

Abstract
Sle2c1 is an NZM2410 and NZB-derived lupus susceptibility locus that induces an expansion of the B1a cell compartment. B1a cells have a repertoire enriched for autoreactivity, and an expansion of this B cell subset occurs in several mouse models of lupus. A combination of genetic mapping and candidate gene analysis presents Cdkn2c, a gene encoding for cyclin-dependent kinase inhibitor p18INK4c (p18), as the top candidate gene for inducing the Slec2c1 associated expansion of B1a cells. A novel SNP in the NZB allele of the Cdkn2c promoter is associated with a significantly reduced Cdkn2c expression in the splenic B cells and Pc B1a cells from Sle2c1-carrying mice, which leads to a defective G1 cell cycle arrest in splenic B cells and increased proliferation of Pc B1a cells. As cell cycle is differentially regulated in B1a and B2 cells, these results suggest that Cdkn2c plays a critical role in B1a cell self-renewal, and that its impaired expression leads to an accumulation of these cells with high autoreactive potential.
Keywords: lupus, B cells, cell cycle, NZB
Sle2 is one of the three major quantitative trait loci that has been associated with an increased susceptibility to lupus nephritis in the NZM2410 mouse model (1). Comparison between combinations of these three loci on a C57BL/6 (B6) genetic background has shown that the presence of Sle2 increased the frequency of fatal lupus nephritis from 41% in B6.Sle1.Sle3 to 98% in B6.Sle1.Sle2.Sle3 mice (2). Sle2 expression by itself is not pathogenic, but it is associated with a number of B cell defects, including an expansion of the B1a cell compartment, especially in the peritoneal cavity (Pc) and an increased production of polyreactive IgM Ab (3). The majority of B1a cells have a fetal origin, are long-lived and undergo self-renewal (4). They differ functionally from conventional B cells (B2) in many ways, such as their constitutively activation of STAT3 (5), and up-regulation of plasma cell differentiation markers (6) associated with the spontaneous secretion of IgM. Recently, the human functional equivalent to the mouse B1a cells has been identified as CD27+ CD43+ CD70− B cells (7). This confers a greater significance to the understanding of the mechanisms that regulate the size and functions of this B cell subset. We have shown that the Sle2-mediated expansion of B1a cells is B cell-intrinsic and results from a combination of factors, which includes an increased spontaneous proliferation of B1a as compared to B2 cells (8). Using congenic recombinants, we mapped the expansion of B1a cells to three regions of Sle2, Sle2a, Sle2b, and Sle2c, but the major contribution was provided by the telomeric sub-locus Sle2c (9).
The repertoire of B1a cells is autoreactive (10) and an expanded B1a cell population has been consistently associated with lupus, but how these cells contribute to autoimmune pathology has been controversial (4). Recent studies have shed a new light on the topic, strongly suggesting a direct pathogenic role of B1a cells. (NZB x NZW)F1 B1a cells migrate to inflamed tissues, notably the kidneys, where they class-switch and produce anti-dsDNA IgG (11). In addition, B1a cells favor the differentiation of CD4+ T cells into Th17 cells (12). Th17 cells have been directly implicated in lupus in that they provide help to autoreactive B cells in mice (13) and lupus patients (14), and contribute to the inflammatory cascade in lupus nephritis (15). Therefore B1a cells contribute to lupus pathology through either Ab production, or antigen-presentation, or both. A complementation analysis of the Sle1.Sle3 combination failed however to show that Sle2c contributed to autoimmune pathology (9). This study was however complicated by the complex structure of Sle2c, which contains, in addition to the B1a-promoting locus now named Sle2c1, a suppressor locus named Sle2c2 (16). In the absence of Sle2c2, Sle2c1 dramatically increased the severity of lupus nephritis induced by Fas-deficiency by enhancing the differentiation of CD4+ T cells into Th17 cells and promoting their infiltration of the kidneys (17). These results support the hypothesis that the expansion of the B1a cell compartment by Sle2c1 contributes to autoimmune pathogenesis at least through their induction of inflammatory T cells.
The present study was conducted to identify the gene in the Sle2c1 interval that is responsible for the expansion of the B1a cell compartment. A combination of genetic mapping and candidate gene analysis presents Cdkn2c, a gene encoding for cyclin-dependent kinase inhibitor p18INK4c (p18), as the top candidate gene for inducing the Slec2c1 phenotype. A novel SNP in the NZB allele of the Cdkn2c promoter is associated with a significantly reduced Cdkn2c expression in the splenic B cells and B1a cells from Sle2c1-carrying mice, which leads to defective G1 cell cycle arrest in splenic B cells and increased proliferation of Pc B1a cells.
