

In nature, cells adhere and detach selectively in circulation with sophisticated signaling mechanisms.[1–4] Selective capture of cells and biological materials using microfluidics has been a rapidly growing field with multiple applications in diagnostics (e.g., CD4 cell count for HIV monitoring,[5–9] capture of circulating tumor cells,[10] pathogen detection,[11–13] isolation of neutrophils from blood,[14] isolation of mixed leucocytes,[15] and separation of T-lymphocytes[16]) and regenerative medicine for stem cell isolation.[17,18] Microfluidic devices offer advantages such as handling small samples volumes, low consumption of expensive reagents, and shorter assay times, enabling portable automated devices.[7,9,17] Although capturing cells in micro-channels has been studied, spatial control of capture and on-demand release of cells in microfluidic channels remains as an unaddressed challenge.
Downstream applications of cell isolation using microfluidic systems currently utilize on-chip lysis of captured cells for genomic and proteomic analyses.[14] However, there are inefficiencies associated with recovering cellular and genetic material from microchannels, due to the large surface area to volume ratio.[19] Additionally, in rare cell applications, captured cells are low in concentration, and they need to be released from micro-channels and expanded in culture for subsequent biological analyses. On-demand release of captured cells with spatial control in microchannels would address the challenges associated with retrieving captured cells from microchannels offering a broadly applicable enabling biotechnology. The advances in stimuli responsive smart interface materials have enabled new functionalities in terms of controlling the material-cell interactions, facilitating a broad range of biological and clinical applications.[20] Here, we present for the first time a microfluidic system integrated with stimuli responsive smart interface material (poly(N-isopropylacrylamide), (PNIPAAm) and thermoelectric local temperature control, enabling both spatial and temporal control over selective capture and on-demand release of cells in microchannels from complex fluids, such as unprocessed whole blood.
In this study, we targeted CD4+ T lymphocyte separation from unprocessed human whole blood for applications such as downstream genomic processing[16] and CD4 counts for HIV monitoring.[5,7–9,12,17] Microfluidic channels were designed to work with manual pipetting to reduce dependence of the system on peripheral equipment and to make the technology broadly accessible.[17] The channel dimensions were designed for optimum flow rates and shear stresses to capture CD4+ T lymphocytes from unprocessed whole blood.[17] We have developed thermoresponsive microfluidic channels using PNI-PAAm, a temperature responsive smart interface material.[21] In this study, we controlled the temperature in selected, localized zones within the channels to achieve region-specific capture and release (Figure 1A and B). Cooled thermoresponsive channels (Figure 1C) and channels heated over the whole surface area were included as controls (Figure 1D). To locally capture cells in pre-determined areas of the microfluidic channel, we integrated miniature thermoelectric modules under predetermined zones in channels and maintained these areas at 37 °C. The remaining areas of channels were kept at room temperature (Figure 1E and F). Using a temperature responsive dye, temperature distribution within channels was monitored (Figure 1G), and quantified using digital imaging and analysis. Figure 1G illustrates a chip indicating the full range of colors displayed by the thermoresponsive dye (Figure 1H). The temperature responsive dye performs in 32 °C to 41 °C range; displays green color at 37 °C (Figure 1H), and black color at temperatures below 32 °C (Figure 1H). Calibration of RGB values corresponding to the color produced by the dye at controlled temperatures in 32 °C to 41 °C range was used in image analysis to quantify temperature distribution in channels (Figure 1I). A similar temperature distribution was observed in all channels and the middle channel was used as a temperature indicator channel during the experiments. Cell capture experiments were performed at 37 °C and the captured cells were released at temperatures below 32 °C.
Figure 1.

Local capture and release of cells in microchannels integrated with smart interface materials. Schematic description of: A) local cooling of channels for local cell release, and B) local warming of channels for local cell capture with thermoelectric module. C) The schematic representation of the cross-section of channels in the absence of heating elements. D) When the whole channel surface was warmed to 37 °C, target cells were captured everywhere on the channel surface. E) Channels can be locally warmed to 37 °C, which enables local capture of cells in a selected region. F) Thermoresponsive channels can be locally cooled using thermoelectric modules to facilitate on-demand local release of a selected set of captured cells in microchannels. G) Local temperature control in thermoresponsive channels. The middle channel on the microchips was stained with a temperature responsive dye to monitor temperature change of the device. A typical image of a chip with a local temperature change in one channel shows the full spectrum of colors. Black area indicates the channel on the microchip that was stained with thermoresponsive dye. The white dashed rectangle shows the area of interest used in image processing. The thermoelectric unit was located below the channel, which resulted in a local temperature control and color change in the channel. H) Temperature responsive dye was responsive in the range of 32 °C to 41 °C, and displayed green color at 37 °C, i.e., the temperature at which cells were captured. The dye displayed black color at temperatures below 32 °C, at which release of cells in the channels was achieved. I) Baseline RGB values represent the colors displayed by the thermosensitive dye, which were used in image processing to quantify temperature distribution in channels.
