

Abstract
Activation of class A G-protein coupled receptors (GPCRs) involves large-scale reorganization of the H3/H6 interhelical network. In rhodopsin (Rh), this process is coupled to a change in the protonation state of a key residue, E134, whose exact role in activation is not well understood. Capturing this millisecond pH-dependent process is a well-appreciated challenge. We developed a scheme combining the Harmonic Fourier Beads (HFB) method and constant pH molecular dynamics with pH-based replica exchange (pH-REX) to gain insight into the structural changes occurring along the activation pathway as a function of change in the protonation state of E134. Our results indicate that E134 protonates as a consequence of H6 tilting by ca. 4.0° with respect to its initial position and simultaneously rotating by ca. 23° along its principal axis. The movement of H6 is associated with the breakage of the E247-R135 and R135-E134 salt bridges and concomitant release of the E134 side chain, which results in the increase of its pKa value above physiological pH. An increase in the hydrophobicity of the environment surrounding E134 leads to further tilt and rotation of H6 and upshift of the E134 pKa. Such atomic-level information, otherwise not accessible to experiments, refines the earlier proposed sequential model of Rh activation (Zaitseva, E.; et al. Sequential Rearrangement of Interhelical Networks Upon Rhodopsin Activation in Membranes: The Meta IIa Conformational Substate. J. Am. Chem. Soc. 2010, 132, 4815) and argues that the E134 protonation switch is both a cause and a consequence of the H6 motion.
A hallmark of the transition between inactive and active forms of heptahelical G-protein coupled receptors (GPCRs) is the large (3–14Å) outward tilt of transmembrane helix 6 (H6) in response to various extracellular stimuli, such as light or ligand binding.1,2 This tilt causes rearrangements in the H3/H6 interhelical network and creates a cavity for the binding of the heterotrimeric G-protein, thus initiating a cascade of signaling events inside a cell.3 In rhodopsin (Rh), the most widely studied member of class A GPCRs, there are several pre- and post-requisites for the H6 motion, including a series of conformational and protonation switches at the late stages of activation.4,5 Following ultrafast 11-cis to all-trans photoisomerization6 of the retinal protonated Schiff base (11-PSB), formed by the chromophore retinal covalently bound to K296, the signal is propagated from the dark state through a series of inactive intermediates to a cytoplasmic receptor domain where a signaling Meta II state capable of binding the G-protein is formed (Figure 1A).3,7
Figure 1.
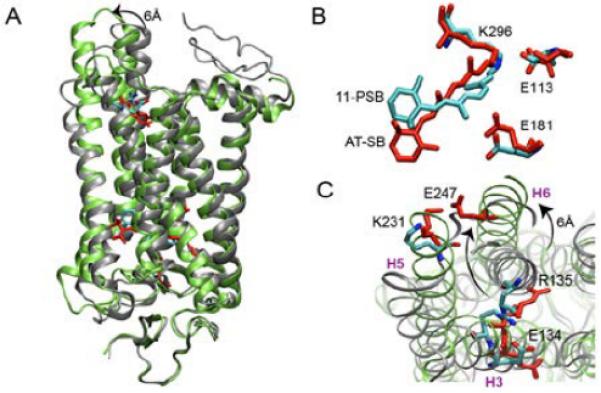
A: Dark (grey; cyan) and Meta II (green; red) states of Rh. B, C: Regions involved in the two protonation-dependent switches.
