
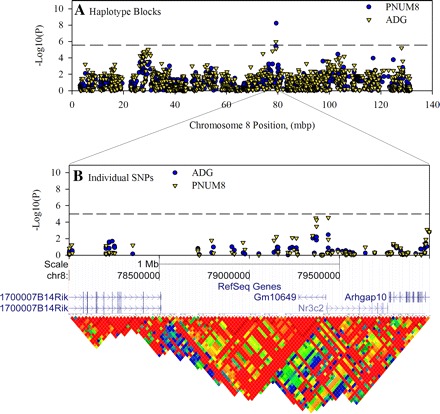
Definition of haplotype blocks on MMU8 that are in close proximity to the gene for Nr3c2. SNP data For MMU8 were loaded into Haploview to confirm the block structure in this region and define the major haplotypes for the region. This same data were also used in an independent single SNP analysis using the efficient mix-model analysis server (http://mouse.cs.ucla.edu/emmaserver/). Association score is plotted against the position for the start of each of 976 haplotype blocks that were defined on this chromosome (A). The region from 15 to 17 Mbp is enlarged to illustrate highly significant single SNP associations in relationship to known genes and LD, which is expressed as r2 in the heat map (B).
The third most highly significant (P = 5×10−9 and P = 1×10−6 for PNUM8 and ADG, respectively) association detected in this analysis was a 64 kbp block at MMU8:79373410 (Table 1, Fig. 6A). This block contained the gene Nr3c2. The analysis of individual SNPs on this region of the chromosome identified three SNPs that approached the genome-wide threshold of P = 10−5 (Fig. 6B). Of these, two were located in the gene for a hypothetical protein, GM10649, and the third was located in the first intron of Nr3c2. The SNPs in this region also had high LD.
Of the remaining haplotype associations that reached the Bonferroni-corrected genome-wide significance threshold (Table 1), there were two adjacent blocks on MMU9 (P = 10−6) that encompassed three genes and reside in a region previously reported to contain Neogq1, a QTL linked to variations in maternal nurturing capacity in the mouse (44). There were also blocks on MMU1, 2, 4, 6, 7, and 16 that encompassed a total of 14 genes including one encoding for the microRNA, namely, miR1902.
To determine if the associations detected for the blocks containing Egfr, Gnaq, Nr3c2, and Sec61g were linked to alterations in mammary gland gene expression, we conducted real-time RT-PCR on a subset of five dams from each strain with mammary tissue that was collected at the end of the study period on day 10 postpartum (Fig. 7). To control for technical variation, the mRNA levels for two reference genes, Ncl and Ppia, were also measured. Comparison of Ct values for these two genes across the sample set demonstrated them to be both stable and correlated. The geometric mean Ct for the reference genes was then employed to calculate a normalized ratio (2−ΔCt) for each gene of interest. Although the mRNAs for all four of these genes were present in mammary RNA from these day 10 lactating dams, the transcript for Sec61g had the greatest average abundance (1.4 ± 09) when normalized to the reference gene mRNAs (Fig. 7D). The average abundance of Gnaq mRNA was 0.03 ± 0.001 (Fig. 7C), while that of Egfr (Fig. 7A) and Nr3c2 (Fig. 7B) was 4 × 10−3 ± 2 × 10−4 and 6.5 × 10−4 ± 3.6 × 10−5, respectively. Abundance for all of these genes varied significantly both across and within strains. However, rather than observing positive correlations between ADG and PNUM8 there were only low negative correlations detected. Significant (P < 0.05) negative Pearson's correlations were detected between ADG and the mRNA levels for Egfr (r = −0.16, P = 0.04) and Nr3c2 (r = −0.20, P = 0.02). A significant negative correlation was also observed for PNUM8 and the mRNA level for Nr3c2 (r = −0.24, P = 0.006). These results support the conclusion that although the haplotype blocks containing these genes are associated with ADG and PNUM8, the effects of these associations are not necessarily mediated through effects on the expression of these genes in the mammary gland during an established lactation.
Fig. 7.
