

Abstract
There is a growing interest in engineering proteins whose function can be controlled with the spatial and temporal precision of light. Here, we present a novel example of a functional light-triggered switch in the Ca-dependent cell–cell adhesion protein E-cadherin, created using a mechanism-based design strategy. We report an 18-fold change in apparent Ca2+ binding affinity upon illumination. Our results include a detailed examination of functional switching via linked changes in Ca2+ binding and cadherin dimerization. This design opens avenues toward controllable tools that could be applied to many long-standing questions about cadherin’s biological function in cell–cell adhesion and downstream signaling.
There has been considerable interest in light-based control of biological systems,1 and successful applications include light-modulation of neuronal ion channels,2 light-switchable signaling proteins,5 and light-controlled protein targeting.6 Light-based methods offer titratable, precise spatial and temporal regulation that has been demonstrated in vitro,7 in cell culture,6,8 and in whole animals.9 Most examples of light-based control fall into one of two categories: (a) those that are genetically encoded using a recombinantly produced protein borrowed from nature,6 and (b) those created via targeted insertion of amino acids into a protein sequence and subsequent reaction with them of an exogenously introduced photoisomerizable small molecule, typically azobenzene based.10 Azobenzene and related molecules undergo a reversible cis–trans isomerization when exposed to specific wavelengths of light, and this change in molecular shape can be coupled to changes in protein function. While in (a) the functional design is already provided naturally, one is limited both by the function (e.g., modulation of protein–protein binding, tuning fluorescence intensity) already encoded by the natural gene and by the requirement to fuse the natural protein with the protein to be modulated. In contrast, the designs in (b) allow many types of functional modulation, such as changes in agonist binding, protein–protein binding, and protein folding. In this work, we used a new strategy where changes in protein–ion affinity couple to protein dimerization, in the cell–cell adhesion protein cadherin (Figure 1A).
Figure 1.
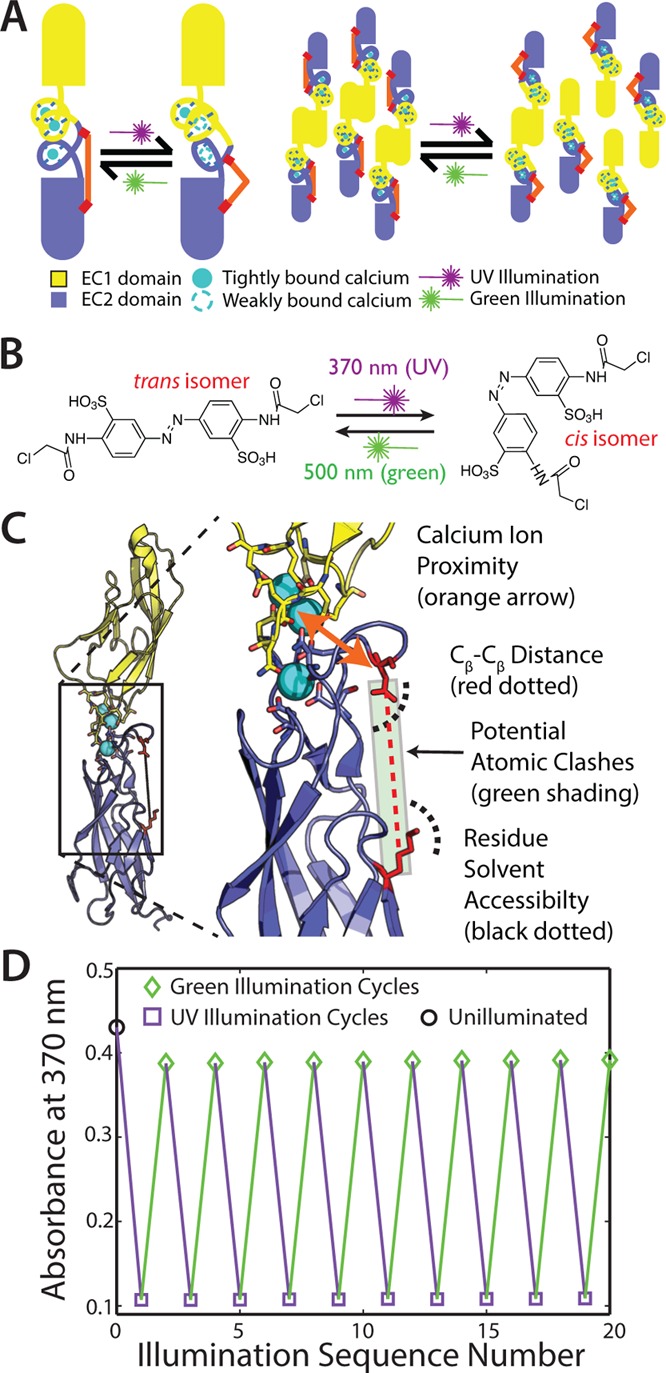
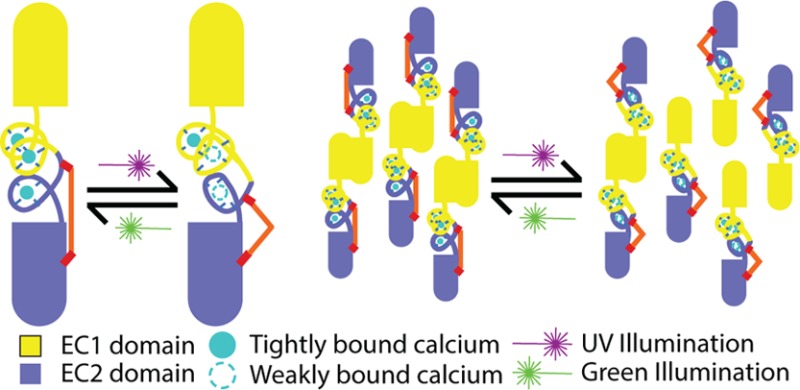