To monitor local capture process, microfluidic channels were divided into four virtual zones defined by where the thermoelectric module is placed. Each zone corresponded to 4.3 mm of the 25 mm full channel length. Controlled capture and release area (17.2 mm2) constituted 17.2% of the total surface area (100 mm2) of a single channel. Figures 1E and 2A illustrate local warming of thermoresponsive channels, enabling local capture of cells in a pre-determined area located close to channel inlet (i.e., zone 1). Control channels were designed to capture cells in all zones (Figure 1D). As expected, control channels typically displayed uniform temperature distribution and cell capture in all zones (Figure 2A and 2B). When we locally controlled the temperature of zone 1, we observed that the majority of cells (79% ±4%, n = 4 channels) were captured in this zone (Figure 2C). A greater (statistically significant, non-parametric Mann-Whitney U test, p < 0.05) number of cells was captured in zone 1 compared to zones 2–4. This result showed that local capture of cells in a selected zone within a channel is possible. Capture specificity of microchannels was 92% (±2%, n = 4 channels, Figure 2D).
Figure 2.

Local cell capture in microchannels. A) Temperature distribution in control and local capture channels. In local capture channels, temperature was controlled locally, enabling local capture of cells in zone 1 only. B) Typical images of cells in channels in zones 1–4 before and after capture of cells (cells were marked with circles). Images indicated a significant difference in number of cells captured in zone 1 compared to zones 2–4. Cells captured in zones 1–4 in control channels displayed a typical distribution pattern for cell capture in microchannels. C) Quantitative analysis of cell capture in zones 1–4, in control and local capture channels. A statistically significant, greater number of cells were captured in zone 1 (79% ±4%, n = 4 channels) compared to zone 1 of control channel and zones 2–4 of local capture channel. Each zone corresponded to 4.3 mm of the 25 mm full channel length. D) Bright field and CD4 immunofluorescent stained locally captured cells indicating the capture specificity of the channels. Capture specificity of the channels was quantified to be 92% (±2%, n = 4 channels). (Brackets connecting individual groups indicate statistically significant difference. Non-parametric Mann-Whitney U test, p < 0.05).
Selective capture and viable (94% ±4%) release of live cells in thermoresponsive microchannels was reported earlier for entire channel surface without spatial control.[17] To locally release the captured cells in microfluidic channels, we placed the cold side of the thermoelectric module underneath zone 1 (Figure 1F and Figure 3). Temperature was reduced locally only in zone 1, which resulted in on-demand local release of captured cells (Figure 3A). Microscope images of channels indicated a significant decrease in number of cells remaining in zone 1 after release (Figure 3B). On the other hand, a statistically significant number (85% ± 4%, n = 4 channels, p < 0.05) of captured cells were released locally in zone 1 (Figure 3C). As reported, protein capture and release of the thermoresponsive PNIPAAm polymer occurs in less than a second.[21] In our microchips, total time elapsed for the overall process of capturing cells, rinsing unbound cells, and release of selectively captured cells was within 10 minutes.
Figure 3.