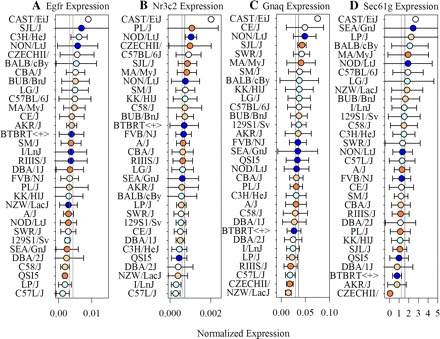
Analysis of mammary mRNA abundance for Egfr, Nr3c2, and Gnaq indicates a low negative correlation with ADG during an established lactation. Total RNA was prepared from mammary tissue collected at the end of the study period on day 10 postpartum from each of 32 inbred strains. Real-time RT-PCR was then conducted with primers specific for Egfr (A), Nr3c2 (B), Gnaq (C), and Sec61g (D). Nucleolin and Ppia mRNAs were measured as reference transcripts. Data are expressed at △ct. Each symbol represents the mean ± 95% CI for 5 animals (with the exception of CAST/EiJ, which had only 1). Symbols are color coded by ADG percentile ranking (see Fig. 1).
DISCUSSION
In the mouse, litter weight gain is a well-documented and robust indicator of milk production (41, 70). The trait is also quite complex with the potential to be influenced not only by the volume of milk produced but also by milk composition, litter size, and maternal behavior. At present, the relative contribution of these factors to litter gain is not well documented. Our own preliminary analysis of these ancillary traits in the MDP suggests that although maternal behavior and milk composition do have some influence on the variability in litter gain, further study of these is necessary to make firm conclusions. A similar case can be made that traits such as maternal food intake and body mass may also contribute to strain-dependent variation in litter gain. Despite this complexity, litter gain in mice is a heritable trait. This fact was illustrated by the analysis of variance results, which demonstrated that strain contributed significantly (P < 0.0001) to the variation in both ADG and PNUM8. Although for PNUM8, the true variation across strains is likely to be larger than that observed in this study. The fact that in eight of the 32 strains studied all of the dams were able to successfully rear all of their cross-foster pups suggests that the upper extreme of the distribution for PNUM8 was not defined. This does not invalidate the trait with regard to the GWA results, but it does suggest that any associations should be viewed as alleles that are linked to decreased maternal rearing ability rather than for higher than average ability.
The heritability of litter weight gain in mice has been reported to be 0.33–0.35 (57, 79). Our own estimates of heritability are not tremendously different from these and suggest a significant genetic contribution to both ADG and PNUM8. In agreement with these observations, studies have demonstrated that pup weight gain during the suckling period, which largely depends on milk production, can be increased through selection (33, 53, 65). As a consequence it was not surprising to observe significant interstrain variation in both pup survival and litter gain. An important point to mention is that three of the strains used in our study, LG/J, SM/J, and KK/HlJ, have also been used for previous work on QTL associated with maternal nurturing ability in mice (71, 79). That these three strains were not in the extremes for ADG illustrates the fact that the additional strains used in this study have documented a broader range of phenotypic diversity than previously appreciated. Interestingly, not all of our highest performing strains came from a similar genetic background. In fact, of the top five strains only three, QSi5, FVB/NJ, and NOD/Ltj have a common origin from the Swiss subgroup of mice (49, 58). The other two, SEA/GnJ and BTBR T + tf/J, have origins that are both distinct from each other and from Swiss mice (58). Of all of these five strains, the QSi5 resulted from direct selection for fecundity. This produced a strain with increased mature body size, larger-than-average litter size, and increased lactation capacity (33, 64, 65, 77). With regard to identifying lactation QTL, the availability of this highly fecund strain, in combination with other strains in the dataset, now provides a great resource from which to expand on previous lactation QTL studies.
Previous lactation QTL studies in mice have revealed the presence of eight QTL intervals. These are on chromosomes 1, 5, 6, 7, and 9. The intervals for these QTL are quite large, ranging in size from 12 to 36 Mbp and containing a total of 1,336 potential candidate genes (44, 57, 71, 72, 79). Consequently the identities of the causal genes underlying these QTL are still unclear. When first introduced in 2001, in silico mapping based on classical inbred strains was suggested to be a computational approach that would markedly accelerate the genetic analysis of murine disease models (24). Subsequent work also suggested that this approach would significantly narrow QTL intervals to <0.5 Mbp (45). A number of studies have also had success with this approach (26, 43, 45, 46, 61, 76). The main advantages of the approach over the traditional linkage mapping are; 1) no requirement for time-consuming matings to generate intercross progeny, 2) no requirement for genotyping, 3) greater resolution due to increased marker density and greater number of historic recombinations, and 4) an ability to capture a broader range of genetic diversity (76). The main disadvantage of the approach is that genetic relatedness among the MDP is high, producing a population structure which can introduce systematic bias leading to false-positive associations if not appropriately accounted for in the analysis (35, 39). A second potential disadvantage is the potential for limited power to detect loci associated with quantitative traits (56). With regard to the power issue, the 32 strains used in our study seem to have provided adequate power to detect potential candidates. With regard to the population structure issue, our use of a similarity matrix as a term in the model should correct for this. There have now been a number of studies using a kinship matrix to correct for population structure in GWAS with classical inbred strains (5, 18, 63, 80). Lastly, the fact that overlap occurred between the current GWAS and that of at least one previously described lactation QTL (44) suggests that the candidate regions discovered here will be well worth pursuing.
