

Abstract
A step towards annotating the mouse genome is to use forward genetics in phenotype-driven screens to saturate the genome with mutations. The purpose of this article is to highlight the new projects in North America that are focused on isolating mouse mutations after ENU mutagenesis and phenotype screening.
Keywords: balancer chromosomes, Human Genome Project, Mouse Genome Project, mouse mutagenesis, phenotype screens
Introduction
As a result of the Human Genome Project, biologists are in the position to assign functions to genes and to identify the genetic lesion leading to many inherited diseases. The laboratory mouse has long been considered an invaluable tool in functional gene discovery, not only because of its genetic malleability, but also because of its striking biological similarity to human systems. In order to keep pace with the wealth of information gained from genomic sequencing, a high throughput method of identifying heritable mutations in the mouse genome is required.
The International Mouse Mutagenesis Consortium (IMMC) has recently published a series of goals for the mouse community in order to define the biological activities of every gene in the human genome using the mouse as a model system (Nadeau et al., 2001). The IMMC proposes combining both genotype and phenotype-driven approaches to achieve this goal. The genotype-driven approach employs ES-cell technology to create targeted mutations by gene knock-out, knock-in or gene-trapping (Evans et al., 1997; Roths et al., 1999). This method has helped define the biological activities of many genes. The genotype-driven approaches are advantageous because the genomic mutation is characterized relatively easily once homologous recombination has occurred. Significantly, this technique has become increasingly powerful when used in combination with the Cre/ loxP system, enabling expression of the mutation in a tissue and/or time specific manner (Lakso et al., 1992; Orban et al., 1992; Gu et al., 1994; Xiaahong et al., 1999). Although the genotype-driven approach is extremely successful, it cannot be achieved on a large scale outside ES cells. As a result, functional genomics using the current genotype-driven approaches alone will continue to lag behind the vast amounts of sequence information available for the mouse and human genomes.
In comparison, the phenotype-driven approach using the supermutagen N-ethyl-N-nitrosurea (ENU) provides a means of performing large-scale screens for phenotypically important mutations (for review, see Justice, 2000). Once the phenotype has been confirmed as heritable, the gene of interest and therefore the mutation is mapped. While genotype-driven approaches typically generate deletions, ENU induces single nucleotide transversions and transitions (Justice et al., 1999). Once a phenotype is confirmed as heritable, the gene of interest (and therefore the mutation) is mapped by positional cloning and sequenced. With continual improvements to mouse linkage maps, the completion of the mouse genome, the use of DNA pooling techniques for mapping (Taylor, 1999) and the introduction of balancer chromosomes, the process of positional cloning is becoming more routine. Therefore, as genotype and phenotype-driven strategies produce completely different types of mutations, they will undoubtedly act together as complementary tools in the in vivo delineation of a gene’s function.
ENU mutagenesis screens are successfully being conducted on a large-scale at Harwell in the United Kingdom (Nolan et al., 2000a, b) and Munich and Neuherberg in Germany (Hrabe de Angelis et al., 2000; Pargent et al., 2000; Rathkolb et al., 2000; Soewarto et al., 2000) in Australia (Fahrer et al., 2001), and in Japan (T. Shiroishi & Y. Gondo, personal communication). In North America a number of centers have recently been established to conduct large-scale ENU mutagenesis screens for a wide variety of phenotypes. In addition to creating large databases of mutants, these centers are devising new and unique ways to examine mutant mice. The purpose of this review is to describe several of the larger ENU projects in North America. We will summarize the biological interest of each center/project, the type of breeding scheme (dominant or recessive) and the phenotypes being surveyed at each site (Table 1). These centers and projects will undoubtedly provide a new and extremely useful resource to the scientific community in order to assign function to some of the 30,000 genes identified in the Human Genome Project.
Table 1.
Summary of ENU mutagenesis centers and projects in North Americaa
| Name | Screen | Emphasis | Website |
|---|---|---|---|
| Mouse Mutagenesis and Phenotyping Center fro Developmental Defects, TMC, Houston | Recessive | Lethal and detrimental to 2 months, size, renal, skeletal, metabolic, pancreatic, hematopoietic | http://www.mouse-genome.bcm.tmc.edu |
| Neuroscience Mutagenesis Center, Jackson Laboratory Maine | Recessive | Motor function, neuromuscular, hearing, visual, taste, olfaction, memory, epilepsy | http://www.jax.org/resources/documents/nmf/documents/about.html |
| Tennessee Mouse Genome Consortium, Tennessee | Recessive | Neurophysiology, and anatomy drugs of abuse, visual, social behavior, ageing | http://tnmouse.org/ |
| CMHD, Toronto, Canada | Dominant, sensitized | Cardiovascular, renal, hematopoietic, neurological, skeletal, metabolic | http://cmhd.mshri.on.ca/ |
| ENU mutagenesis at Northwestern University | Dominant, recessive | Circadian rhythms, fear conditioning, vision, neuroendocrine hormones, response to psychostimulants | http://www.northwestern.edu/neurobiology/faculty/takahashi.html |
| ENU mutagenesis, McLaughlin Research Institute, Montana | Dominant, recesssive | Prion disorders, Alzehimers | www.montana.edu/wwwmri |
| ENU mutagenesis at Case Western Reserve University/University Hospitals, Cleveland | Dominant, sensitized | Testicular cancer, X-inactivation, methylation, obesity, diabetes, colon cancer, vision | Not available |
| The Jackson Laboratory Center for Mouse Models of Heart, Lung, Blood and Sleep Disorders, Maine | Recessive | Atherosclerosis, hypertension, lung function, cardiac structure and function, blood formation, clotting and thrombosis, obesity, and sleep | http://pga.jax.org |
The projects listed here were established shortly after the Symposium. In addition to these projects, Dr. Kathryn Anderson of Sloan-Kettering Cancer Center, New York, NY, is carrying out a project designed to isolate a variety of defects in embryonic patterning and organ development. Dr. Bruce Beutler, The Scripps Institute, San Diego, CA, is carrying out a project to isolated mutations in the innate immune response.
Mutagenesis at the Texas Medical Center
The Mouse Mutagenesis and Phenotyping Center for Developmental Defects at the Texas Medical Center draws expertise from Baylor College of Medicine and the University of Texas MD Anderson Cancer Center where the focus is on a broad range of phenotypes affecting mammalian development. One of the most exciting phenotypic screens conducted at this center is for recessive lethal or detrimental mutations using balancer chromosomes (Zheng et al., 1999; Justice, 2000; Kile et al., 2003). In addition, viable screens are also conducted looking for developmental defects affecting hematopoeisis, metabolism, pancreatic function, urogenital anomalies and bone remodeling and growth. The center is currently using balancer chromosomes in three generation mating schemes to screen for recessive lethal, visible, and blood cell or blood biochemistry mutations. These mutations may segregate anywhere in the genome, or may be linked to the balancer, which provides rapid chromosomal localization. The center provides screening and primary characterization of the mutations, and access to these mutations can be obtained at <http://www.mouse-genome.bcm. tmc.edu>. The current screen in this center is the balancer mutagenesis scheme for chromosome 11 (Figure 1). In this scheme, male C57BL/6 mice are treated with three doses of ENU and allowed to recover fertility. A three generation mating scheme is employed where the test-class G3 animals are identified by their black coat color indicating that they are homozygous for a mutation on chromosome 11. Four test-class animals (homozygous mutant) and one carrier animal are phenotypically surveyed for each recessive pedigree. Using this scheme, the center expects to classify 30–50 embryo lethal mutations and at least 30 visible mutations each year.