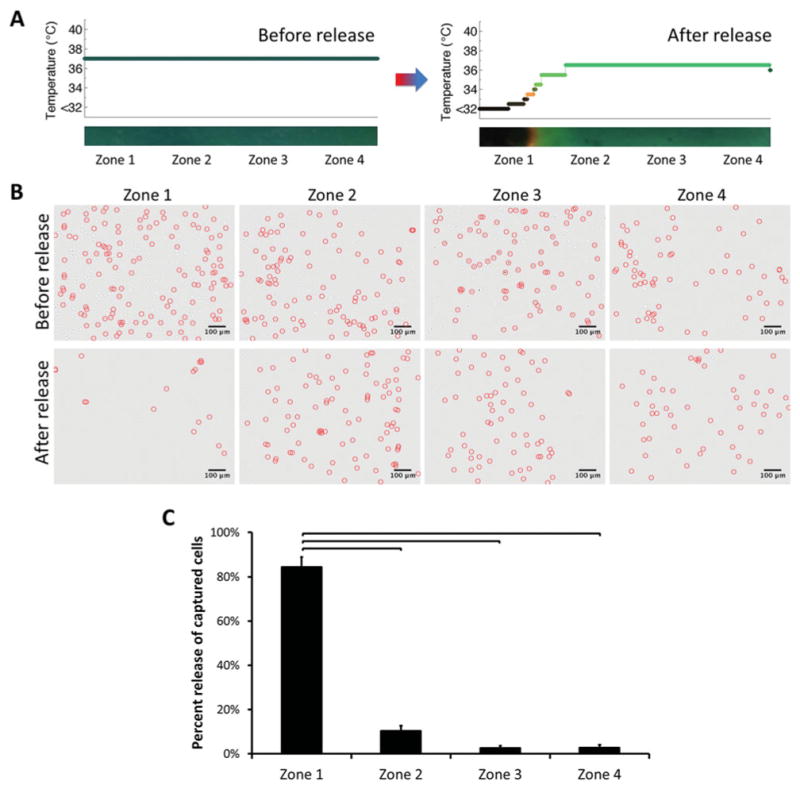
Local release of captured cells in microchannels. A) Temperature distribution in channels during capture and for subsequent local release. Temperature was controlled locally, which resulted in release of captured cells in zone 1 only. B) Typical images of cells within channels in zones 1–4, before and after release of captured cells (cells were marked with circles). The cells were released from zone 1. Images indicated a significant difference in number of cells remaining in zone 1 after release. On the other hand, the number of cells remained similar before and after release in zones 2–4. C) Quantitative analysis of cell numbers in channels in zones 1–4 before and after release. A statistically significant number (85% ± 4%, n = 4 channels) of captured cells was released locally in zone 1. Each zone corresponded to 4.3 mm of the 25 mm full channel length. (Brackets connecting individual groups indicate statistically significant difference. Non-parametric Mann-Whitney U test, p < 0.05)
Selective release of locally captured cells in microchannels was also performed (Figure 4). In a predetermined zone, cells were captured and subsequently released on demand. Temperature was controlled locally in zone 1 during both capture and release steps (Figure 4A), which resulted in capture and release of cells in this zone only (Figure 4B). A significant number (93% ± 2%, n = 4 channels, p < 0.05) of captured cells were released locally in zone 1 (Figure 4C). The number of cells remained similar before and after capture/release steps in zones 2, 3 and 4.
Figure 4.
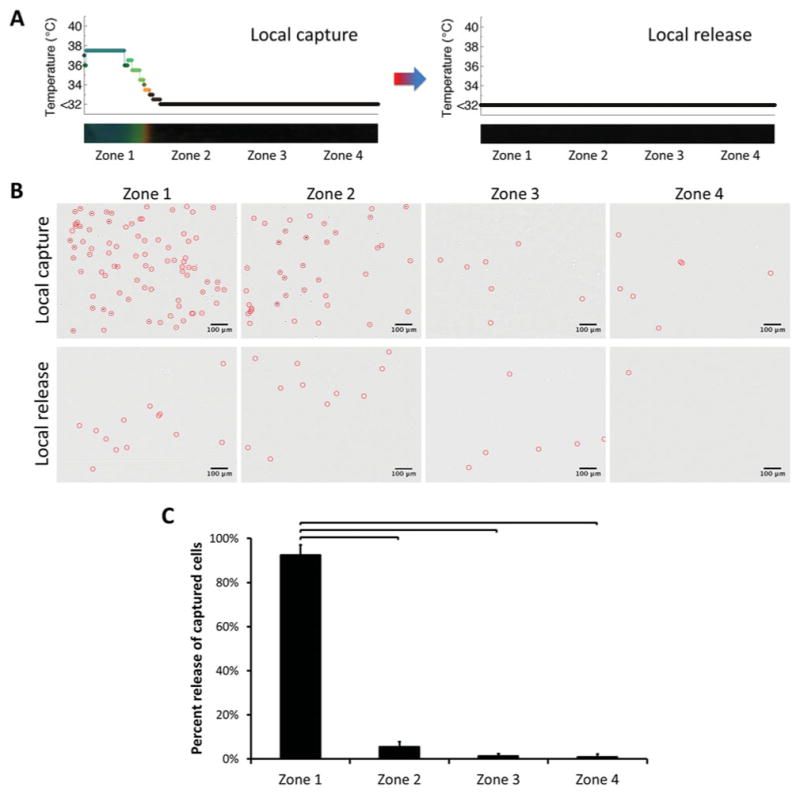
Release of locally captured cells in microchannels. A) Temperature change in channels before and after local capture/release. Temperature was controlled locally, which resulted in local capture and release of cells in zone 1 only. B) Typical images of cells in channels in zones 1–4 before and after local release of locally captured cells (cells were marked with circles). The cells were captured specifically in zone 1, followed by on-demand release from the same area. Images indicated a significant difference in number of cells in zone 1 after capture and release. As designed, the number of cells remained similar before and after capture/release steps in zones 2–4. C) Quantitative analysis of cell numbers in channels in zones 1–4 before and after release. A statistically significant number (93% ± 2%, n = 4 channels) of captured cells was released locally in zone 1, at which local temperature control was performed. Each zone corresponded to 4.3 mm of the 25 mm full channel length. (Brackets connecting individual groups indicate statistically significant difference. Non-parametric Mann-Whitney U test, p < 0.05)
To demonstrate local capture and release in other zones closer to the middle of channels, we performed temperature control in zone 2 (Figure 5). A statistically significant number (65% ± 8%, n = 4 channels, p < 0.05) of captured cells were released locally from zone 2 (Figure 5A). When local capture was performed followed by local release, a significant number (86% ± 7%, n = 4 channels) of captured cells were released locally from zone 2 (Figure 5B), as designed. These results indicated control over cell capture and release in various zones in microchannels.