(A) A cartoon showing the basis of our design. As designed, our photoswitch reduces Ca2+ binding affinity, which, in turn, reduces homodimer affinity. (B) BSBCA undergoes a reversible cis/trans isomerization when illuminated with specific wavelengths of light. (C) EC12 structure showing the region targeted for photoswitchability. Labels indicate the design considerations. (D) Photoswitchability and reversibility measured by absorbance after many cycles of illumination of X-EC12.
Cadherins are a key family of Ca-dependent cell–cell adhesion proteins and are divided into several subtypes, including the most commonly studied subtype, the classical cadherins. Classical cadherins, which include E-, N-, P-, R-, and C-cadherin,11 are composed of an intracellular domain, a transmembrane helix, and five, repeated, extracellular domains labeled EC1 (N-terminal, membrane distal) to EC5 (C-terminal, membrane proximal), along with three Ca2+ binding sites present in the loops at each extracellular domain boundary.12,13 Calcium binding is required for cadherin function, as depletion of Ca2+ disrupts cadherin-mediated cell adhesion;14 the presence of Ca2+ is suggested to rigidify the cadherin structure, allowing it to multimerize.15 Knockdowns of cadherin significantly slow cell–cell adhesion,16 and in a classic experiment, cadherin-free, nonadherent cells transfected with cadherin acquire morphological similarities to naturally adherent cells.18
Our approach to creating a light-switchable cadherin aimed to modulate its Ca2+ binding affinity. Because Ca2+ binding is essential for cadherin multimerization, we reasoned that reversibly changing Ca2+ binding affinity would be an effective way to also modulate cadherin adhesive function (Figure 1A). We designed cysteine residues into the protein to serve as conjugation sites for an azobenzene-based photoisomerizable chromophore, BSBCA (Figure 1B). BSBCA has been used in previous applications,7,20 demonstrating reversible switching between the cis and trans states when exposed to 370 nm (near UV) and 550 nm (green) light, respectively.7 Our strategy involved conjugating both ends of the chromophore to the Ca-binding loops between cadherin domains EC1 and EC2, as these Ca2+ sites have previously been shown to be most critical for function.13 In addition, because the Ca2+ binding sites are located in loop regions and bind Ca2+ with relatively weak affinities near 20 μM,22 we reasoned it would be easier to induce conformational changes affecting Ca2+ binding there than in more rigid secondary structural elements or well-packed core regions of the protein.