Before further discussing the biological significance of the associations detected in this study, is it is important to note that the GWA analysis described here is based only on SNP. Consequently some of the variation in phenotype may have been due to genomic variation that was unaccounted for in the present analysis. In addition, the possibility exists that SNPs detected in this study, though associated with the phenotype may not be causative, but simply in high LD with other genomic variants that are causative. This will be discussed further.
The use of the Bonferroni correction screened out all but the most significant associations producing a list of 25 blocks containing a total of 19 genes. These were prioritized for follow-up based on two criteria, P value and the presence of overlap between the two traits. The most interesting among these was the one at MMU11:16509601, which was in close proximity to the gene for Egfr. Previous work comparing mammary gene expression between QSi5 and CBA suggested that MAPK signaling and EGF may contribute to differences in lactation performance (64). Consequently, the regions identified on MMU11 in close proximity to Egfr are very interesting. The other gene close to this association was Sec61g, which encodes a subunit of the Sec61 complex a major component of the protein translocation apparatus within the endoplasmic reticulum (22, 31). Although not well studied in mammalian systems, variations in Sec61 function within the lactating mammary cell could impact milk production through potential effects on protein trafficking. Egfr is a strong candidate from the standpoint that EGF family members are known to be important to mammary gland development and lactation. Targeting of AR/EGF/TGF-α produced a lactation defect that was linked to both reduced mammary development and decreased secretory cell differentiation (48). The genes for all of the Erbb family members are expressed in the lactating mammary gland and the receptors are phosphorylated, indicating that these pathways are highly active (67). In addition, a natural mutation in the EGF receptor (waved-2) trans-membrane domain has been demonstrated on at least one genetic background to produce a lactation defect, making the region on MMU11 near the Egfr a very strong candidate region for follow up (19).
The associated block on MMU19 was also a very attractive candidate because it was detected for both ADG and PNUM8. The only gene within this block, Gnaq, encodes for an α-subunit in the Gq class that is an integral component of heterotrimer G protein signaling through G protein-coupled receptors. Recently, variations in a haplotype within the human Gnaq promoter have been linked to insulin resistance in mammary adipocytes isolated from obese women with polycystic ovary syndrome (40). In addition, studies in mice have demonstrated that targeted deletion of Gnaq within the forebrain abolishes maternal nurturing behavior in a Gα11-deficient mouse line (78). These observations support the notion that the block on MMU19 could play a significant role in maternal nurturing ability either through potential effect on insulin signaling within the mammary gland or possibly through impacting maternal behavior. This will be addressed in future studies.
The association on MMU8 was also very interesting from the standpoint that Nr3c2, which encodes for the mineralocorticoid receptor, has been implicated as playing a potential role in the regulation of mammary cell differentiation. In this regard both glucocorticoids and mineralocorticoids have been known for some time to play an important role in mammary cell differentiation and lactation (1, 15). More recent work in lactating mice has also illustrated the importance of glucocorticoids as well as the potential for the mineralocorticoid receptor to play a role in lactation (13, 38). These observations suggest that a region on MMU8 near Nr3c2 may also be important with regard to variation in milk production.
An important consideration with all of the above genes is that although the blocks they were contained in had highly significant P values that were also confirmed with individual SNP analysis, our estimates of effect sizes did not rank them at the top. It is also important to note that none of the SNPs detected in our top three regions were present in known regulatory or coding regions. The fact that none were in coding regions was not surprising given that previous work cataloging the locations of allelic variants suggests that these tend to be overrepresented in intergenic regions (25, 36). It was, however, puzzling that mRNA levels for our four most promising candidate genes were not correlated to ADG. This lack of correlation could be interpreted in one of several ways. Firstly, it may be that day 10 of lactation was not the correct time to study the expression of these genes and that differences in expression during earlier stages of mammary gland development may have been more strongly correlated to ADG. For Egfr, this is clearly a possibility, since this receptor family plays a major role in earlier stages of mammary gland development. Secondly, for some of the candidate blocks, it may be that the polymorphism is acting in an organ other than the mammary gland. Although we only collected mammary tissue from the animals in the current study we hope to address this issue in future studies with a subset of animals taken from the extremes of our litter gain distribution. Lastly it is possible that the SNPs identified here are not causative, but in LD with as of yet unidentified polymorphisms. A number of other types of genomic variation have increasingly come under scrutiny with regard to both disease traits and quantitative traits. In this regard recent sequencing work has not only catalogued >12 million SNP in classical inbred mouse strains, but also >2 million structural variants including insertions, deletions, and transposable elements (36). In addition, studies have now linked the presence of copy number variants (CNV) to effects not only on gene expression in various tissues, but also on metabolic traits such as blood metabolites and body composition (9, 55). In this regard, further analysis of the sequence surrounding the regions of interest identified in this study would be helpful as would microarray analysis of mammary tissue gene expression from these inbred strains. A more extensive database of CNV within the inbred mouse strains would also provide a useful tool for follow-up on the results presented here.