Figure 1.

C57BL/6J males are treated with a 3 · 100 mg/kg dose of ENU and allowed to recover fertility. Fertile males are mated to females carrying a balancer chromosome (ARROW) to generate G1 animals that may carry a new mutation (*). G1 animals (males or females) are mated to animals carrying a balancer chromosome and a marker for the non-mutagenized chromosome (Re = Rex, which gives a dominant curly coat: ZIG ZAG LINES). Informative G2 animals are identified by their yellow coat color and intercrossed to look for new mutations in the G3 offspring. The Test Class (black) animals are examined for mutations.
Phenotypic screens
Mutations that cause lethal phenotypes in mice are easy to identify in targeted genotype-driven approaches; however, in the collection of spontaneous mutations that develop a heritable phenotype in mice, the cohort of lethal mutations is absent. By using the balancer chromosome marked with a coat color and a recessive lethal gene, any ENU induced mutation on chromosome 11 that causes early death can be identified. Lethal mutations identified in this manner are surveyed by establishing timed matings to identify the exact embryonic day of resorption. Once the time of death is established, an appropriate panel of molecular markers is applied by the in situ core facility to examine the mutation in more detail.
At weaning, all mice are examined for abnormalities of size. Small mice at weaning and adulthood may have defects in the hypothalamic– pituitary axis (H–P axis) or its target endocrine organs. Furthermore, obesity can be a reflection of mutations in the H–P axis or genes expressed in the pancreas or adipose tissue. As a primary screen for abnormalities in size, animals are weighed at weaning and stocks of small or obese mice from a single pedigree are established with their weights recorded from birth to 8 weeks. Mice that are two standard deviations outside the normal growth curve for age-matched controls are further characterized by serum analysis of IGF-1, T3/T4 and glucose.
Skeletal abnormalities or bone density are identified using X-ray analysis. This technique is a non-invasive method for identifying skeletal patterning, osteoporosis and osteopetrosis. All test-class animals are X-rayed at 8 weeks of age as a high throughput screen for skeletal abnormalities. Once abnormalities are detected by X-ray, a more detailed analysis using decalcification histology and alizaren red skeleton preparations is performed as a secondary screen to further characterize the abnormalities.
In humans, proteinuria is the hallmark of renal disease and occurs when the glomerulus is no longer capable of providing a sufficient barrier for filtration of plasma proteins. Unfortunately, it is difficult to use this approach to identify renal insufficiency in mice as they excrete low molecular weight proteins called major urinary proteins (MUP) under normal conditions. In recent reports, the problem of using total urinary protein as a measure of renal function has been addressed by calculating the ratio of urinary albumin to urinary creatinine (O’Bryan et al., 2000; Ziyadeh et al., 2000). Using this approach, the identification of renal insufficiency in both diabetic and non-diabetic models of renal disease have been achieved. The high throughput screen currently being devised for examining renal insufficiency is to use urinary dipstick and ELISA analysis for albumin levels as a primary screen, and then to survey mutants with secondary screens for albumin/creatinine ratios, urine volumes and creatinine clearance.
In order to screen for possible mutations in glucose metabolism, spot urine tests for glucose (glucosuria) are conducted using urinary dipsticks. In order to identify mutations that affect glucose metabolism and renal glucose resorption, mutants detected in the primary screen as having glucosuria are surveyed in secondary screens using intraperitoneal glucose tolerance tests (IPGTT). Abnormalities in the reproductive system that result in infertility or subfertility are also surveyed in all pedigrees. In order to identify mutations that affect reproductive capacity, the test-class mice are surveyed in primary screens by fertility matings with siblings from the same pedigree.
Recessive mutations that induce blood cell disorders, such as thalassemia, iron deficiency, mal-absorption, vitamin deficiency, aplastic bone marrow, polycythemia and endocrine disorders, are identified by performing a complete blood count (CBC) with differential. Once a mutant is identified in the primary screen, the mutation is further characterized by bone marrow histopathology for abnormalities in cell populations. Tandem mass spectrometry on plasma and urine is used to screen to diagnose inborn errors of metabolism, cholesterol related developmental disorders, abnormalities in vitamin metabolism, mitochondrial and peroxisomal disorders, as well as multiple mental retardation and growth anomalies.
Neuroscience Mutagenesis Center at the Jackson Laboratory
The goal of this facility is to produce mouse models of human neurological diseases in the areas of motor function, learning and memory, hearing, eye and vision, epilepsy, taste and olfaction. This sophisticated facility combines state-of-the-art phenotype screening approaches with detailed further characterization once the mutations are confirmed as heritable. The center aims to produce 50 novel mouse mutations per year for the study of human neurological disorders, and a robust information management system is in place to ensure that investigators gain up-to-date internet access to mutants generated by the facility. Information on the types of screens conducted in this center can be found at <http://www.jax.org/resources/documents/nmf/documents/about.html>. The ENU mutagenesis scheme employed by this facility involves a three generation backcross breeding scheme to identify recessive mutations.
Phenotypic screens
The mutant animals identified in the behavioral screen represent neuromuscular disease phenotypes with a disruption in motor neuron, cerebellar or neuromuscular activity. As a primary screen, mice are observed at weaning and after 1 year of aging for gait, posture, wasting and paralysis. Once a mutant phenotype is observed, additional testing is performed by rotorod tests, pawprinting or histopathology of the cerebellum, depending upon the suspected phenotype. Additional screening of the neuromuscular mutants are performed by measuring in situ muscle contractile properties and histopathology of the neuronal tissue within the spinal cord.
By using simple behavioral systems such as the fear potentiated startle (FPS) as primary screens, mice can be easily identified as having mutations in learned responses. Upon identification and confirmation of a mutant phenotype, a more comprehensive analysis is conducted by analyzing the individual performance records of the mice in pre-training and post training periods of this test. Once heritability is established the mutant mice are screened for mutations that affect short and/or long-term memory by combining tone plus shock training trials together with a freezing technique.
Hearing impairment in humans has a strong genetic component and affects one in every 1000 children. To screen for mice that have mutations associated with hearing impairment, mice are analyzed using the acute startle response (ASR), and a smaller proportion of mice are screened using the auditory brainstem response (ABR). The acute startle response employs an auditory signal with subsequent detection of the Preyer reflex (ear flick). However, given that this screen will only detect severely hearing impaired or deaf mice, novel phenotypic screens are being developed combining the hearing prepulse inhibition and the FPS to establish an auditory profile. Following identification in the primary screen, the mutant mice are tested using the ABR, which measures auditory evoked activity in the brain stem. When hearing impairment is confirmed and the ABR is shown to be heritable, the middle and inner ears are examined by histopathology and the surface of the cochlea is examined by scanning electron microscopy.