Because BSBCA spontaneously cross-links cysteine residues,23 the design challenge presented here can be generalized as the problem of finding the best pair of residues to mutate to cysteine. In practice, however, an enormous number of pairs are possible, the overwhelming majority of which are likely to be nonfunctional. We took a sequential and computational approach to identifying likely functional pairs (Figure S1 and Supporting Information [SI]). First, we used the program Rosetta24 to computationally mutate all residues in four representative E-cadherin structures (PDB identifiers: 1FF5, 1EDH, 2O72, and 1Q1P) to alanine (the simplest mutation) and then calculated the predicted change in fold stability using a protocol we developed previously.25,26 Residues with predicted destabilization >1 kcal/mol were not considered, as mutations to these residues were presumed to be disruptive. Next, we narrowed the pairs to those that would be geometrically compatible with the small molecule. We calculated pairwise Cβ–Cβ distances between the residues remaining using the 1FF5 structure and kept those pairs that fell in the range 17–20 Å (appropriate for the BSBCA trans isomer). Finally, the remaining pairs were ranked for an additional set of structural and geometric constraints (Figure 1C and SI). Eleven high-ranking pairs (Table S1) were cloned, expressed, and tested for conjugability (Figure S2), photoswitchability, and functionality (SI). One pair, K129C/D138C (Figure 1C, red residues) showed the best switchability and functionality, and was further characterized in detail.
We focused on, and expressed, the first two domains of E-cadherin (EC12), because they contain the homodimeric binding interfaces27 and are the specificity determining domains,28 making them most principally responsible for cadherin’s function. Additionally, the shortened EC12 construct can be readily produced in high yields in E. coli. EC12 contains a single native cysteine residue, which we mutated to alanine (C9A), previously shown not to affect cadherin function.29
We first sought to show that K129C/D138C conjugated with trans BSBCA (termed X-EC12) could undergo isomerization to cis. Unconjugated BSBCA in the trans state has an absorbance peak near 370 nm that decreases when illuminated at this wavelength, resulting in a population that is 80–90% cis; subsequent illumination at 500–550 nm will reverse the isomerization and produce a population that is >90% trans. Illuminating our conjugated protein (X-EC12) at 365 nm with a hand-held LED showed the characteristic reduction in absorbance of the trans state. The reverse isomerization (pan-visual illumination, including 500–550 nm bands) also behaved as expected, leading to a reappearance of the absorbance band of the trans state. We illuminated X-EC12 for 10 complete UV-green illumination cycles without any apparent loss of absorbance or switchability (Figure 1D); the switchability was also titratable via shorter illumination times (Figure S3).
We next tested whether isomerization changes Ca2+ binding affinity. To do so, we used a previously described mass spectrometry based assay22 to directly measure the Ca2+ binding affinity of WT C9A as well as trans and cis X-EC12 (Figures 2A and S4; these assays used a cadherin concentration of 2 μM, significantly below a previously measured homodimeric Kd of WT cadherin (98.6 ± 15.5 μM),27 to avoid potential complications due to cadherin dimerization). If isomerization alters Ca2+ binding, the cis X-EC12 should have weaker affinity than trans. In addition, because EC12 binds three Ca2+ ions, any of which could be interfered with, a decrease in apparent cooperativity as measured by the Hill coefficient (Nh) would be expected. WT C9A cadherin specifically bound three Ca2+ ions with a dissociation constant Kd = 28.5 ± 1.9 μM (throughout the text, errors are the boundaries of a 95% confidence interval unless otherwise indicated) and extensive cooperativity with Nh = 2.85 ± 0.47, close to previously reported numbers22 of Kd = 20 ± 0.7 μM and Nh = 2.6 ± 0.2. Trans X-EC12 showed 2-fold weaker affinity and less cooperativity, with Kd = 55.2 ± 5.8 μM and Nh= 1.80 ± 0.35, but also bound three Ca2+ ions. In contrast, Ca2+ binding to cis X-EC12 was dominated by nonspecific binding. By using quadruple and higher Ca-bound states from trans X-EC12 (Figure S5) as a reference for nonspecific binding, we subtracted the estimated contribution of nonspecific Ca2+ binding from the measured average Ca2+ occupancy for cis X-EC12 (SI). The resulting line shows significantly reduced binding compared to trans. (A quantitative fit was not possible due to required Ca2+ concentrations being higher than the dynamic range of the assay.)
Figure 2.