In summary, the application of previously existing SNP datasets in conjunction with the inbred mouse mapping panel has allowed for the identification of several new regions within the mouse genome that contain genes of potential importance to the variation observed in lactation performance. Follow-up studies are needed on these regions to confirm this analysis and further our understanding of factors that contribute to variations in lactation and possibly other aspects of mammary gland biology.
GRANTS
This work was funded by grants from the National Institute of Child Health and Human Development Grant #1R21HD-059746-01A1 and the USDA/ARS (cooperative agreement #6250-51000-054).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: D.L.H., J.W., and P.W. conception and design of research; D.L.H., J.W., W.O., and L.A.H. performed experiments; D.L.H., J.W., W.O., L.A.H., A.R., P.C.T., and M.S. analyzed data; D.L.H. and P.W. interpreted results of experiments; D.L.H. prepared figures; D.L.H. drafted manuscript; D.L.H., P.C.T., and P.W. edited and revised manuscript; D.L.H., J.W., P.C.T., and P.W. approved final version of manuscript.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Ian Martin for providing the QSi5 strain. The authors thank Dr. Junfeng Tong and Valerie Lee for assistance in collecting the phenotype data. Thanks to Dr. Mathew Pletcher for providing the heritability calculator. Thanks also goes to Dr. Monique Rijnkels for a critical reading of the manuscript and to Linda Kemper for editorial suggestions.
Footnotes
The online version of this article contains supplemental material.
REFERENCES
- 1.Anderson RR, Turner CW. Aldosterone and corticosterone in maintenance of lactation in adrenalectomized rats. Proc Soc Exp Biol Med 112: 997–1000, 1963 [DOI] [PubMed] [Google Scholar]
- 2.Ashwell MS, Van Tassell CP. Detection of putative loci affecting milk, health, and type traits in a US Holstein population using 70 microsatellite markers in a genome scan. J Dairy Sci 82: 2497–2502, 1999 [DOI] [PubMed] [Google Scholar]
- 3.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21: 263–265, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Blott S, Kim JJ, Moisio S, Schmidt-Kuntzel A, Cornet A, Berzi P, Cambisano N, Ford C, Grisart B, Johnson D, Karim L, Simon P, Snell R, Spelman R, Wong J, Vilkki J, Georges M, Farnir F, Coppieters W. Molecular dissection of a quantitative trait locus: a phenylalanine-to-tyrosine substitution in the transmembrane domain of the bovine growth hormone receptor is associated with a major effect on milk yield and composition. Genetics 163: 253–266, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bopp SE, Ramachandran V, Henson K, Luzader A, Lindstrom M, Spooner M, Steffy BM, Suzuki O, Janse C, Waters AP, Zhou Y, Wiltshire T, Winzeler EA. Genome wide analysis of inbred mouse lines identifies a locus containing Ppar-gamma as contributing to enhanced malaria survival. PloS One 5: e10903, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cervino AC, Darvasi A, Fallahi M, Mader CC, Tsinoremas NF. An integrated in silico gene mapping strategy in inbred mice. Genetics 175: 321–333, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cervino AC, Gosink M, Fallahi M, Pascal B, Mader C, Tsinoremas NF. A comprehensive mouse IBD database for the efficient localization of quantitative trait loci. Mamm Genome 17: 565–574, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Cervino AC, Li G, Edwards S, Zhu J, Laurie C, Tokiwa G, Lum PY, Wang S, Castellini LW, Lusis AJ, Carlson S, Sachs AB, Schadt EE. Integrating QTL and high-density SNP analyses in mice to identify Insig2 as a susceptibility gene for plasma cholesterol levels. Genomics 86: 505–517, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Chaignat E, Yahya-Graison EA, Henrichsen CN, Chrast J, Schutz F, Pradervand S, Reymond A. Copy number variation modifies expression time courses. Genome Res 21: 106–113, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen HY, Zhang Q, Yin CC, Wang CK, Gong WJ, Mei G. Detection of quantitative trait loci affecting milk production traits on bovine chromosome 6 in a Chinese Holstein population by the daughter design. J Dairy Sci 89: 782–790, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Chesler EJ, Rodriguez-Zas SL, Mogil JS. In silico mapping of mouse quantitative trait loci. Science 294: 2423, 2001 [DOI] [PubMed] [Google Scholar]