The majority of human eye disorders are genetic in nature. As a primary screen for mice that have mutations in eye morphology and function, the eyes are thoroughly inspected visually, followed by dilatation with 1% atropine to facilitate a biomicroscopic examination to examine the lens for cataracts and an indirect opthalmoscopic examination for signs of retinal degeneration. However, new procedures including gonioscopy pattern, electroretinogram (ERG), visual evoked potential (VEP), contralateral pupillary response assay and intraocular pressure (IOP) measurements are currently being assessed as potential primary screens. Once a mutation has been identified as heritable, a more thorough investigation is conducted using the sophisticated techniques of electroretinograms, photodocumentation, fluorescein angiography or intraocular pressure measurements and histology.
High-throughput electroconvulsive threshold screens are performed as primary screens to identify epileptic mouse mutants. Following identification of a heritable mutation, the animals are further characterized by their susceptibility to other stimuli or to spontaneous seizures, their responses to antiepileptic drugs, and an analysis of brain regions involved in seizure activity.
Human taste sensations can be divided into four basic categories including bitter, sweet, salty and sour. Interestingly, these categories can also be applied to mice with an additional glutamate-like taste quality and an irritation mechanism transduced by the trigemminal nerve. Primary screening for taste mutations are conducted using the two bottle test where 100 mice are tested simultaneously. These tests have previously been standardized for C57BL/6 mice and a typical screening cycle takes 1–1.5 months. The olfactory system provides an excellent model system to study neuronal patterning. Primary screens for mutations in the olfactory system involve a simple behavioral task where the fasted tester mouse finds a piece of food buried under bedding in a mouse cage within 5 min. This test is then repeated over 5 sequential days to confirm the mutation. More thorough screening of the confirmed mutant animals are conducted to distinguish between mutations in the olfactory epithelium or within the brain.
Targeted mutagenesis of the mouse genome and neural phenotypes: Tennessee Mouse Genome Consortium
Researchers across the state of Tennessee have combined their expertise in ENU mutagenesis of the mouse genome and experience in the neurosciences to screen for deficits in neural function and structure and thereby lay the basis for a large-scale analysis of the functional genomics of the nervous system. A profile of neural function is obtained from all potentially mutant mice by high throughput screens that examine basic behavioral, sensori-motor function and the functional anatomy of the nervous system. Five domains including responses to alcohol and drugs of abuse, vision, social behavior and aging are assessed in mature mice from each pedigree. Test class mice that are flagged by demonstrating aberrant results in a primary screen are tested for heritability of the trait, and moved into appropriate secondary screens that explore performance in the domains of learning and memory, audition, and nociception.
The guiding force behind the Tennessee mouse genome consortium effort is to promote the widespread use of genetic mutant mice to understand the functional genomics of the nervous system. The genetic system used by the Tennessee consortium is similar to the TMC in that mice carrying the inversion are mated in genetic schemes (see Rinchik, this issue). Furthermore, like the TMC and the Neuroscience Mutagenesis Center at The Jackson Laboratory, the Tennessee consortium offers phenotyping expertise to researchers who would like to characterize a mutant mouse and encourages researchers to obtain their mutant mice for hypothesis-driven research.
Phenotypic screens
In order to evaluate primary behavior in all animals, a screening set composed of multiple assessment tools is performed. This screening set detects basic alterations in nervous system function. The assessment targets include (1) motor function/control centers (righting, reaching, vi-brissa-placing reflexes, negative geotaxis and grip strength at weaning; horizontal and vertical activity in the open field at 7 weeks) (2) brainstem-spinal cord reflex as measured through startle reflex parameters (magnitude, duration, habituation, and pre-pulse inhibition), (3) learning and memory via simple one-time associations with sound and spatial cues (fear conditioning test), (4) anxiety/ explorativity (light/dark preference), (5) despair (tail suspension test), (6) nociception (heat response), and other sensory pathways (click box, pupillary reflex, olfaction). For each pedigree examined, four mice go through the behavioral screen. In addition, one mouse undergoes whole-body imaging by microCT, two mice undergo hematology analysis (Cell-Dyne 3500), and gametes are cryopreserved from two males. Finally, one male and one female are dissected for cryopreservation of representative tissues. For the aging studies, each pedigree has eight additional mice that are aged to 18 months for the assessment of late-onset abnormalities. Once mutants are identified in the primary behavior screen, secondary screening for learning, memory, nociception and auditory defects are conducted.
In order to examine the histological brain architecture of test class mice from each pedigree, a number of special staining techniques are employed. These stains encompass a broad spectrum of nervous system parameters including cytoarchitectonics, myelinated fiber pathways, terminal fields, astroglia and neuronal populations, activity state, proliferative populations and neuropathology in brain. A brain from a test-class animal from each pedigree is chosen as the representative of the pedigree and stained with Cresyl violet, osmium tetroxide, acetylcholinesterase, anti-glia fibrillary acidic protein and anti-NeuN. In addition, histochemistry for cytochrome oxidase, cell birthdating with BrdU immunohistochemistry, and neuropathology using anti-ubiquitin immunohistochemistry are carried out. In addition, new stains are being assessed in the phenotyping protocol that will serve as markers of aging. These include PAS and immunocytochemistry using anti-laminin, anti-chondroitin sulfate proteoglycan, and stains to label dying cells.
In addition to behavior as a function of central nervous system abnormalities, the Center is also interested in identifying mutations with comparatively subtle effects on eye and retinal architecture. A stratified search beginning with simple quantitative screens in eye weight, lens weight, and retinal dimensions are used. More refined histological and immunohistochemical methods are subsequently used on identified mutants to characterize cellular targets. Eyes are phenotypically surveyed for shape, pigmentation and vascular patterning, and mutants are chosen that have eye weights that deviate by more than one standard deviation from the mean for the strain background. Additional parameters in the primary screen include lens weight (post fixation), corneal dimensions (post fixation), optic disk diameters (lm in two axes), INL and ONL cell density, and retinal/pigment epithelium/choroid thickness. Primary screening for eye nerve head abnormalities include immunohistochemistry for Calbindin-28 kD (AII and horizontal cells), Choline acetyltransferase (starburst amacrine cells), Protein kinase C (rod bipolar cells) and COS-1 or OS-2 (cone subtypes).
Within the human population, both ethanol consumption and the response to ethanol have been shown to have a genetic component. In mice, moderate doses of ethanol typically cause motor incoordination, hyperactivity and hypothermia, as well as acting as an anxiolytic agent. The level of anxiety of the mouse is measured in the elevated plus maze test. Motor incoordination is measured by comparing performance of a mouse before and after ethanol on an accelerating rotorod test and the Majchrowicz rating scale, which measures the degree of intoxication ranging from unaffected to comatose.