While early transitions can be exclusively described by the small-scale rearrangements in the retinal binding pocket adjusting to accommodate the isomerized chromophore,8 the major functional and structural changes of the α-helical bundle occur during the pH-dependent Meta I to Meta II transition.9,10 According to the activation scheme, originally proposed by Hofmann and Hubbell and colleagues11,12 and later corroborated and extended by Vogel and co-workers,13,14 this transformation occurs sequentially via a series of metastable states Meta IIa, Meta IIb, and Meta IIb-H+, with the latter two collectively representing the Meta II form. Based on this scheme, Meta IIa is formed upon disruption of the salt bridge between the all-trans protonated Schiff base (AT-PSB) and the primary counterion E113 via an internal proton transfer between the two moieties with the formation of an unprotonated Schiff base (AT-SB; first protonation switch; Figure 1B); Meta IIb stems from the 6 Å outward tilt of H6; and Meta IIb-H+ is a result of the proton uptake by E134 of the conserved E(D)RY motif located on H3 near the cytoplasmic water/lipid interface (second protonation switch; Figure 1C). The suggested reaction scheme is supported by a number of experimental studies,9,11,12,14 however a precise dynamic picture of how the proposed structural changes occur is still missing. Molecular dynamics (MD) simulations can provide a detailed answer to this question and, thus, have been widely exploited by different research groups.15 However, capturing this millisecond transition is beyond the reach of standard MD simulations. Several attempts have been made to overcome the timescale limitation and to arrive at the Meta II state by running long MD guided by the NMR distance restraints starting from either the dark state16 or, most recently, from lumirhodopsin.17 These calculations, however, all assumed fixed protonation states during the dynamics and did not consider on-the-fly coupling between pH and conformational switching, as we do in the current work. Here, we delineate a sequence of late activation events in Rh by probing a conformational transition between its putative semi-active Meta IIa and active Meta II forms as a function of change in the protonation state of E134.
We used the 4.15 Å (PDB entry 2I37) and the 3.0Å (3PXO) X-ray structures to construct initial models of the Meta IIa and Meta II states, respectively. We utilized the Generalized Born with a Simple Switching (GBSW)18,19 implicit solvent/implicit membrane model to determine an optimal position of each structure in the membrane and used the corresponding lowest energy configurations as the starting points to generate a transition pathway between the initial Meta IIa and final Meta II state using the Harmonic Fourier Beads method (HFB).20 Prior to the pathway generation, constant pH molecular dynamics21 with pH-based replica exchange (pH-REX)22 was used to determine the protonation states of E113, E181, and E134 in the starting Meta IIa and final Meta II states. E113 and E181 were found to be protonated in both states, so these residues were modeled as neutral in all subsequent simulations. Once the path was generated, pH-REX was used to capture the protonation of E134 along the conformational transition pathway. All calculations were performed using the CMAP-corrected23 all-atom CHARMM22 force field for proteins24. For details of the computational methods and analysis see the Supporting Information.
Our results show that the pKa of E134 changes from 5.6 to 8.3 upon transition with a protonation switch (pKa=7.4) occurring early along the computed path (Figure 2A). This process is associated with the breakage of the E134–R135 and R135–E247 salt bridges, holding Rh in the semi-active conformation, and a subsequent formation of the new E247–K231 ionic lock stabilizing the Meta II state. The R135–E247 breaks first, releasing the side chain of E134 and allowing its pKa value to rise, as confirmed by the pKa switch at 7.4 shortly afterwards. The outward tilt of H6, monitored as the angle between the helical axis at each new point of the pathway relative to its initial position, is depicted in Figure 2B. The observed tilt is sequential (segments a–b', b'–c', and c'–d; green line) and is partially coupled to the rotation of H6 along its principal axis, which also occurs in a sequential manner (segments a–b, b–c, and c–d; red line). As shown, the E134 protonation switch, associated with breakage of the R135-E247 and E134-R135 salt bridges, is a consequence of H6 rotating by ca. 23° along the helical axis (segment a–b) and simultaneously tilting outwards by ca. 4°. Later (region b–c), the H6 tilt and rotation are partially decoupled with H6 continuing to tilt but not rotating anymore, while their coupled motion becomes apparent again along the c–d and c'–d segments. The latter changes, characterized by an additional ca. 30° helical rotation and ca. 10° outward tilt, bringing the H5 and H6 extracellular ends together, accompanies the shift of the pKa of E134 from 7.4 to 8.3.