- 13.Chida D, Miyoshi K, Sato T, Yoda T, Kikusui T, Iwakura Y. The role of glucocorticoids in pregnancy, parturition, lactation, and nurturing in melanocortin receptor 2-deficient mice. Endocrinology 152: 1652–1660, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Cohen-Zinder M, Seroussi E, Larkin DM, Loor JJ, Everts-van der Wind A, Lee JH, Drackley JK, Band MR, Hernandez AG, Shani M, Lewin HA, Weller JI, Ron M. Identification of a missense mutation in the bovine ABCG2 gene with a major effect on the QTL on chromosome 6 affecting milk yield and composition in Holstein cattle. Genome Res 15: 936–944, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cowie AT, Tindal JS. Adrenalectomy in the goat; replacement therapy and the maintenance of lactation. J Endocrinol 16: 403–414, 1958 [DOI] [PubMed] [Google Scholar]
- 16.Darvasi A. In silico mapping of mouse quantitative trait loci. Science 294: 2423, 2001 [PubMed] [Google Scholar]
- 17.de Koning DJ, Schulmant NF, Elo K, Moisio S, Kinos R, Vilkki J, Maki-Tanila A. Mapping of multiple quantitative trait loci by simple regression in half-sib designs. J Anim Sci 79: 616–622, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Farber CR, Bennett BJ, Orozco L, Zou W, Lira A, Kostem E, Kang HM, Furlotte N, Berberyan A, Ghazalpour A, Suwanwela J, Drake TA, Eskin E, Wang QT, Teitelbaum SL, Lusis AJ. Mouse genome-wide association and systems genetics identify Asxl2 as a regulator of bone mineral density and osteoclastogenesis. PLoS Genet 7: e1002038, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fowler KJ, Walker F, Alexander W, Hibbs ML, Nice EC, Bohmer RM, Mann GB, Thumwood C, Maglitto R, Danks JA. A mutation in the epidermal growth factor receptor in waved-2 mice has a profound effect on receptor biochemistry that results in impaired lactation. Proc Natl Acad Sci USA 92: 1465–1469, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freyer G, Sorensen P, Kuhn C, Weikard R, Hoeschele I. Search for pleiotropic QTL on chromosome BTA6 affecting yield traits of milk production. J Dairy Sci 86: 999–1008, 2003 [DOI] [PubMed] [Google Scholar]
- 21.Georges M, Nielsen D, Mackinnon M, Mishra A, Okimoto R, Pasquino AT, Sargeant LS, Sorensen A, Steele MR, Zhao X, Womack JE, Hoeschele I. Mapping quantitative trait loci controlling milk production in dairy cattle by exploiting progeny testing. Genetics 139: 907–920, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greenfield JJ, High S. The Sec61 complex is located in both the ER and the ER-Golgi intermediate compartment. J Cell Sci 112: 1477–1486, 1999 [DOI] [PubMed] [Google Scholar]
- 23.Grisart B, Coppieters W, Farnir F, Karim L, Ford C, Berzi P, Cambisano N, Mni M, Reid S, Simon P, Spelman R, Georges M, Snell R. Positional candidate cloning of a QTL in dairy cattle: identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res 12: 222–231, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Grupe A, Germer S, Usuka J, Aud D, Belknap JK, Klein RF, Ahluwalia MK, Higuchi R, Peltz G. In silico mapping of complex disease-related traits in mice. Science 292: 1915–1918, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Gunther T, Schmitt AO, Bortfeldt RH, Hinney A, Hebebrand J, Brockmann GA. Where in the genome are significant single nucleotide polymorphisms from genome-wide association studies located? Omics 15: 507–512, 2011 [DOI] [PubMed] [Google Scholar]