A different primary screen analyzes voluntary alcohol consumption in mice. Mice are given free access to two water bottles, one that contains water and one that contains an ethanol solution. Both the amount of ethanol solution that is consumed and the percentage of the consumed liquid, that is ethanol, are monitored daily. Once a particular pedigree has been designated as mutant with respect to the drug abuse screens, mutant mice are tested in secondary screens to determine whether an abnormal response to cocaine can be generalized to other psychoactive drugs. Amphetamine and MDMA have been chosen because they elicit behavioral effects through different mechanisms.
Screens are currently being conducted that are concerned with the socio-sexual behavior of test class mice and the socio-sexual responses of control mice to test class mice. The primary screens for social behavior consist of eight 1-min tests. These tests monitor odor preference, self grooming, nonspecific and same sex intruder investigation, habituation, scent marking anogenital distance and sexual behavior.
There are three endpoints to the screening of test class mice that are aged for 18 months after birth. These are determining morbidity and mortality in mutant mice pedigrees followed by examining behavioral changes resulting from neurological dysfunction. To identify morbidity, any mouse with body weight changes of 20% from mean body weight for age-matched controls is considered morbid. To examine mortality, aged pedigrees are monitored daily for death. In control mice, significant mortality accrues after 12 months and reaches approximately 25% at 18 months. All mice that die before 12 months of age are evaluated post mortem.
All mouse pedigrees identified as having aberrant aging are evaluated by specific stains that highlight central nervous system abnormalities. In addition, mice flagged in the neurohistology core will be examined using these techniques to determine the consequences of aging on the abnormalities observed at younger ages.
Both the acute and sensitized behavioral response to many drugs of abuse are thought to involve the activation of dopamine in the mesocorticolimbic system and/or dopamine dependent or independent elements in the cell body or terminal regions of this system. It follows directly that appropriate screens should directly assess genetic modifications of the functional output of the mesocorticolimbic dopamine system as well as potential conditioned incentive influences. In order to examine these mechanisms, locomotion elicited by acute and repeated administration of cocaine (i.e. sensitization), and conditioned place preference (CPP) in response to cocaine will be used. In addition to behavioral responses to drugs, vulnerability to addiction will also be monitored. In these screens, the behavioral responsiveness to a stressor is examined. In particular, locomotion in a novel environment as well as locomotion in response to an acute injection of saline will be used as ‘markers’ of drug vulnerability.
In humans, elevated novelty and sensation seeking are consistently associated with illicit drug use as well as other risky behaviors. Furthermore, impaired self-restraint or impulsivity is a prominent feature of alcoholism. As a result, behavioral screens can be used to assess aspects of temperament including novelty seeking and risk taking. These aspects of temperament will be examined using screens measuring investigation of a novel object and novel food neophobia.
The Center for Modeling Human Disease, Toronto, Canada
The Center for Modeling Human Disease (CMHD) is the first of its kind in Canada to perform large-scale ENU mutagenesis screens. The CMHD represents a major collaborative effort of investigators in the Toronto area with the goal of producing mouse models of human developmental and clinical disorders. Two types of ENU mutagenesis screens are currently being established in the center. The first screen is a genome wide dominant screen that involves treating C57BL/6J males with ENU and mating these mutagenized males to C3H females once fertility is recovered. All resulting viable G1 offspring are tested for the acquisition of dominant mutations affecting cardiovascular, renal, hematopoietic, neurological, skeletal and metabolic systems. The second type of screen is the genome wide sensitized forward genetic screen. In this screen, the G1 C57BL/6J ENU mutant males from the first screen are mated with females from a genetically sensitized strain of choice. The resulting G2 progeny are assayed by a set of rapid specialized focused screens.
Phenotypic screens
In order to detect abnormalities in the cardiovascular system, non-terminal bleeds are performed and blood smears and hematology samples are prepared and analyzed. In addition, blood pressure, heart rate, ECG and Doppler aortic wave forms are performed on the same animal. Mutations that predispose towards diabetes are identified by examining fasting blood glucose and insulin levels after a glucose challenge. Behavior and appearance anomalies are identified by analyzing morphology and general behavior of the mice. In addition, hearing, vision and righting reflex are also tested. Mutations that result in renal dysfunction are identified by abnormalities in blood electrolytes and serum creatinine levels combined with urinalysis. Body composition is tested with the use of a small animal densitometer, which measures density of bone and percentage of fat. Learning and memory mutants are sought using the fear-conditioning assay, and a battery of behavioral screens has been designed to detect later-onset neurodegenerative conditions. All mice, whether they have an obvious living phenotype or not, go through a thorough necropsy at termination. Appropriate tissues are stored for histology, other tissue is stored for DNA, and sperm or ovaries are cryopreserved for possible future recovery. These types of primary screens have been generated to suit the research interests of current collaborators involved with the project. Additional investigators who are interested in new projects are encouraged to visit the web site at <http://cmhd.mshri.on.ca/>. All screening and services of the center are also available to Canadian investigators on a fee-for-service basis to assist in phenotypic analysis of specific mutant mice.
Currently the center is funded to screen 60 mice a week in this dominant G1 screen. Sensitized screens are now being initiated. These include:
Glomerulosclerosis, diabetes, inflammation, limb patterning, using mice heterozygous for the Oligosyndactyly (Os) mutation with pronounced predisposition to kidney disease (He et al., 1995).
Limb patterning defects, holoprosencephaly, and tumorigenesis, using the Ptch1lacZ mice (Aszterbaum et al., 1999a, b).
Learning, memory, and behavior, using mutants or agonists of the metabotropic glutamate receptor, mGluR5, which have partial deficits in hippocampal based learning.
Osteoporosis, using knockouts of a2-HS-glycoprotein (Ahsg), which show modest changes in bone phenotype. This sensitized background is particularly relevant because serum AHSG declines with age in women and two common allelic forms of human AHSG gene correlate with a difference in bone phenotype.
Diabetes and glucose intolerance: A number of genetic mouse models of mild glucose intolerance and diabetes are suitable for genetically sensitized screens. If required, complementary approaches for unmasking diabetogenic genes involving use of b-cell toxins, such as streptozotocin (STZ) and high fat diet, may be applied.
ENU mutagenesis at Northwestern University
Northwestern University is in the process of establishing a Center that will act as a national neuroscience resource. The center has the goal of applying the same forward genetics strategy (Takahashi et al., 1994) which resulted in the identification of the mouse Clock mutant (Vitaterna et al., 1994), and subsequently the Clock gene (Antoch et al., 1997; King et al., 1997), to five phenotypic domains that are relevant to the nervous system and behavior. Mutations are identified from both dominant one-generation screens and recessive three-generation screens using the C57BL/6J strain. The Center aims to analyze 8000–10,000 mice every year in the primary phenotypic screens. Mutants confirmed as heritable will be mapped at low resolution and made available to any interested researchers.