Figure 2.
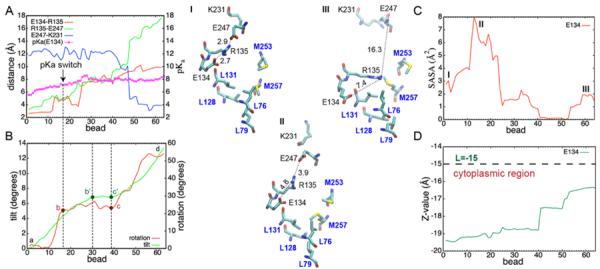
Values computed along the Meta IIa to Meta II pathway: pKa of E134 and average salt bridge distances measured between the closest oxygens and nitrogens of the corresponding acidic (E134, E247) and basic (R135, K231) residues, respectively (A); H6 tilt and rotation angles (B); SASA of the E134 side chain (C); and Z-value showing position of the E134 center of mass relative to the center of the lipid bilayer (D). Changes in the E(D)RY motif with respect to a hydrophobic barrier at different points of the pathway are shown for the end states (I and II) and in the region of the pKa switch (III).
Our results are in apparent disagreement with the experimental interpretation suggesting that proton uptake by E134 in Rh does not happen unless H6 has fully moved.12–14 These experimental conclusions are based on the observed pH independence of the EPR signal arising from the immobilization of the nitroxide side chain in the spin-labeled V227C Rh mutant (R227) in response to the outward tilt of H6.12 In this mutant, R227 belongs to the neighboring helix H5 and senses the changes in the surrounding environment. Once the outward tilt of H6 in the direction of H5 becomes sufficiently large, the spin-label can sense the change in the environment and gives rise to the EPR signal. Since the maximum amplitude of the EPR signal remained the same in the entire pH range from 5 to 9, it was concluded that the E134 proton uptake followed H6 motion. Based on our results, we provide an alternative explanation of the observed pH independence of the EPR signal. Along segment a–b', an outward tilt of H6 is expected to be strongly pH as depicted in Figure S7. Thus, our results call for the re-interpretation of the EPR data on Rh and reconsideration of the existing activation scheme.
To understand the origin of the pKa shift, the solvent accessible surface area (SASA) of E134 was computed along the transition pathway (Figure 2C). It increases up to the point where the E134 protonation switch occurs, in agreement with the observed breakage of the E247-R135 and E134-R135 salt bridges releasing the side chain of E134 and exposing it to the solvent. Then, along the portion of the pathway corresponding to the shift of the pKa from 7.4 to 8.3, the SASA gradually drops indicating an increase in hydrophobicity of the environment surrounding the side chain of E134. As Figure 2D illustrates, this is in part due to a desolvation effect and displacement of the side chain from the water bulk dependent due to a large shift of the pKa of E134 (5.6 to 7.4), but the small tilt angle (ca. 4°) associated with it is insufficient to cause the immobilization of the spin label and a saturation of the EPR signal. The latter is confirmed by the small number of contacts that R227 makes with H6 along that portion of pathway (Figure S7). In contrast, along segment b'–d, associated with an easily detectable H6 tilt (ca. 10°), the pKa shift is significantly smaller (7.4 to 8.3) and the expected pH dependence of the tilt is small as well. Accordingly, the computed number of contacts along b'–d substantially increases, closer to the lipid/water interface, from L=−19.4 to L=−17.5, where L=−15 indicates a position of the lower membrane plane lying on the cytoplasmic side of the 30Å-thick implicit lipid bilayer used in our study.