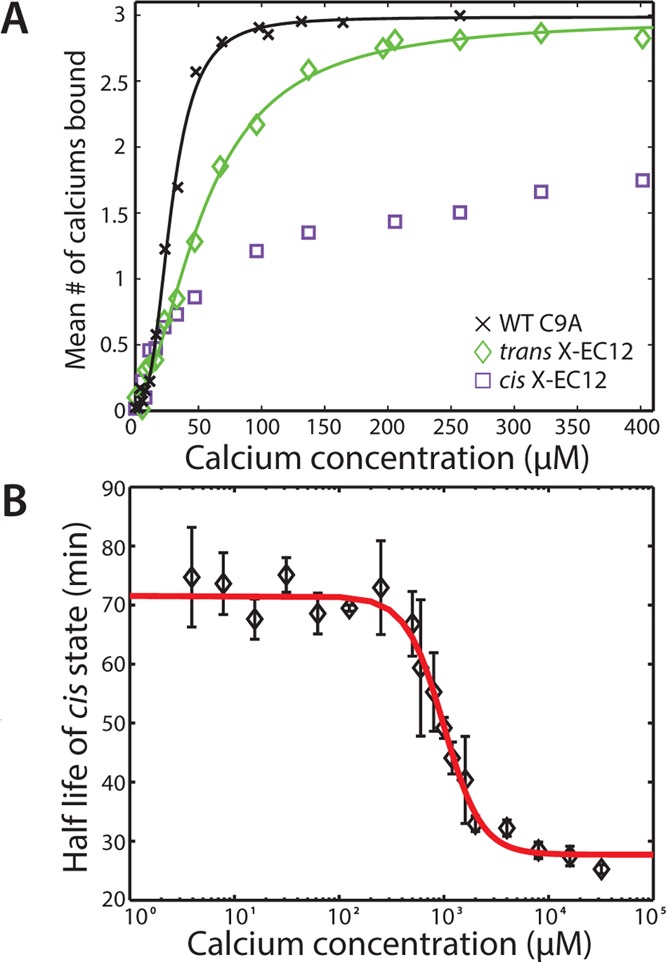
Characterization of photoswitchable Ca2+ binding affinity. (A) Ca2+ binding as monitored by mass spectrometry. While WT and trans X-EC12 bound three Ca2+ ions specifically, cis X-EC12 showed considerably weaker, predominantly nonspecific binding (Figure S4 and SI). Fits are based on a model of a single class of binding site for a maximum of three specifically bound Ca2+ ions. (B) The half-life of the cis state as a function of Ca2+ concentration, as measured by absorbance. Error bars are +1 SD from three independent experiments.
To more directly measure the Ca2+ binding of cis X-EC12, we turned to a different assay that determined the cis half-life as a function of Ca2+ concentration. One general caveat inherant to azobenzene-based strategies is that switching to the cis state is generally incomplete; i.e., the cis state always contains a minor trans population.10,23 However, the entirety of any change observed in half-life experiments is due only to the cis subpopulation, allowing measurement of pure cis properties unaffected by the small fraction that remains trans. Therefore, if chromophore isomerization significantly affects Ca2+ binding in our conjugated constructs, with stronger binding of Ca2+ to trans X-EC12, by thermodynamic coupling we would expect to see a change in the cis X-EC12 half-life with Ca2+ (Figure 2B). The cis state is thermodynamically unstable, and cis BSBCA relaxes back to the stable trans state in the dark with a half-life of ∼20 min at 25 °C,23 although conjugation to proteins can alter chromophore half-lives.7,31 By observing the increase in absorbance at 370 nm during relaxation of our conjugated constructs back to trans, one can compute the half-life of the process (Figures S6 and S7; SI; these assays used a protein concentration of 12 μM to minimize potential changes in half-life due to protein dimerization). The half-life should decrease with increasing Ca2+ concentration as trans X-EC12 becomes stabilized by Ca2+ binding. As expected, we observed a half-life decrease from ∼72 to 28 min, with an EC50 of 996 ± 135 μM Ca2+. This represents a nearly 18-fold change in apparent Ca2+ binding affinity from the 55 μM for trans X-EC12 (mass spectrometry analysis, Figure 2A).
We also observed a cooperative transition in half-life duration, with a measured Nh of 2.4 ± 0.74. In showing interdependence between isomerization and Ca2+ binding, these results indicate that, as expected, isomerization of the chromophore significantly weakens Ca2+ binding. In addition, the observed cooperative nature indicates multiple Ca2+ ions are binding simultaneously during the transition from cis to trans, hinting that the cis state likely weakens multiple binding sites.