Figure 5.

Local capture and release of cells in zones close to the middle of channels. Temperature was controlled locally, which resulted in local capture and release of cells in zone 2 only. A) A statistically significant number (65% ± 8%, n = 4 channels) of captured cells was released locally in zone 2, at which temperature control was performed. B) Release of locally captured cells in zone 2. A statistically significant number (86% ± 7%, n = 4 channels) of captured cells was released locally in zone 2, at which local capture and release was performed. These results indicated the capability of controlling local capture and release of cells towards the middle of the channels. Each zone corresponded to 4.3 mm of the 25 mm full channel length. (Brackets connecting individual groups indicate statistically significant difference. Non-parametric Mann-Whitney U test, p < 0.05)
A small number of cells captured in other zones (i.e., zones 2, 3 and 4 in the case of zone 1 as a control, and zones 1, 3 and 4 in the case of zone 2 as a control) may be resulting from non-specific capture due to heat diffusion to adjacent zones. The temperature control in channels is limited by the heat diffusion from the heated area, to where the thermoelectric module is coupled (Figure 1). The footprint of the target zone could be reduced by decreasing the thermoelectric module size or the contact area at the coupling interface.
To better understand mechanism of local release and potential effect of heat diffusion in channels, we developed a computational model of thermal distribution and fluid flow inside a microchannel to evaluate temperature distribution as a function of time, and to investigate coupled effects of thermal circulation and fluid flow. Computational model provided temperature distribution as a function of time on a microchannel surface. When the cold side of thermoelectric module (5 °C) was placed in contact with a microchannel (initial temperature: 37 °C), thermal gradients occurred along the microchannel surface. Here, we assumed that release of cells from the polymer film was correlated with temporal thermal gradients on the microchannel surface, based on the reported results on protein capture and release on thermoresponsive PNIPAAm polymer.[21] We evaluated temporal temperature distributions of predetermined locations on microchannel surface (Figure 6A). More frequent locations were selected near the thermoelectric module where the thermal gradients were steeper. To measure the rate of temperature changes at these locations, we calculated variances,[22] , of these temporal distributions,
Figure 6.

Theoretical understanding of local cell release in microchannels and comparison to experimental results. A) Schematic for 2D computational modeling of fluid flow and heat transfer inside a microchannel. Thermoelectric module was placed onto locations: x1 = 1 mm for zone 1, and x1 = 6 mm for zone 2. Top surface was 3.5 mm thick polymethyl-methacrylate (PMMA) and bottom surface was 0.65 mm thick polystyrene. To model heat loss from top and bottom surfaces, thermal conductivity of PMMA was set as, k = 0.19 W/m.K, and thermal conductivity of polystyrene was set as, k = 0.08 W/m.K. Channel height is H = 80 μm, and length is L = 25 mm. BC stands for boundary condition. Width of inlet flow was w2 = 250 μm, and width of thermoelectric module was w1 = 4.3 mm. Steady-state surface temperature (°C) distribution as a function of channel location (mm) were plotted in the presence of thermoelectric module at: B) x1 = 1 mm (zone 1), and C) x1 = 6 mm (zone 2). Results from local release experiments were compared to the results from computational model for (D) zone 1, and E) zone 2.