Primary phenotypic screens
The five phenotypic domains under analysis include: (1) circadian rhythms, (2) fear conditioning, (3) vision, (4) neuroendocrine hormones, and (5) response to psychostimulants. These each represent complex behaviors or physiological processes that can be altered by mutation, making a forward genetics approach to assign gene function possible. High-throughput assays exist for each phenotype. Furthermore, interrelationships can be drawn between the various phenotypes. For example, photoreception regulates circadian rhythms, which in turn regulates neuroendocrine hormones (Lowrey & Takahashi, 2000). Furthermore, circadian Clock mutant mice have a deficit in contextual fear conditioning (Kolker et al., in preparation), while Drosophila circadian mutants are desensitized to cocaine (Andretic et al., 1999). Emotionality, anxiety, stress, and novelty influence both fear conditioning and responses to psychostimulants (Gross et al., 2000). Consequently, the five phenotypic screens together establish a mutually reinforcing set, and therefore the combined results of the set give a more definitive interpretation of the mutant phenotypes. A summary of the five phenotypic domains and the primary screens to identify each phenotype are given in Table 2.
Table 2.
Phenotypic domains and the corresponding assay to identify mutant phenotypes in the Northwestern University screen
| Phenotypic domain | Assay |
|---|---|
| Circadian rhythmicity | Record the circadian rhythm of wheel-running activity under both entrained and free-running conditions |
| Learning and memory | Context-dependent and sound-cued fear conditioning |
| Vision | Electroretinography (ERG), visually evoked potential (VEP) recording, and photography of the fundus |
| Neuroendocrine | Selected hormone samples targeting the Hypothalamic-pitui tary-adrenal (HPA) and Hypothalamic-pituitary-thyroid (HPT) axes at basal conditions and following a 5-min restraint stress |
| Psychostimulant | Naïve behavioral responsiveness: hyperlocomotion and stereotypy |
In addition, all animals are subject to a series of ‘preliminary assessment’ tests which include body weight at two different ages, open field behavior, elevated plus maze, rotarod, and Preyer reflex. These preliminary assessments are aimed at (1) providing supplemental information to aid in the interpretation of data in the primary screens, (2) identify individuals that may have impairments affecting their performance in a primary screen so that data is interpreted appropriately, and (3) identifying individuals that may respond or behave differently in a primary screen and whose data should accordingly be viewed differently.
ENU mutagenesis at the McLaughlin Research Institute
Due to the nature of mouse husbandry, mutations relevant to human diseases of later life are underrepresented. The goal of the ENU mutagenesis screen at the McLaughlin Research Institute is to correct this shortage and identify both dominant and recessive mutations that can be used to advance our understanding of at least two neurodegenerative diseases, prion disorders and Alzheimer’s disease (AD). Currently, a three generation recessive screen for ENU-induced mutations is being conducted in collaboration with Celltech R&D, Inc. The phenotyping surveys performed on behalf of Celltech R&D are aimed at detecting mutations affecting immunity and inflammation. The screens conducted at McLaughlin Research Institute on this same G3 population are based on the primary screen called SHIRPA employed in the UK mutagenesis program in Harwell (Rogers et al., 1997). Currently, the screen is performed at weaning and again at 16 weeks. This screen assesses muscle and lower motor neuron, spinocerebellar, sensory, neuropsychiatric, and autonomic functions. Although these screens were designed as a series of semi-quantitative tests, researchers at the McLaughlin Research Institute modified most of these tests, recording a score only when the animal was abnormal. For example, the visual placing test involves lowering the mouse held by its tail from a height of ~15 cm above a wire grid. Extension of the forelimbs was recorded as: 0 = none, 1 = upon nose contact, 2 = upon whisker contact, 3 = before whisker contact (18 mm), and 4 = early vigorous extension. In testing 800 mice, all scored either three or four with no correlation of these scores with those in other tests. Therefore, for this and many other tests, no score is recorded unless the animal deviates from the norm; this was found to save considerable time allowing more animals to be tested.
Another deviation from the SHIRPA protocol is that behavior is not observed for 5 min in a viewing jar, instead including similar observations in the home cage and after transferring to the arena. After transfer to a rat cage, with the floor of the cage being divided into 16 rectangles of approximately 55 cm2, the mouse is observed for 30 s and the number of rectangles entered by all 4 ft is recorded. The mean and standard deviation for the locomotor score was 14.4 ± 6.6. In every instance so far, where the locomotor score was two or less, other abnormalities were noted. Additional tests for heat sensation (55 °C) and for swimming ability have also been added to the protocol. Some of the phenodeviants identified in the studies would have been observed during routine cage changing; this class includes coat color, skin lesions, severe ataxia, and some bizarre behaviors. However, careful observation and testing was required to detect the majority of the behavioral or neurological abnormalities. Phenodeviants that are observed two or more times in a single pedigree and proven mutants from our neurological/ behavioral screen are freely available and will be posted on the following web site: <www.montana.edu/wwwmri>.
The Jackson Laboratory Center for Mouse Heart, Lung, Blood, and Sleep Disorders
The National Heart, Lung, and Blood Institute (NHLBI) launched the Programs for Genomic Applications (PGAs) in October, 2000. Eleven PGAs were funded. A description of the overall goals of each PGA can be viewed at <http://www.nhlbi.nih.gov/resources/pga/index.htm>. These programs constitute a major initiative to advance functional genomic research related to heart, lung, blood, and sleep. The overall goal of each PGA is to develop the tools and resources required to link genes to biological function on a genomic scale. All the information, reagents, and tools developed in the PGAs are available without restriction to the research community. In addition, each PGA sponsors courses and workshops to assist the scientific community in the most effective use of these tools and to directly demonstrate their utility in the process of gene discovery.
The Jackson Laboratory PGA (JAX PGA) includes collaborations with Boston University, the University of Pennsylvania, and Duke University. The overall goal of the JAX PGA is to identify single genes and interacting gene networks (QTL) that underlie the physiology and pathophysiology of heart, lung, blood, and sleep diseases. To achieve these goals, the JAX PGA is (1) generating new mutations in B6 mice by chemical mutagenesis combined with high throughput screening for heart, lung, blood, and sleep phenotypes, and (2) characterizing an extensive set of common inbred and recombinant inbred strains to identify the existing variation in heart, lung, blood, and sleep phenotypes. Appropriate crosses are then established between inbred strains that differ in multiple phenotypes to locate the major QTL affecting each phenotype. Information about all aspects of the JAX PGA (e.g., strain characterization, availability of mutants, phenotyping and mutagenesis protocols, courses and workshops, Visiting Investigator Program) can be accessed via the JAX PGA website: <http://pga.jax.org/>.