Another important factor that contributes to the pKa shift is the exposure of the E134 side chain to the so-called “hydrophobic barrier,25 consisting of L76, L79, L128, L131, M253, M257. Structural rearrangements of the E(D)RY motif with respect to this region along the Meta IIa to Meta II transition pathway are shown in Figure 2 (structures I, II and III). Upon breakage of the E247-R135 and E134-R135 salt bridges preceding the E134 protonation switch, the released E134 side chain makes close contacts with L128 and L131 and becomes deeply buried inside the protein. The more hydrophobic environment stabilizes the neutral form of E134 and leads to a shift of its pKa from 7.4 to 8.3. Notably, no experimental data exists for the pKa values of the ionizable residues in Rh, however, our predictions agree well with the change of the E134 protonation state determined experimentally,26,27 as well as with the electrostatic MM-SCP calculations by Periole et al.28 It is also worth noting that CPHMD calculations based on the GBSW implicit solvent model have been successfully applied to many proteins29–32 where comparison between experimental and computed values could be rigorously made yielding the RMSE of 0.6 pH units for proteins containing few buried ionizable side chains.29 Given the precision of the pKa values in our simulations, the shift in pKa from 7.4 to 8.3 that we observe is statistically significant at the 95% confidence interval and so the observed change reflects conformational dynamics of the protein and allows us to delineate a sequence of activation events using the protonation state of E134 as a reaction coordinate.
Overall, the results of our study combining the HFB method and pH-REX simulations enable us to derive a detailed mechanistic picture of the late activation events in Rh and elucidate mechanistic details of coupling between the change in the protonation state of E134 and conformational dynamics of the protein. The HFB made it possible to explore the reaction pathway without making any prior assumptions about the reaction mechanism, and the pH-REX performed at every point of the pathway allowed us to incorporate the information about the protonation state of E134 on-the-fly and, thus, probe an existing sequential Meta IIa–Meta IIb–Meta IIb-H+ reaction scheme.10–12 While the experimental data suggests that proton uptake by Glu134 is a consequence of H6 motion, our simulations offer an alternative explanation. We show that E134 protonates early along the computed path as a consequence of H6 tilting by ca. 4.0° with respect to the membrane normal and simultaneously rotating by ca. 23° along the helical axis. This process is associated with the breakage of the E247-R135 and R135-E134 salt bridges releasing the side chain of E134 and allowing it to raise its pKa value above physiological pH. This, in turn, leads to a further ca. 10° tilt and ca. 30° rotation of H6 together with the 2–3 Å translation of the E134 side chain towards the center of the membrane causing the upshift of its pKa. In the first segment of the pathway, the expected pH dependence of the H6 tilt, associated with the significant difference in the pKa of E134 (5.6 to 7.4), is large, but the small value of the tilt is, we argue, insufficient to cause an immobilization of the spin label in the EPR experiment and saturate the EPR signal. In the second region, however, the tilt is large enough to be observed, but the expected pH dependence of the tilt is negligible owing to a two-fold smaller modulation of the pKa shift.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (GM037554 and GM057513).