| (1) |
where, Ti, j is temperature at ith time step, and jth spatial location, N is the number of time steps (= 100), μj is temporal mean of temperature distribution at jth point, . Then, variance values at predetermined channel locations, , were integrated and their area weighted means were evaluated for different zones (Figure 6B-E), which provided percent occurrence of thermal gradients. Here, we demonstrated that local cell release profiles agreed with the variances of temporal temperature distributions occurring on a microchannel surface. As only zone 1 was cooled down, 89% of thermal gradients occurred in zone 1 and experimentally 84% of cells were released from this zone (Figure 6B, D). This result can be attributed to coupling of thermal circulation and fluid flow (Figure 6B). However, the same effect (i.e., domination of thermal gradients in a single zone) was not observed, when only zone 2 was cooled down (Figure 6C). This result can be explained by the lower temperature of inlet flow (25 °C) compared to initial temperature of microchannel (37 °C) yielding thermal gradients near zone 1. High level of local release in zone 1 (Figure 6D) can be explained by the combined effects of thermal circulation and fluid flow, as the model suggested. On the other hand, a more evenly distributed cell release profile was observed between zone 1 and 2 (Figure 6E), when only zone 2 was cooled down.
Isolation and enrichment of cells from heterogeneous cell suspensions have been demonstrated in earlier studies with microfluidic systems.[23] We have previously shown that thermoresponsive microfluidics achieve up to 25 times enrichment of CD4+ T lymphocytes (enrichment from 3.6% to greater than 90% of mononuclear blood cells), and up to 180 times enrichment of CD34+ endothelial progenitor cells (enrichment from 0.5% to greater than 90% of mononuclear blood cells) from blood.[17] Here, we have demonstrated, for the first time, local capture and on-demand local release of cells in microfluidic channels. We have demonstrated the ability to release a small number of cells at a time, for downstream processing, as opposed to releasing all cells in a channel. On the other hand, as microfluidic cell capture methods suffer from non-specific binding events, the ability to selectively release either undesired or desired cells in channels might improve the specificity of isolation, especially for subsequent applications including genomic/proteomic analyses,[24] and biomarker discovery.[25]
In this study, we demonstrated that stimuli responsive, smart interface materials can be integrated with microfluidic technologies, enabling a broad range of biological and clinical applications by controlling the material and cell interactions. We have demonstrated for the first time controlled on-demand local capture and local release of selected cell populations in a microfluidic system. Microfluidic local capture and release of cells have broad applications in multiple areas, including rare cell isolation, stem cell purification, regenerative medicine, downstream proteomic/ genomic analyses, clonal studies, biomarker discovery, cancer research, and multivariate diagnostics.
Experimental Section
Design and Fabrication of Microchips for Local Capture and Release of Cells from whole Blood
In each microfluidic chip, three channels with PNIPAAm treatment on a layered polystyrene lower surface (dimensions: 25 mm × 4 mm × 80 μm) were employed, as detailed in our previous work.[17] Briefly, simple microfabrication steps were employed to assemble commercially available materials to form a microfluidic chip: 3.5 mm thick PMMA sheets (McMaster Carr, Santa Fe Springs, CA), 80 μm double sided adhesive (DSA, iTapeStore, Scotch Plains, NJ), and PNIPAAm polymer layer (Upcell, Nalge Nunc International, Rochester, NY). Using a laser-cutter (Versalaser, Universal Laser Systems Inc., Scottsdale, AZ), channels were cut into the DSA and inlet and outlet ports were cut into the PMMA. Tygon tubing with external diameter of 0.762 mm (Cole-Parmer, Vernon Hills, IL) was inserted into each outlet, and epoxy glue (5-Minute Epoxy, Devcon, Danvers, MA) was used to seal the final assembled microfluidic chips. The chips were designed to be single use and disposable due to inexpensive material cost and simplicity of fabrication. In addition, cross-contamination between samples could be an issue with reusable chips. Therefore, disposable chip design presents a feasible and practical solution. On the other hand, it would be possible to reuse these channels by repeating the surface chemistry, since the thermoresponsive behavior of the interface material has been shown to be reversible and repeatable.[22]
Local Temperature Control in Microchannels
The middle channel in each microchip was stained with temperature sensitive liquid crystal dye (Edmund Scientific, Tonawanda, NY) to monitor and quantify the channel temperature distribution throughout the experiment. For local capture, we controlled temperature in a 17.2 mm2 area (4 mm width × 4.3 mm length), of the full 100 mm2 microfluidic channel surface area, using thermoelectric modules (TE-7-0.6-1.0, Peltier Module, TE Technology, Traverse City, MI) placed below each channel (Figure 1A-F). Each thermoelectric module had dimensions of 4.3 mm × 4.3 mm × 2.75 mm, and featured a heated side and a cooled side. The temperature of two sides can be adjusted by changing the current flowing through the module terminals. To keep warm side of the modules at 37 °C, we applied 0.3 V, and 610 mA current to the terminals (Figure 1G). In this state, a 32 °C temperature difference between the warm and the cold sides of the module was achieved, where the temperature of the warm side was 37 °C and the temperature of the cold side was 5 °C. Control microchips were placed on a temperature controlled heating pad (Omega Engineering Inc., Stamford, CT) maintained at 37 °C to uniformly heat the whole channel surface to capture cells in all zones in microchannels.