After successfully demonstrating photoswitchable Ca2+ binding in our engineered cadherin, we next asked whether the change in Ca2+ binding affinity also results in the expected change in protein binding activity. We used surface plasmon resonance (SPR) to measure protein homodimerization as a function of Ca2+ concentration (Figures 3, S8, and S9; SI). In this assay, similar to that of Harrison et al.,27 biotinylated WT C9A cadherin was immobilized to the SPR chip and WT C9A, trans or cis X-EC12 were flowed over it. Direct measurements of both Ca2+ affinity and homodimeric protein affinity in SPR are difficult due to solution homodimers competing with those on the surface, reducing the effective protein concentration; to minimize solution homodimerization, we used a protein concentration (40 μM) below the Kd for homodimerization of WT EC12 cadherin.27 Additionally, cis measurements are of mixed populations due to the inability of reaching full conversion to the cis state and some thermal relaxation to trans during the experiment, limiting the observable fold change in affinity (SI). We observed a Ca2+ binding EC50 for WT C9A cadherin, as measured by a single Hill fit, of 72.0 μM (with mean fit values and ±2 SD error, as measured by a bootstrapping analysis of the data, of 71.2 ± 14 μM; see SI and Figures S10, S11) and Nh = 2.24 (2.45 ± 1.7). In comparison, trans X-EC12 has an EC50 of 156 μM (170 ± 33 μM), with Nh = 1.38 (1.28 ± 0.28). These EC50 values are higher than those measured in the mass spectrometry assay (Figure 2A), which is due to cadherin binding multiple Ca2+ ions to function, causing any measured EC50 to necessarily be, at a minimum, a multiple of the protein concentration used, which here was 40 μM. Strikingly, cis X-EC12 showed substantially weakened binding (Figure 3A), with EC50 = 619 μM (611 ± 180 μM) and Nh = 0.76 (0.77 ± 0.15), demonstrating a nearly 4-fold change in Ca2+ affinity under these conditions. (Note: nonspecific protein binding to the SPR chip appeared at Ca2+ concentrations >2 mM for WT C9A and trans X-EC12; see SI.) The change in protein–protein binding was also reversible as measured over multiple illumination cycles with 40 μM protein and 1 mM Ca2+ (Figure 3B).
Figure 3.
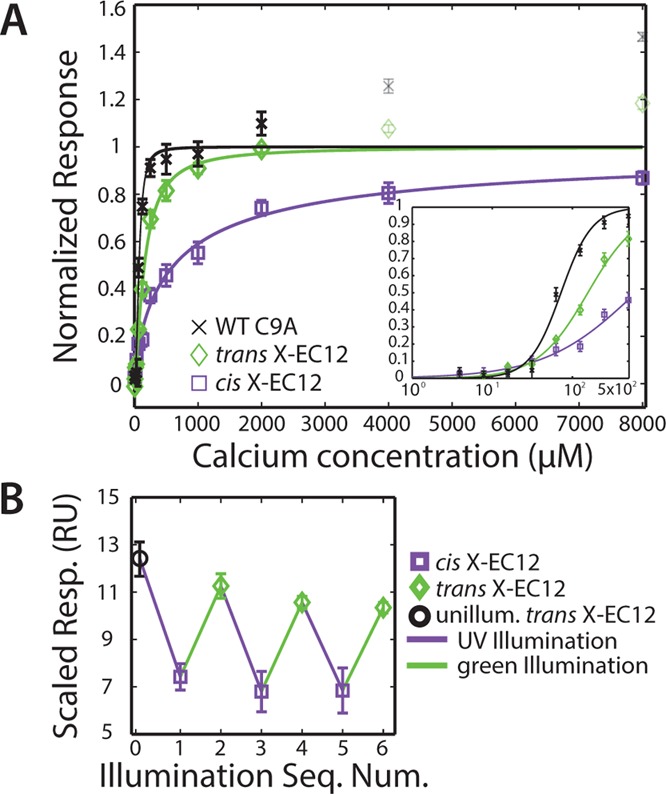
Characterization of photoswitch homodimeric binding. (A) Homodimeric binding monitored in SPR as a function of Ca2+ concentration. The data were fit to a Hill equation. Faded points contain significant nonspecific binding and were not used in the fits. Responses between flow cells were scaled to minimize a least-squares difference, and then mean values were normalized such that the fit value at [Ca2+] = ∞ was 1.0 (SI). Error bars are ±1 SD of the three active flow cells in the instrument after scaling and normalization. Inset shows fits at low Ca2+ concentrations. (B) Homodimeric binding monitored in SPR at 1 mM Ca2+, after repeated illumination cycles. Responses between flow cells were scaled to minimize a least-squares difference. Error bars are ±1 SD of the three active flow cells in the instrument after scaling.