The JAX PGA mutagenesis component includes traditional whole animal ENU-mutagenesis as well as mutagenesis of embryonic stem (ES) cells using the alternative mutagen, ethyl methane sulfonate (EMS). While ENU preferentially targets A–T base pairs, EMS primarily targets G–C base pairs and thus has the potential to provide access to ENU-refractory genes (O’Brien, 2002). The JAX mutagenesis strategy is designed to detect recessive mutations in a broad range of phenotypic ‘domains’ relevant to heart, lung, blood and sleep diseases (Table 3). These domains are blood formation, coagulation and thrombosis, plasma chemistry/atherosclerosis, hypertension, cardiac structure and function, lung function, obesity, and sleep/metabolism. The specific phenotypes being screened within each domain are described below. The goal is to screen 4000 G3 mice per year as well as both males and females of 42 inbred and two recombinant inbred (AXB, BXA) strains. The JAX PGA strain characterization component is an extension of the Mouse Phenome Project; all strain survey data are deposited in the Mouse Phenome Database (http://www.jax.org/phenome).
Table 3.
Summary of the major phenotypic domains and the primary screens used to detect phenodeviants at The Jackson Laboratory Center for Mouse Models of Heart, Lung, Blood, and Sleep Disorders
| Phenotypic domain | Screening tests |
|---|---|
| Blood formation | Complete blood count |
| Coagulation and thrombosis | PT, PTT, fibrinogen, ATIII, FVIII, D-dimer, TAT |
| Blood chemistry/atherosclerosis | Triglycerides, cholesterol, HDL-cholesterol |
| Hypertension | Blood pressure, heart rate |
| Cardiac structure and function | ECG, cardiac ultrasound |
| Lung function | RR, TV, 10% CO2 challenge, methacholine challenge, DLCO |
| Obesity | Body composition, plasma lipids, glucose, insulin, leptin |
| Sleep/metabolic activity | Sleep/wake cycles (activity monitoring), ingestive behavior, energy expenditure |
Primary phenotypic screens
All G3 mice are put through a sequential battery of tests beginning at 6 weeks of age (Figure 2). Potential deviants are removed from the protocol upon detection and re-tested for confirmation and, if indicated, heritability testing is initiated. In most cases, secondary testing to further classify the mutant phenotype and genetic mapping are performed once heritability is established. Most of the testing is done in the context of a high fat diet, a significant risk factor for cardiovascular disease and sleep disorders.
Figure 2.
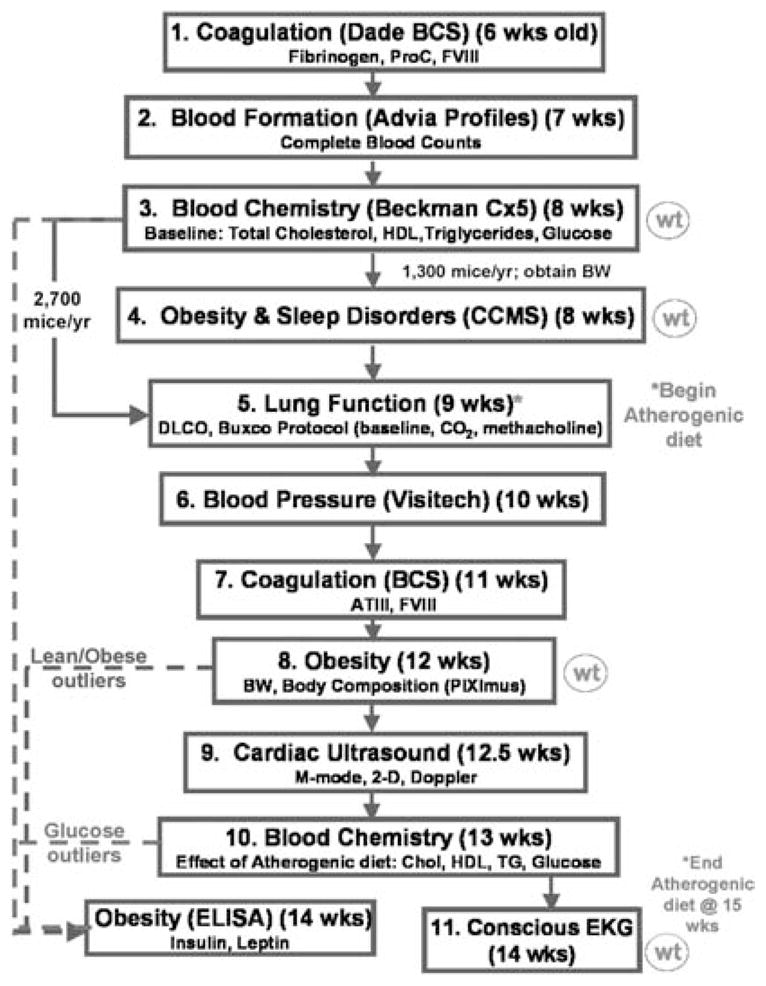
Current sequential phenotyping protocol for G3 mice generated at The Jackson Laboratory. Mice are screened sequentially, beginning at 6 weeks of age. As indicated on the right, weight measurements are taken periodically, and all mice consume an atherogenic diet beginning at 9 weeks of age. As shown on the left, only a subset of the mice can be screened for obesity and sleep using the CCMS due to space and cost considerations. Also, certain phenotypic deviants are subjected to additional tests not performed for each mouse. For example, glucose outliers are screened for insulin and leptin levels.
Blood formation is assessed by performing CBCs using an Advia 120 Multispecies Hematology Analyzer (Bayer Corporation, Tarrytown, NY). Coagulation studies (prothrombin time, PT; partial thromboplastin time, PTT; fibrinogen, fib; antithrombin III, ATIII) are performed using a Dade-Behring Corporation (Marburg, Germany) automated blood coagulation system (BCS). Of note, the BCS programming was successfully modified to reduce the sample and reagent volume requirements per test (Peters et al., 2002). Additional tests indicative of thrombotic risk (D-dimer; thrombin-antithrombin complexes, TAT) are being developed using an ELISA format.
Blood chemistries (total cholesterol, HDL cholesterol, triglycerides, glucose) are measured with a Beckman Instruments Synchron Cx5 analyzer (Beckman Instruments, Fullerton, CA). Blood pressure and heart rate are determined by the tail cuff method using the Visitech BP-2000 system (Visitech Systems, Cary, NC). For some strains (e.g., wild-derived), catheterization procedures are being used to measure blood pressure. Cardiac structure and function is assessed via non-invasive electrocardiogram (ECG) recordings (Mouse Specifics, Inc., Boston, MA) and cardiac ultrasound imaging (Sonos 5500 Cardiac Ultrasound, Philips Technologies, Andover, MA). Lung phenotyping includes measurements of respiratory rate (RR) and tidal volume (TV), a 10% CO2 challenge to assess central control mechanisms, and a methacholine challenge to screen airway responsiveness. These determinations are performed using barometric plethysmography (‘Buxco box’) technology (Buxco Electronics, Inc., Troy NY). In addition, DLCO (diffusion limit of carbon monoxide) is measured in a carbon monoxide uptake system (Columbus Instruments, Columbus, OH) to assess the air–blood interface. The obesity and atherosclerosis domains include leptin and insulin determinations in addition to plasma lipids and glucose levels. Leptin and insulin are determined by ELISA using kits from Crystalchem, Inc., (Chicago, IL) and Alpco (Windham, NH), respectively. Obesity is typically associated with lipid abnormalities and predisposition to type II diabetes and is a major risk factor for sleep apnea. In addition, intra-abdominal obesity is a strong risk factor for insulin resistance and heart disease. Therefore, regional fat distribution is being determined by CT scanning using the PIXImus dual energy X-ray absorptiometer (LUNAR, Madison, WI).