Quantification of Temperature Distribution in Microchannels
Microchip images were taken using a Sony Alpha 700 digital camera and were recorded in “.RAW” format to quantify temperature distribution (Figure 1G and H). Black and white calibration targets were used to adjust the dynamic range in each frame to capture colors consistently. Reflectance spectra of the thermosensitive dye at temperatures between 32–41 °C were quantified using a USB2000 spectrometer (Ocean Optics, Dunedin, FL) (Figure 1I). For each image, a rectangular mask was made outlining the channel filled with the thermoresponsive dye to select the area where the color change is quantified (Figure 1G). An average value was computed for each column in the part of the image within the rectangular mask. This step was done in red (R), green (G) and blue (B) channels separately. These average values were combined to obtain a “line image” (1 row by M columns for each of the red, green and blue layers) showing the mean colors across the rectangular region. For the initial baseline calibration, the microchip was imaged at predetermined temperatures (from 32 °C to 41 °C, in 0.2 °C increments) and RGB values for each was quantified (Figure 1I). For each experimental image, the RGB value of each pixel in the “line image” is compared to the table of baseline RGB values and associated with the bin to which it is closest. Each RGB triplet approximates the colors shown by the dye at certain temperatures. Local temperature control was performed in zone 1 (Figures 2–4) and zone 2 (Figure 5) to capture and release cells within these regions.
Functionalization of Microchannels for CD4+ Cell Capture
We fabricated microfluidic channels and we applied biotinylated antibody based surface chemistry (Figure 1C) at 37 °C.[17] Briefly, channels were first washed with 30 μL Phosphate Buffered Saline (PBS, Mediatech, Herndon, VA). Then, 100 μg/mL NeutrAvidin (Thermo Fisher Scientific Inc., Rockford, IL) in PBS was injected into the channels. The channels were kept in dark during an hour incubation. Next, surface passivation was performed with 1% Albumin from Bovine Serum (BSA) solution (Sigma- Aldrich Co., St. Louis, MO) in PBS for an hour. Then, the capture antibody (biotinylated Anti-CD4 antibody) was injected into the channels and incubated for 30 minutes.
Local Capture of CD4+ Cells from Human Blood in Microfluidic Channels
Discarded human whole blood samples were obtained from Brigham and Women’s Hospital (Boston, MA) daily, under the approval of institutional review board. Blood cell concentrations were verified using a hematology system (Drew-3, Drew Scientific Group, Dallas, TX) before each experiment. At 37 °C, 15 μL of unprocessed human whole blood sample was injected into the control and local capture channels with a 100 μL manual pipette, which was shown to result in flow rates of 45 ± 3 μl/min for blood and 63 ± 3 μl/min for PBS. Next, PBS was gently pipetted through channels to wash away unbound cells. Then, 100 μL red blood cell lysis solution (BD Biosciences, San Jose, CA) in HyPure Cell Culture Grade Water (HyClone Laboratories Inc., Logan, UT) was injected into the channels and incubated for 5 minutes to lyse the remaining erythrocytes. Channels were washed with PBS before release experiments or microscope imaging.
Local Release of Captured Cells in Microchannels
To release the captured CD4+ cells locally, thermoelectric modules were applied to the chip surface for 10 minutes to cool specific areas of the microfluidic channels, as shown in Figure 1F. Then, each channel was rinsed with PBS to wash away released cells and microscope imaging was performed on the channels. The released cells were collected at the channel outlet, which was reported to result in released cell viability of 94% ± 4%.[17]
Capture Specificity and Imaging of Captured Cells in Microchannels
To determine capture specificity, cells in microchannels were stained with anti-Human CD4 antibody conjugated with Alexa Fluor 488 (eBioscience Inc., San Diego, CA) and imaged (Carl Zeiss Observer D1 Model Axio Inverted Microscope) at 4 predetermined zones (zones 1–4) in bright field and in fluorescent modes. CD4 stained cell counts were divided by the cell counts in brightfield mode to determine capture specificity (Figure 1D). Cells were marked with a circle and automatically identified using matched filtering implemented in MATLAB (Mathworks, Natick MA). The intensity of each image was reduced by taking the fourth root of its value at every pixel. The original image was convolved with a template, which represented the circular structure of the cells (a circle of 8-pixel radius surrounded by a light ring approximately 4 pixels thick). The result of this convolution was a grayscale map which was used to yield the exact locations of cells. These locations were then overlaid onto the reduced-intensity image, and a circle was placed at each location a cell was found.