- 26.Guo Y, Lu P, Farrell E, Zhang X, Weller P, Monshouwer M, Wang J, Liao G, Zhang Z, Hu S, Allard J, Shafer S, Usuka J, Peltz G. In silico and in vitro pharmacogenetic analysis in mice. Proc Natl Acad Sci USA 104: 17735–17740, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hadsell D, George J, Torres D. The declining phase of lactation: peripheral or central, programmed or pathological? J Mammary Gland Biol Neoplasia 2: 59–70, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Hadsell DL, Torres D, George J, Capuco AV, Ellis SE, Fiorotto ML. Changes in secretory cell turnover, and mitochondrial oxidative damage in the mouse mammary gland during a single prolonged lactation cycle suggest the possibility of accelerated cellular aging. Exp Gerontol 41: 271–281, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Hadsell DL, Torres DT, Lawrence NA, George J, Parlow AF, Lee AV, Fiorotto ML. Overexpression of des(1–3) insulin-like growth factor 1 in the mammary glands of transgenic mice delays the loss of milk production with prolonged lactation. Biol Reprod 73: 1116–1125, 2005 [DOI] [PubMed] [Google Scholar]
- 30.Harder B, Bennewitz J, Reinsch N, Thaller G, Thomsen H, Kuhn C, Schwerin M, Erhardt G, Forster M, Reinhardt F, Kalm E. Mapping of quantitative trait loci for lactation persistency traits in German Holstein dairy cattle. J Anim Breed Genet 123: 89–96, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Hartmann E, Sommer T, Prehn S, Gorlich D, Jentsch S, Rapoport TA. Evolutionary conservation of components of the protein translocation complex. Nature 367: 654–657, 1994 [DOI] [PubMed] [Google Scholar]
- 32.Henderson CR. Best linear unbiased estimation and prediction under a selection model. Biometrics 31: 423–447, 1975 [PubMed] [Google Scholar]
- 33.Holt M, Nicholas FW, James JW, Moran C, Martin IC. Development of a highly fecund inbred strain of mice. Mamm Genome 15: 951–959, 2004 [DOI] [PubMed] [Google Scholar]
- 34.Jonas E, Thomson PC, Hall EJ, McGill D, Lam MK, Raadsma HW. Mapping quantitative trait loci (QTL) in sheep. IV. Analysis of lactation persistency and extended lactation traits in sheep. Genet Sel Evol 43: 22, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E. Efficient control of population structure in model organism association mapping. Genetics 178: 1709–1723, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, Furlotte NA, Eskin E, Nellaker C, Whitley H, Cleak J, Janowitz D, Hernandez-Pliego P, Edwards A, Belgard TG, Oliver PL, McIntyre RE, Bhomra A, Nicod J, Gan X, Yuan W, van der Weyden L, Steward CA, Bala S, Stalker J, Mott R, Durbin R, Jackson IJ, Czechanski A, Guerra-Assuncao JA, Donahue LR, Reinholdt LG, Payseur BA, Ponting CP, Birney E, Flint J, Adams DJ. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477: 289–294, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khatkar MS, Thomson PC, Tammen I, Raadsma HW. Quantitative trait loci mapping in dairy cattle: review and meta-analysis. Genet Sel Evol 36: 163–190, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kingsley-Kallesen M, Mukhopadhyay SS, Wyszomierski SL, Schanler S, Schutz G, Rosen JM. The mineralocorticoid receptor may compensate for the loss of the glucocorticoid receptor at specific stages of mammary gland development. Mol Endocrinol 16: 2008–2018, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Kirby A, Kang HM, Wade CM, Cotsapas CJ, Kostem E, Han B, Furlotte N, Kang EY, Rivas M, Bogue MA, Frazer KA, Johnson FM, Beilharz EJ, Cox DR, Eskin E, Daly MJ. Fine mapping in 94 inbred mouse strains using a high-density haplotype resource. Genetics 85: 1081–1095, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klenke S, Tan S, Hahn S, Mann K, Hauner H, Manthey I, Peters J, Siffert W, Frey UH. A functional GNAQ promoter haplotype is associated with altered Gq expression and with insulin resistance and obesity in women with polycystic ovary syndrome. Pharmacogenet Genomics 20: 476–484, 2010 [DOI] [PubMed] [Google Scholar]
- 41.Knight CH, Maltz E, Docherty AH. Milk yield and composition in mice: effects of litter size and lactation number. Comp Biochem Physiol A 84A: 127–133, 1986 [DOI] [PubMed] [Google Scholar]
- 42.Krol E, Speakman JR. Limits to sustained energy intake. VI. Energetics of lactation in laboratory mice at thermoneutrality. J Exp Biol 206: 4255–4266, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Liao G, Wang J, Guo J, Allard J, Cheng J, Ng A, Shafer S, Puech A, McPherson JD, Foernzler D, Peltz G, Usuka J. In silico genetics: identification of a functional element regulating H2-Ealpha gene expression. Science 306: 690–695, 2004 [DOI] [PubMed] [Google Scholar]