Sleep/wake cycles are measured using a comprehensive cage monitoring system (CCMS) from Columbus Instruments (Columbus, OH) that has two levels of electronic infrared beams capable of 24 h monitoring of ambulatory, horizontal, and vertical movements. An algorithm was developed based on this activity data to estimate sleep patterns as a primary screen to detect potential sleep deviants. Data comparing estimated total sleep using the CCMS activity algorithm closely approximates that obtained by electroencephalogram (EEG) recordings. In addition, the CCMS provides readouts of ingestive behavior and energy expenditure.
Long term goals of the JAX PGA mutagenesis program include regionally directed mutagenesis screens using marked loci and chromosomal aberrations to detect ‘missing classes’ of progeny as a means to recover heart and lung developmental mutations. Moreover, as phenotype expression is highly dependent upon genetic background, ‘sensitized’ screens will be conducted in which alternate inbred strains are mutagenized to gain access to phenodeviants not recovered on the B6 background. This will provide a larger collection of mouse models for understanding the complex basis of heart, lung, blood and sleep phenotypes. The JAX PGA strain characterization studies described above will identify mice that are genetically predisposed (‘sensitized’) to specific heart, lung, blood, or sleep phenotypes and will provide valuable insight for the selection of the strains to be mutagenized.
ENU mutagenesis at Case Western Reserve University/University Hospitals of Cleveland
This ENU mutagenesis project is focused towards identifying mouse models of human disease. In particular, the emphasis is placed upon cancers, cardiovascular diseases and metabolic diseases. The types of ENU mutagenesis schemes currently employed include identification of mutations with dominant effects as well as some sensitized screens. Future ENU projects will involve screening for phenotypic mutations with recessive effects.
The specific phenotypes being surveyed in the primary screens include identification of testicular cancer, X-inactivation and methylation, obesity, diabetes, blood amino acid levels, colon cancer and vision phenotypes. A number of novel screens are being developed which will no doubt lead to new mutant phenotype discoveries in the areas of interest.
Summary
Large-scale ENU mutagenesis screens for clinically relevant phenotypes are currently being conducted in North America. The screens conducted here analyze both dominant and recessive modes of inheritance in a variety of biological systems. This review demonstrates that there is a strong emphasis towards studying neuronal and behavioral phenotypes including learned responses, vision, olfaction, and hearing as well as mutations that affect size. In all centers and projects, the majority of primary screens are conducted on live animals; once the mutations are confirmed as heritable, analysis becomes progressively invasive to fully characterize the mutant phenotype. Taken together, the ENU mutagenesis centers and projects in North America cover an impressively broad range of phenotypic analysis from embryo development to mutations that affect fertility and aging. These centers will undoubtedly provide important resources to the scientific community.
References
- Andretic R, Chaney S, Hirsh J. Requirement of circadian genes for cocaine sensitization in Drosophila. Science. 1999;285(5430):1066–1068. doi: 10.1126/science.285.5430.1066. [DOI] [PubMed] [Google Scholar]
- Antoch MP, Song EJ, Chang AM, Vitaterna MH, Zhao Y, Wilsbacher LD, Sangoram AM, King DP, Pinto LH, Takahashi JS. Functional identification of the mouse circadian Clock gene by transgenic BAC rescue. Cell. 1997;89(4):655–667. doi: 10.1016/s0092-8674(00)80246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aszterbaum M, Epstein J, Oro A, Douglas V, LeBoit PE, Scott MP, Epstein EH., Jr Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat Med. 1999a;5(11):1285–1291. doi: 10.1038/15242. [DOI] [PubMed] [Google Scholar]
- Aszterbaum M, Beech J, Epstein EH., Jr Ultraviolet radiation mutagenesis of hedgehog pathway genes in basal cell carcinomas. J Invest Derm Symp Proc. 1999b;4(1):41–45. doi: 10.1038/sj.jidsp.5640179. Review. [DOI] [PubMed] [Google Scholar]
- Evans MJ, Carlton MB, Russ AP. Gene trapping and functional genomics. Trends Genet. 1997;13:370–374. doi: 10.1016/s0168-9525(97)01240-7. [DOI] [PubMed] [Google Scholar]
- Gross C, Santarelli L, Brunner D, Zhuang X, Hen R. Altered fear circuits in 5-HT(1A) receptor KO mice. Biol Psychiat. 2000;48:1157–1163. doi: 10.1016/s0006-3223(00)01041-6. [DOI] [PubMed] [Google Scholar]
- Fahrer AM, Bazan JF, Papathanasiou P, Nelms KA, Goodnow CC. A genomic view of immunology. Nature. 2001;409(6822):836–838. doi: 10.1038/35057020. [DOI] [PubMed] [Google Scholar]
- Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of DNA polymerase b gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- He C, Zalups RK, Henderson DA, Striker GE, Striker LJ. Molecular analysis of spontaneous glomerulosclerosis in Os/+ mice, a model with reduced nephron mass. Am J Physiol. 1995;269(2 Pt 2):F266–273. doi: 10.1152/ajprenal.1995.269.2.F266. [DOI] [PubMed] [Google Scholar]
- Hrabe de Angelis MH, Flaswinkel H, Fuchs H, Rathkolb B, Soewarto D, Marschall S, Heffner S, Pargent W, Wuensch K, Jung M, Reis A, Richter T, Alessandrini F, Jakob T, Fuchs E, Kolb H, Kremmer E, Schaeble K, Rollinski B, Roscher A, Peters C, Meitinger T, Strom T, Steckler T, Holsboer F, Klopstock T, Gekeler F, Schindewolf C, Jung T, Avraham K, Behrendt H, Ring J, Zimmer A, Schughart K, Pfeffer K, Wolf E, Balling R. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat Genet. 2000;25(4):444–447. doi: 10.1038/78146. [DOI] [PubMed] [Google Scholar]
- Justice MJ. Capitalizing on large-scale mouse mutagenesis screens. Nat Rev Genet. 2000;1:111–115. doi: 10.1038/35038549. [DOI] [PubMed] [Google Scholar]
- Justice MJ, Noveroske JK, Weber JS, Zheng B, Bradley A. Mouse ENU mutagenesis. Hum Mol Genet. 1999;8(10):1955–1963. doi: 10.1093/hmg/8.10.1955. [DOI] [PubMed] [Google Scholar]