Statistical Analysis
Data obtained in this study were reported as mean ± standard error of the mean. Experimental results were analyzed using non-parametric Mann-Whitney U test with Bonferroni correction for multiple comparisons. Statistical significance was set at 95% confidence level for all tests (p < 0.05). All results reflect experimental data obtained from at least 4 channels.
Computational Modeling of Thermal Distribution and Fluid Flow
Flow inside microfluidic chip was modeled by Navier-Stokes equations[26] (Figure 6A):
| (2) |
| (3) |
| (4) |
where, u is velocity in x direction, υ is velocity in γ direction, ρ is density, μ is viscosity, t is time, p is pressure. The governing equation for thermal distribution along the channel is:[27]
| (5) |
where, T is temperature, α is thermal diffusivity (= k/ρcp), k is thermal conductivity, and cp is specific heat capacity. For upper and lower surfaces of the chip, we included thermal conductivity of PMMA (k = 0.19 W/m.K) and polystyrene (k = 0.08 W/m.K), respectively.
Acknowledgments
We would like to acknowledge the W.H. Coulter Foundation Young Investigator Award, NIH R01 A1081534, R21 AI087107, R21 HL112114, and R21 HL095960. Dr. Demirci acknowledges that this material is based in part upon work supported by the National Science Foundation under NSF CAREER Award Number 1150733. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. This work was supported by the Center for Integration of Medicine and Innovative Technology (CIMIT) under U.S. Army Medical Research Acquisition Activity Cooperative Agreements DAMD17-02-2-0006, W81XWH-07-2-0011, and W81XWH-09-2-0001. This work was also made possible by a research grant that was awarded and administered by the U.S. Army Medical Research and Materiel Command (USAMRMC) and the Telemedicine and Advanced Technology Research Center (TATRC), at Fort Detrick, MD.
Contributor Information
Dr. Umut Atakan Gurkan, Postdoctoral Research Fellow in Medicine, Harvard Medical School, Brigham and Women’s Hospital, Harvard-MIT Health Sciences & Technology, 65 Landsdowne St. PRB 252, Cambridge, MA 02139, USA
Dr. Savas Tasoglu, Postdoctoral Research Fellow in Medicine, Harvard Medical School, Brigham and Women’s Hospital, Harvard-MIT Health Sciences & Technology, 65 Landsdowne St. PRB 252, Cambridge, MA 02139, USA
Derya Akkaynak, Massachusetts Institute of Technology (MIT) Department of Mechanical Engineering, Woods Hole Oceanographic Institution (WHOI) Joint Program in Oceanography, Applied Ocean Science and Engineering, Cambridge, MA, USA.
Oguzhan Avci, Harvard Medical School, Brigham and Women’s Hospital, 65 Landsdowne St. PRB 252, Cambridge, MA 02139, USA.
Sebnem Unluisler, Harvard Medical School, Brigham and Women’s Hospital, 65 Landsdowne St. PRB 252, Cambridge, MA 02139, USA.
Serli Canikyan, Harvard Medical School, Brigham and Women’s Hospital, 65 Landsdowne St. PRB 252, Cambridge, MA 02139, USA.
Noah MacCallum, Harvard Medical School, Brigham and Women’s Hospital, 65 Landsdowne St. PRB 252, Cambridge, MA 02139, USA.