- 44.Liljander M, Sallstrom MA, Andersson S, Wernhoff P, Andersson A, Holmdahl R, Mattsson R. Identification of genetic regions of importance for reproductive performance in female mice. Genetics 173: 901–909, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu P, Vikis H, Lu Y, Wang D, You M. Large-scale in silico mapping of complex quantitative traits in inbred mice. PLoS One 2: e651, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu P, Wang Y, Vikis H, Maciag A, Wang D, Lu Y, Liu Y, You M. Candidate lung tumor susceptibility genes identified through whole-genome association analyses in inbred mice. Nat Genet 38: 888–895, 2006 [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Jansen GB, Lin CY. Quantitative trait loci mapping for dairy cattle production traits using a maximum likelihood method. J Dairy Sci 87: 491–500, 2004 [DOI] [PubMed] [Google Scholar]
- 48.Luetteke NC, Qiu TH, Fenton SE, Troyer KL, Riedel RF, Chang A, Lee DC. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development 126: 2739–2750, 1999 [DOI] [PubMed] [Google Scholar]
- 49.Lynch CJ. The so-called Swiss mouse. Lab Anim Care 19: 214–220, 1969 [PubMed] [Google Scholar]
- 50.McClurg P, Janes J, Wu C, Delano DL, Walker JR, Batalov S, Takahashi JS, Shimomura K, Kohsaka A, Bass J, Wiltshire T, Su AI. Genomewide association analysis in diverse inbred mice: power and population structure. Genetics 176: 675–683, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McLachlan S, Lee SM, Steele TM, Hawthorne PL, Zapala MA, Eskin E, Schork NJ, Anderson GJ, Vulpe CD. In silico QTL mapping of basal liver iron levels in inbred mouse strains. Physiol Genomics 43: 136–147, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller BH, Schultz LE, Gulati A, Su AI, Pletcher MT. Phenotypic characterization of a genetically diverse panel of mice for behavioral despair and anxiety. PLoS One 5: e14458, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nagai J, Sarkar NK. Relationship between milk yield and mammary gland development in mice. J Dairy Sci 61: 733–739, 1978 [DOI] [PubMed] [Google Scholar]
- 54.Olsen HG, Gomez-Raya L, Vage DI, Olsaker I, Klungland H, Svendsen M, Adnoy T, Sabry A, Klemetsdal G, Schulman N, Kramer W, Thaller G, Ronningen K, Lien S. A genome scan for quantitative trait loci affecting milk production in Norwegian dairy cattle. J Dairy Sci 85: 3124–3130, 2002 [DOI] [PubMed] [Google Scholar]
- 55.Orozco LD, Cokus SJ, Ghazalpour A, Ingram-Drake L, Wang S, van Nas A, Che N, Araujo JA, Pellegrini M, Lusis AJ. Copy number variation influences gene expression and metabolic traits in mice. Hum Mol Genet 18: 4118–4129, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Payseur BA, Place M. Prospects for association mapping in classical inbred mouse strains. Genetics 175: 1999–2008, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peripato AC, De Brito RA, Vaughn TT, Pletscher LS, Matioli SR, Cheverud JM. Quantitative trait loci for maternal performance for offspring survival in mice. Genetics 162: 1341–1353, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petkov PM, Ding Y, Cassell MA, Zhang W, Wagner G, Sargent EE, Asquith S, Crew V, Johnson KA, Robinson P, Scott VE, Wiles MV. An efficient SNP system for mouse genome scanning and elucidating strain relationships. Genome Res 14: 1806–1811, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett 26: 509–515, 2004 [DOI] [PubMed] [Google Scholar]
- 60.Plante Y, Gibson JP, Nadesalingam J, Mehrabani-Yeganeh H, Lefebvre S, Vandervoort G, Jansen GB. Detection of quantitative trait loci affecting milk production traits on 10 chromosomes in Holstein cattle. J Dairy Sci 84: 1516–1524, 2001 [DOI] [PubMed] [Google Scholar]