An alternative explanation for the observed decrease in the SPR signal upon isomerization to the cis state could be an increase in cis homodimerization in solution, reducing the effective concentration of X-EC12 cadherin monomers available to bind to the WT cadherin immobilized on the chip. To exclude that possibility, we analyzed X-EC12 cadherin homodimerization via gel filtration. The observed decrease in the dimer/monomer ratio after UV illumination additionally confirms the expected weaker cis homodimerization upon illumination (Figure S12).
Questions remain about the structure of the functional cadherin multimers, including evidence that cadherin forms strand-swapped dimers.27,32,33 While we cannot directly determine the structure of the interacting species formed in our SPR experiments, each set of SPR traces for a given cadherin variant can be fit to a single off rate returning to baseline levels, even at higher Ca2+ concentrations (Figure S13). This behavior is consistent with a single dimer type formed for each assayed cadherin variant.
Taken together, our results demonstrate the successful design of a reversibly photoswitchable cadherin. When illuminated with light, its Ca2+ binding affinity changes from 56 to 996 μM, a nearly 18-fold change, and this change in affinity is coupled to a change in protein–protein binding. One constraint on our current design is the inability of BSBCA and other currently available chromophores to switch completely to cis. Several new chromophores have become available31,35,37 that possess either more complete isomerization or longer half-lives that could allow for isolation and use of the pure cis state.
When applied in cell culture experiments, the light-modulatable cadherin could help answer several outstanding questions about cadherin’s function. One way to introduce this engineered molecule into a cellular context is via cadherin-coated substrata. Coated substrata have been used to study cell–cell adhesion38 and stem cell pluripotency.40 Creation of coated surfaces allows spatial control of cadherin patterning and fine control over cadherin concentration, which could help maximize the switchability of cadherin-mediated adhesion.38
Although interest in photoswitchable proteins has increased in recent years, relatively few examples exist in the literature, perhaps because finding good cysteine attachment points remains difficult. Compared to high-throughput and other library techniques, we were able to create a successful conjugate using a rational design strategy and a small library of constructs. In our design, we chose to focus on modulating loop structures that may have a lower activation energy barrier to transition compared to more rigid parts of protein domains. While not all proteins could be modified in this way, we believe our combined rational/computational selection method and our focus on loops can be generalized to create other photoswitchable designs.
Acknowledgments
The authors would like to thank Laura Lavery and Rebeca Choy for contributing to the original idea, Elizabeth Tanner, Drs. Matthew Banghart, Andrew Woolley, James Nelson, and Nicolas Borghi for helpful discussions, Dr. Woolley separately for a gift of BSBCA, Alec Nielsen for assistance with assay development and the Mass Spectrometry Resource at UCSF (funded by NIH P41GM103481, PI A.L. Burlingame). R.R. and K.K. were partially funded by NIH Grant T32 GM008284. Research funding for this project came from NSF Grant 1134127 to T.K. and the Synthetic Biology Engineering Research Center (NSF EEC-0540879, PI J. Keasling).