Dr. Utkan Demirci, Email: udemirci@rics.bwh.harvard.edu, Assistant Professor, Harvard Medical School, Brigham and Women’s Hospital, Harvard-MIT Health Sciences & Technology, 65 Landsdowne St. PRB 252, Cambridge, MA 02139, USA
References
- 1.Tanaka Y, Adams DH, Hubscher S, Hirano H, Siebenlist U, Shaw S. Nature. 1993;361:79. doi: 10.1038/361079a0. [DOI] [PubMed] [Google Scholar]
- 2.Rabelink TJ, de Boer HC, van Zonneveld AJ. Nat Rev Nephrol. 2010;6:404. doi: 10.1038/nrneph.2010.65. [DOI] [PubMed] [Google Scholar]
- 3.Lee J, Ishihara A, Oxford G, Johnson B, Jacobson K. Nature. 1999;400:382. doi: 10.1038/22578. [DOI] [PubMed] [Google Scholar]
- 4.Evans EA, Calderwood DA. Science. 2007;316:1148. doi: 10.1126/science.1137592. [DOI] [PubMed] [Google Scholar]
- 5.Moon S, Keles HO, Ozcan A, Khademhosseini A, Haeggstrom E, Kuritzkes D, Demirci U. Biosens Bioelectron. 2009;24:3208. doi: 10.1016/j.bios.2009.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang S, Xu F, Demirci U. Biotechnol Advances. 2010;28:770. doi: 10.1016/j.biotechadv.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gurkan UA, Moon S, Geckil H, Xu F, Wang S, Lu TJ, Demirci U. Biotechnol J. 2011;6:138. doi: 10.1002/biot.201000427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alyassin MA, Moon S, Keles HO, Manzur F, Lin RL, Haeggstrom E, Kuritzkes DR, Demirci U. Lab Chip. 2009;9:3364. doi: 10.1039/b911882a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moon S, Gurkan UA, Blander J, Fawzi WW, Aboud S, Mugusi F, Kuritzkes DR, Demirci U. PloS One. 2011;6:1409. doi: 10.1371/journal.pone.0021409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Nature. 2007;450:1235. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang SQ, Esfahani M, Gurkan UA, Inci F, Kuritzkes DR, Demirci U. Lab on a Chip. 2012;12:1508. doi: 10.1039/c2lc20706k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim YG, Moon S, Kuritzkes DR, Demirci U. Biosens Bioelectron. 2009;25:253. doi: 10.1016/j.bios.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang SQ, Inci F, Chaunzwa TL, Ramanujam A, Vasudevan A, Subramanian S, Ip AC, Sridharan B, Gurkan UA, Demirci U. Int J Nanomed. 2012 doi: 10.2147/IJN.S29629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kotz KT, Xiao W, Miller-Graziano C, Qian WJ, Russom A, Warner EA, Moldawer LL, De A, Bankey PE, Petritis BO, Camp DG, Rosenbach AE, Goverman J, Fagan SP, Brownstein BH, Irimia D, Xu W, Wilhelmy J, Mindrinos MN, Smith RD, Davis RW, Tompkins RG, Toner M. Nat Med. 2010;16:1042. doi: 10.1038/nm.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sethu P, Moldawer LL, Mindrinos MN, Scumpia PO, Tannahill CL, Wilhelmy J, Efron PA, Brownstein BH, Tompkins RG, Toner M. Anal Chem. 2006;78:5453. doi: 10.1021/ac060140c. [DOI] [PubMed] [Google Scholar]
- 16.Rosenbach AE, Koria P, Goverman J, Kotz KT, Gupta A, Yu M, Fagan SP, Irimia D, Tompkins RG. Clin Translat Sci. 2011;4:63. doi: 10.1111/j.1752-8062.2010.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gurkan UA, Anand T, Tas H, Elkan D, Akay A, Keles HO, Demirci U. Lab Chip. 2011;11:3979. doi: 10.1039/c1lc20487d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plouffe BD, Kniazeva T, Mayer JE, Murthy SK, Sales VL. FASEB J. 2009;23:3309. doi: 10.1096/fj.09-130260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeo LY, Chang HC, Chan PPY, Friend JR. Small. 2011;7:12. doi: 10.1002/smll.201000946. [DOI] [PubMed] [Google Scholar]
- 20.Sun T, Qing G. Advanced Materials. 2011;23:H57. doi: 10.1002/adma.201004326. [DOI] [PubMed] [Google Scholar]
- 21.Huber DL, Manginell RP, Samara MA, Kim BI, Bunker BC. Science. 2003;301:352. doi: 10.1126/science.1080759. [DOI] [PubMed] [Google Scholar]
- 22.Hardle W, Simar L. Applied Multivariate Statistical Analysis. Springer-Verlag; Berlin/Heidelberg: 2003. [Google Scholar]
- 23.Green JV, Murthy SK. Lab Chip. 2009;9 doi: 10.1039/b906768j. [DOI] [PubMed] [Google Scholar]
- 24.Lion N, Rohner TC, Dayon L, Arnaud IL, Damoc E, Youhnovski N, Wu ZY, Roussel C, Josserand J, Jensen H, Rossier JS, Przybylski M, Girault HH. Electrophoresis. 2003;24:3533. doi: 10.1002/elps.200305629. [DOI] [PubMed] [Google Scholar]
- 25.Tarasow TM, Penny L, Patwardhan A, Hamren S, McKenna MP, Urdea MS. Bioanalysis. 2011;3:2233. doi: 10.4155/bio.11.224. [DOI] [PubMed] [Google Scholar]
- 26.Batchelor GK. An Introduction to Fluid Dynamics. Cambridge University Press; Cambridge UK: 2000. [Google Scholar]
- 27.Incropera FP, DeWitt DP. Fundamentals of Heat and Mass Transfer. John Wiley & Sons; New York: 2007. [Google Scholar]