Footnotes
Supporting Information. Details of the computational protocol and analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- (1).Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- (2).Deupi X, Standfuss J. Curr. Opin. Struct. Biol. 2011;21:541–551. doi: 10.1016/j.sbi.2011.06.002. [DOI] [PubMed] [Google Scholar]
- (3).Hubbell WL, Altenbach C, Hubbell CM, Khorana HG. In: Membrane Proteins. Chemistry DCRBT-A, P., editors. Volume 63. Academic Press; 2003. pp. 243–290. [Google Scholar]
- (4).Trzaskowski B, Latek D. Curr. Med. Chem. 2012;19:1090–1109. doi: 10.2174/092986712799320556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mahalingam M, Martínez-Mayorga K, Brown MF, Vogel R. Proc. Natl. Acad. Sci. USA. 2008;105:17795–17800. doi: 10.1073/pnas.0804541105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Schoenlein RW, Peteanu LA, Mathies RA, Shank CV. Science. 1991;254:412–415. doi: 10.1126/science.1925597. [DOI] [PubMed] [Google Scholar]
- (7).Choe H, Kim Y, Park J, Morizumi T. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- (8).Struts AV, Salgado GFJ, Tanaka K, Krane S, Nakanishi K, Brown MF. Journal of Molecular Biology. 2007;372:50–66. doi: 10.1016/j.jmb.2007.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Altenbach C, Kusnetzow A. Proc. Natl. Acad. Sci. USA. 2008;105:7439–7444. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- (11).Arnis S, Hofmann KP. Proc. Natl. Acad. Sci. USA. 1993;90:7849–7853. doi: 10.1073/pnas.90.16.7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Knierim B, Hofmann KP, Ernst OP, Hubbell WL. Proc. Natl. Acad. Sci. USA. 2007;104:20290–20295. doi: 10.1073/pnas.0710393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mahalingam M, Martinez-Mayorga K, Brown MF, Vogel R. Proc. Natl. Acad. Sci. USA. 2008;105:17795–17800. doi: 10.1073/pnas.0804541105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zaitseva E, Brown MF, Vogel R. J. Am. Chem. Soc. 2010;132:4815–4821. doi: 10.1021/ja910317a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Grossfield A. Biochim. Biophys. Acta: Biomembr. 2011;1808:1868–1878. doi: 10.1016/j.bbamem.2011.03.010. [DOI] [PubMed] [Google Scholar]
- (16).Hornak V, Ahuja S, Eilers M. J. Mol. Biol. 2010;396:510–527. doi: 10.1016/j.jmb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tikhonova I, Best R. J. Am. Chem. Soc. 2008;130:10141–10149. doi: 10.1021/ja0765520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chen J, Im W, Brooks CL., III J. Am. Chem. Soc. 2006;128:3728–3736. doi: 10.1021/ja057216r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Im W, Feig M, Brooks CL., III Biophys. J. 2003;85:2900–2918. doi: 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Khavrutskii IV, Arora K, Brooks CL., III J. Chem. Phys. 2006;125:174107–174108. doi: 10.1063/1.2363379. III. [DOI] [PubMed] [Google Scholar]
- (21).Khandogin J, Brooks CL., III Constant pH Molecular Dynamics with Proton Tautomerism. Biophysical journal. 2005;89:141–157. doi: 10.1529/biophysj.105.061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sugita Y, Okamoto Y. Chem. Phys. Lett. 1999;314:141–151. [Google Scholar]
- (23).Mackerell AD, Feig M, Brooks CL., III J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- (24).MacKerell AD, Bashford D, Bellott, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- (25).Standfuss J, Edwards PC, D'Antona A, Fransen M, Xie G, Oprian DD, Schertler GFX. Nature. 2011;471:656–660. doi: 10.1038/nature09795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yan ECY, Kazmi MA, Ganim Z, Hou J-M, Pan D, Chang BSW, Sakmar TP, Mathies RA. Proc. Natl. Acad. Sci. USA. 2003;100:9262–9267. doi: 10.1073/pnas.1531970100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Fahmy K, Sakmar TP, Siebert F. Biochemistry. 2000;39:10607–10612. doi: 10.1021/bi000912d. [DOI] [PubMed] [Google Scholar]
- (28).Periole X, Ceruso M, Mehler E. Biochemistry. 2004;43:6858–6864. doi: 10.1021/bi049949e. [DOI] [PubMed] [Google Scholar]
- (29).Khandogin J, Brooks CL., III Biochemistry. 2006;45:9363–9373. doi: 10.1021/bi060706r. [DOI] [PubMed] [Google Scholar]
- (30).Khandogin J, Chen J, Brooks CL., III Proc. Natl. Acad. Sci. USA. 2006;103:18546–18550. doi: 10.1073/pnas.0605216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zhang BW, Brunetti L, Brooks CL., III J. Am. Chem. Soc. 2011;133:19393–19398. doi: 10.1021/ja2060066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Law SM, Zhang Bin W, Brooks CL., III Protein Sci. 2013:595–604. doi: 10.1002/pro.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.