Supporting Information Available
Data for other library members; detailed experimental methods; descriptions of, and figures for, data analysis methodology; additional figures for the unconjugated mutant. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Krauss U.; Drepper T.; Jaeger K. E. Chemistry 2011, 17, 2552. [DOI] [PubMed] [Google Scholar]
- Banghart M.; Borges K.; Isacoff E.; Trauner D.; Kramer R. H. Nat. Neurosci. 2004, 7, 1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. I.; Frey D.; Lungu O. I.; Jaehrig A.; Schlichting I.; Kuhlman B.; Hahn K. M. Nature 2009, 461, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levskaya A.; Weiner O. D.; Lim W. A.; Voigt C. A. Nature 2009, 461, 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley G. A.; Jaikaran A. S. I.; Berezovski M.; Calarco J. P.; Krylov S. N.; Smart O. S.; Kumita J. R. Biochemistry 2006, 45, 6075. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Muller K. M.; Woolley G. A.; Arndt K. M. Methods Mol. Biol. 2012, 813, 195. [DOI] [PubMed] [Google Scholar]
- Wyart C.; del Bene F.; Warp E.; Scott E. K.; Trauner D.; Baier H.; Isacoff E. Y. Nature 2009, 461, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beharry A. A.; Woolley G. A. Chem. Soc. Rev. 2011, 40, 4422. [DOI] [PubMed] [Google Scholar]
- Ivanov D. B.; Philippova M. P.; Tkachuk V. A. Biokhimiya/Biochemistry 2001, 66, 1174. [DOI] [PubMed] [Google Scholar]
- Koch A.; Pokutta S.; Lustig A.; Engel J. Biochemistry 1997, 36, 7697. [DOI] [PubMed] [Google Scholar]
- Prakasam A.; Chien Y.; Maruthamuthu V.; Leckband D. Biochemistry 2006, 45, 6930. [DOI] [PubMed] [Google Scholar]
- van Roy F.; Berx G. Cell. Mol. Life Sci. 2008, 65, 3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokutta S.; Herrenknecht K.; Kemler R.; Engel J. Eur. J. Biochem. 1994, 223, 1019. [DOI] [PubMed] [Google Scholar]
- Capaldo C. T.; Macara I. G. Mol. Biol. Cell 2007, 18, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nose A.; Nagafuchi A.; Takeichi M. Cell 1988, 54, 993. [DOI] [PubMed] [Google Scholar]
- Guerrero L.; Smart O. S.; Woolley G. A.; Allemann R. K. J. Am. Chem. Soc. 2005, 127, 15624. [DOI] [PubMed] [Google Scholar]
- Courjean O.; Chevreux G.; Perret E.; Morel A.; Sanglier S.; Potier N.; Engel J.; van Dorsselaer A.; Feracci H. Biochemistry 2008, 47, 2339. [DOI] [PubMed] [Google Scholar]
- Burns D. C.; Zhang F.; Woolley G. A. Nat. Protoc. 2007, 2, 251. [DOI] [PubMed] [Google Scholar]
- Rosetta - The premier software suite for macrommolecular modeling. http://www.rosettacommons.org (accessed May 16th 2013).
- Kortemme T.; Baker D. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 14116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortemme T.; Kim D.; Baker D. Sci. Signaling 2004, 2004, l2. [Google Scholar]
- Harrison O. J.; Bahna F.; Katsamba P. S.; Jin X.; Brasch J.; Vendome J.; Ahlsen G.; Carroll K. J.; Price S. R.; Honig B.; Shapiro L. Nat. Struct. Mol. Biol. 2010, 17, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nose A.; Tsuji K.; Takeichi M. Cell 1990, 61, 147. [DOI] [PubMed] [Google Scholar]
- Troyanovsky R. B.; Sokolov E.; Troyanovsky S. M. Mol. Cell. Biol. 2003, 23, 7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta S.; Woolley G. A. ChemBioChem 2011, 12, 1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanovsky R. B.; Laur O.; Troyanovsky S. M. Mol. Biol. Cell 2007, 18, 4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häussinger D.; Ahrens T.; Aberle T.; Engel J.; Stetefeld J.; Grzesiek S. EMBO J. 2004, 23, 1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Lomas M.; Samanta S.; Campos P. J.; Woolley G. A.; Sampedro D. J. Am. Chem. Soc. 2012, 134, 6960. [DOI] [PubMed] [Google Scholar]
- Samanta S.; Qin C.; Lough A. J.; Woolley G. A. Angew. Chem., Int. Ed. 2012, 51, 6452. [DOI] [PubMed] [Google Scholar]
- Borghi N.; Lowndes M.; Maruthamuthu V.; Gardel M. L.; Nelson W. J. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka M.; Koshimizu U.; Yuasa S.; Hattori F.; Chen H.; Tanaka T.; Okabe M.; Fukuda K.; Akaike T. PLoS ONE 2006, 1, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.