- Kile BT, Hentges KE, Clark AT, Nakamura H, Salinger AP, Liu B, Box N, Stockton DW, Johnson RL, Behringer RR, Bradley A, Justice MJ. Functional genetic analysis of mouse Chromosome 11. Nature. 2003;485:81–86. doi: 10.1038/nature01865. [DOI] [PubMed] [Google Scholar]
- King DP, Zhao Y, Sangoram AM, Wilsbacher LD, Tanaka M, Antoch MP, Steeves TD, Vitaterna MH, Kornhauser JM, Lowrey PL, Turek FW, Takahashi JS. Positional cloning of the mouse circadian Clock gene. Cell. 1997;89(4):641–653. doi: 10.1016/s0092-8674(00)80245-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M, Sauer B, Mosinger B, Jr, Lee EJ, Manning RW, Yu SH, Mulder KL, Westphal H. Targeted oncogene activation by site specific recombination in transgenic mice. Proc Natl Acad Sci USA. 1992;89:6232–6236. doi: 10.1073/pnas.89.14.6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey PL, Takahashi JS. Genetics of the mammalian circadian system: photic entrainment, circadian pacemaker mechanisms, and post translational regulation. Annu Rev Genet. 2000;34:533–562. doi: 10.1146/annurev.genet.34.1.533. [DOI] [PubMed] [Google Scholar]
- Nadeau JH, Balling R, Barsh G, Beier D, Brown SD, Bucan M, Camper S, Carlson G, Copeland N, Eppig J, Fletcher C, Frankel WN, Ganten D, Goldowitz D, Goodnow C, Guenet JL, Hicks G, de Angelis MH, Jackson I, Jacob HJ, Jenkins N, Johnson D, Justice MJ, Kay S, Kingsley D, Lehrach H, Magnuson T, Meisler M, Poustka A, Rinchik EM, Rossant J, Russell LB, Schimenti J, Shiroishi T, Skarnes WC, Soriano P, Stanford W, Takahashi JS, Wurst W, Zimmer A. Functional annotation of mouse genome sequences. Science. 2001;291:5507, 1251–1255. doi: 10.1126/science.1058244. [DOI] [PubMed] [Google Scholar]
- Nolan PM, Peters J, Strivens M, Rogers D, Hagan J, Spurr N, Gray IC, Vizor L, Brooker D, Whitehill E, Washbourne R, Hough T, Greenaway S, Hewitt M, Liu X, McCormack S, Pickford K, Selley R, Wells C, Tymowska-Lalanne Z, Roby P, Glenister P, Thornton C, Thaung C, Stevenson JA, Arkell R, Mburu P, Hardisty PR, Kiernan A, Erven A, Steel KP, Voegeling S, Guenet JL, Nickols C, Sadri R, Nasse M, Isaacs A, Davies K, Browne M, Fisher EM, Martin J, Rastan S, Brown SDM, Hunter J. A systematic, genome-wide, phenotype-driven mutagenesis program for gene function studies in the mouse. Nat Genet. 2000a;25(4):440–443. doi: 10.1038/78140. [DOI] [PubMed] [Google Scholar]
- Nolan PM, Peters J, Vizor L, Strivens M, Washbourne R, Hough T, Wells C, Glenister P, Thornton C, Martin J, Fisher E, Rogers D, Hagan JC, Gray I, Wood J, Spurr N, Browne M, Rastan S, Hunter J, Brown SD. Implementation of a large-scale ENU mutagenesis program: towards increasing the mouse mutant resource. Mamm Genome. 2000b;11(7):500–506. doi: 10.1007/s003350010096. [DOI] [PubMed] [Google Scholar]
- O’Brien TP. Mutagenesis and genetic screens in the mouse. In: Smith RS, editor. Systematic Evaluation of the Mouse Eye. CRC Press; New York: 2002. pp. 93–108. [Google Scholar]
- O’Bryan T, Weiher H, Rennke HG, Kren S, Hostetter TH. Course of renal injury in the Mpv17deficient transgenic mouse. J Am Soc Nephrol. 2000;11(6):1067–1074. doi: 10.1681/ASN.V1161067. [DOI] [PubMed] [Google Scholar]
- Orban PC, Chui D, Marth JD. Tissue and site-specific DNA recombination in transgenic mice. Proc Natl Acad Sci USA. 1992;89:6861–6236. doi: 10.1073/pnas.89.15.6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pargent W, Heffner S, Schable KF, Soewarto D, Fuchs H, Hrabe de Angelis M. MouseNet database: digital management of a large-scale mutagenesis project. Mamm Genome. 2000;11:590–593. doi: 10.1007/s003350010112. [DOI] [PubMed] [Google Scholar]
- Peters LL, Cheever EM, Ellis HR, Magnani P, Svenson KL, Smith RV, Bogue M. Large scale, high throughput screening for coagulation and hematologic phenotypes in mice. Physiol Genomics. 2002;11:185–193. doi: 10.1152/physiolgenomics.00077.2002. [DOI] [PubMed] [Google Scholar]
- Rathkolb B, Decker T, Fuchs E, Soewarto D, Fella C, Heffner S, Pargent W, Wanke R, Balling R, Hrabe de Angelis M, Kolb HJ, Wolf E. The clinical-chemical screen in the Munich ENU Mouse Mutagenesis Project: screening for clinically relevant phenotypes. Mamm Genome. 2000;11:543–546. doi: 10.1007/s003350010104. [DOI] [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SDM, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8(10):711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- Roths JB, Foxworth WB, McArthur MJ, Montgomery CA, Kier AB. Spontaneous and engineered mutant mice as models for experimental and comparative pathology: history, comparison and developmental technology. Lab Anim Sci. 1999;49:12–34. [PubMed] [Google Scholar]
- Soewarto D, Fella C, Teubner A, Rathkolb B, Pargent W, Heffner S, Marschall S, Wolf E, Balling R, Hrabe de Angelis M. The large-scale Munich ENU-mousemutagenesis screen. Mamm Genome. 2000;11:507–510. doi: 10.1007/s003350010097. [DOI] [PubMed] [Google Scholar]
- Taylor BA. Mouse Mapping. In: Jackson Ian, Abbott Cathy., editors. Mouse Genetics and Transgenics: A Practical Approach. Oxford University Press; Oxford: 1999. [Google Scholar]
- Takahashi JS, Pinto LH, Vitaterna MH. Forward and reverse genetic approaches to behavior in the mouse. Science. 1994;264(5166):1724–1733. doi: 10.1126/science.8209253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, Dove WF, Pinto LH, Turek FW, Takahashi JS. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994;264(5159):719–725. doi: 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiaohong M, Fujiwara Y, Orkin SH. Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc Natl Acad Sci USA. 1999;96:5037–5042. doi: 10.1073/pnas.96.9.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Sage M, Cai WW, Thompson DM, Tavsanli BC, Cheah YC, Bradley A. Engineering a mouse balancer chromosome. Nat Genet. 1999;22(4):375–378. doi: 10.1038/11949. [DOI] [PubMed] [Google Scholar]
- Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, Chen S, McGowan TA, Sharma K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerularmesangial matrix expansion by treatment with monoclonal anti-transforming growth factor-beta antibody in db/db diabetic mice. P Natl Acad Sci USA. 2000;97:8015–8020. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]