- 61.Pletcher MT, McClurg P, Batalov S, Su AI, Barnes SW, Lagler E, Korstanje R, Wang X, Nusskern D, Bogue MA, Mural RJ, Paigen B, Wiltshire T. Use of a dense single nucleotide polymorphism map for in silico mapping in the mouse. PLoS Biol 2: e393, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raadsma HW, Jonas E, McGill D, Hobbs M, Lam MK, Thomson PC. Mapping quantitative trait loci (QTL) in sheep. II. Meta-assembly and identification of novel QTL for milk production traits in sheep. Genet Sel Evol 41: 45, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Radovanovic I, Mullick A, Gros P. Genetic control of susceptibility to infection with Candida albicans in mice. PloS One 6: e18957, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramanathan P, Martin IC, Gardiner-Garden M, Thomson PC, Taylor RM, Ormandy CJ, Moran C, Williamson P. Transcriptome analysis identifies pathways associated with enhanced maternal performance in QSi5 mice. BMC Genomics 9: 197, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Riley LG, Zubair M, Thomson PC, Holt M, Xavier SP, Wynn PC, Sheehy PA. Lactational performance of Quackenbush Swiss line 5 mice. J Anim Sci 84: 2118–2125, 2006 [DOI] [PubMed] [Google Scholar]
- 66.Rosner B. Percentage points for a generalized ESD many-outlier procedure. Technometrics 25: 165–172, 1983 [Google Scholar]
- 67.Schroeder JA, Lee DC. Dynamic expression and activation of ERBB receptors in the developing mouse mammary gland. Cell Growth Differ 9: 451–464, 1998 [PubMed] [Google Scholar]
- 68.Schrooten C, Bink MC, Bovenhuis H. Whole genome scan to detect chromosomal regions affecting multiple traits in dairy cattle. J Dairy Sci 87: 3550–3560, 2004 [DOI] [PubMed] [Google Scholar]
- 69.Sheehy PA, Riley LG, Raadsma HW, Williamson P, Wynn PC. A functional genomics approach to evaluate candidate genes located in a QTL interval for milk production traits on BTA6. Anim Genet 40: 492–498, 2009 [DOI] [PubMed] [Google Scholar]
- 70.Shipman LJ, Docherty AH, Knight CH, Wilde CJ. Metabolic adaptations in mouse mammary gland during a normal lactation cycle and in extended lactation. Q J Exp Physiol 72: 303–311, 1987 [DOI] [PubMed] [Google Scholar]
- 71.Suto J, Sekikawa K. Further mapping and characterization of Naq1, a quantitative trait locus responsible for maternal inferior nurturing ability in RR mice. J Vet Med Sci 66: 1033–1038, 2004 [DOI] [PubMed] [Google Scholar]
- 72.Suto J, Yamanaka H, Sekikawa K. Genetic analysis of inferior nurturing ability in RR mice. Reproduction 123: 52–58, 2002 [DOI] [PubMed] [Google Scholar]
- 73.Tang PL, Cheung CL, Sham PC, McClurg P, Lee B, Chan SY, Smith DK, Tanner JA, Su AI, Cheah KS, Kung AW, Song YQ. Genome-wide haplotype association mapping in mice identifies a genetic variant in CER1 associated with BMD and fracture in southern Chinese women. J Bone Miner Res 24: 1013–1021, 2009 [DOI] [PubMed] [Google Scholar]
- 74.Team RDC. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; http://www.R-project.org/. [Google Scholar]
- 75.Viitala SM, Schulman NF, de Koning DJ, Elo K, Kinos R, Virta A, Virta J, Maki-Tanila A, Vilkki JH. Quantitative trait loci affecting milk production traits in Finnish Ayrshire dairy cattle. J Dairy Sci 86: 1828–1836, 2003 [DOI] [PubMed] [Google Scholar]
- 76.Wang J, Liao G, Usuka J, Peltz G. Computational genetics: from mouse to human? Trends Genet 21: 526–532, 2005 [DOI] [PubMed] [Google Scholar]
- 77.Wei J, Ramanathan P, Thomson PC, Martin IC, Moran C, Williamson P. An integrative genomic analysis of the superior fecundity phenotype in QSi5 mice. Mol Biotechnol [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 78.Wettschureck N, Moers A, Hamalainen T, Lemberger T, Schutz G, Offermanns S. Heterotrimeric G proteins of the Gq/11 family are crucial for the induction of maternal behavior in mice. Mol Cell Biol 24: 8048–8054, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wolf JB, Vaughn TT, Pletscher LS, Cheverud JM. Contribution of maternal effect QTL to genetic architecture of early growth in mice. Heredity 89: 300–310, 2002 [DOI] [PubMed] [Google Scholar]
- 80.Yang IV, Wade CM, Kang HM, Alper S, Rutledge H, Lackford B, Eskin E, Daly MJ, Schwartz DA. Identification of novel genes that mediate innate immunity using inbred mice. Genetics 183: 1535–1544, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.