Materials and Methods
Mice
The B6.Sle2, B6.Sle2c1 and BcN/LmJ (B6.TC) congenic strains have been previously described (2,9,18). B6.Sle2c1 recombinants obtained from a backcross to B6 were identified and fine-mapped with a panel of microsatellite markers and SNPs that are polymorphic between NZB and B6 (Fig. 1). These recombinants were then expanded in out-crosses to B6 and bred to homozygosity to generate three Sle2c1 recombinant strains. B6, NZM2410, NZB and NZW mice were originally obtained from the Jackson Laboratories. The phenotypes of B6.Sle2c1 and its recombinants were analyzed in both males and females at the age indicated. Cohorts of (NXB X B6.Sle2)F1 (NxSle2), (NZB X B6.Sle2c1)F1 (NxSle2c1) and (NZB X B6)F1 (NxB) females were aged to 12 months of age. Kidney histopathology was performed and glomerulonephritis (GN) was scored as previously described (17) by combining a 0-4 semi-quantitative scale for severity and a description of the type of lesion (in order of severity: hyaline, mesangial matrix, mesangial cellular, and proliferative). All experiments were conducted according to protocols approved by the University of Florida Institutional Animal Care and Use Committee.
Figure 1.
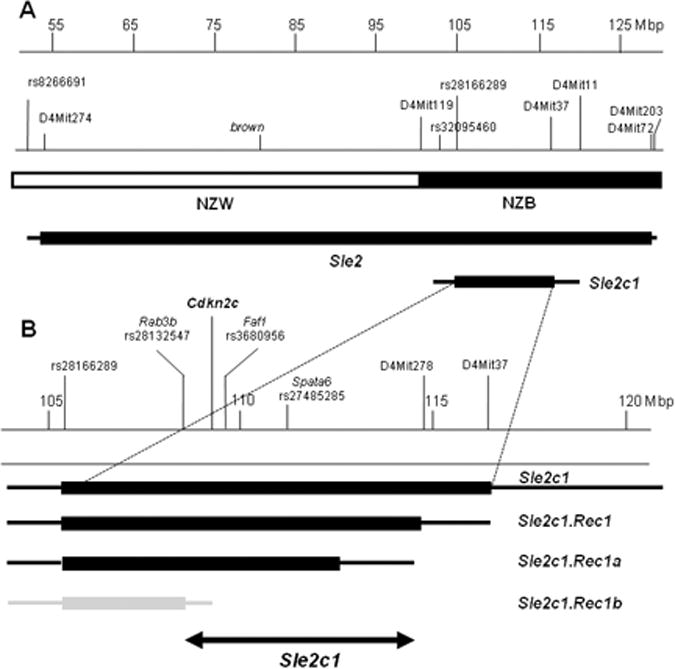
Map of the Sle2c1 locus. A, Location of Sle2c1 relative to Sle2 on chromosome 4. The NZB/NZW derivation of the region is shown, as well as the markers that define the Sle2 and Sle2c1 termini. B, Sle2c1 and its three recombinants define the critical interval (double-arrowed line) associated with Pc B1a cell expansion. The location of Cdkn2c and that of the markers defining the termini of each recombinant are indicated on the top. NZM2410 (NZW or NZB)-derived intervals are indicated by solid boxes, with the area of recombination between the NZM2410 and B6 genomes indicated by the lines on each side. Intervals associated with an expanded Pc B1a cell phenotype are shown in black, while the Sle2c1.Rec1b interval associated with a B6-like B1a phenotype is indicated in grey.
Flow cytometry and proliferation assays
Pc lavages and splenic single cell suspensions were prepared by lysing RBCs with 0.83% NH4Cl. Cells were first blocked with saturating amounts of anti-CD16/CD32 (2.4G2) and then stained with FITC-, PE-, or biotin-conjugated mAbs: CD4 (RM4–5), CD5 (53-7.3), CD19 (1D3), CD21 (7G6), CD23 (B3B4), CD93 (AA4.1), B220 (RA3-6B2), and IgM (IgH6), all purchased from BD Pharmingen. Biotinylated mAbs were revealed by Streptavidin-PerCP-Cy5.5. Mononuclear live cells were gated on the basis of forward and side scatters characteristics. At least 30,000 events were acquired per sample on a FACSCalibur cytometer (BD Biosciences). For in vitro proliferation assays, CFSE-labeled Pc cells or splenocytes were cultured with 10 μg/ml of LPS in complete RPMI for 72 h. For ex vivo proliferation, fresh splenic and Pc cells were stained with Ki67 (M-19, Santa-Cruz). Both CFSE dilution and Ki67 staining were determined in B1a and B2 cells gated on the basis of anti-CD5 and anti-B220 staining.
Gene expression analysis
Global gene expression was compared between Pc B1a and splenic B (sB) cells from B6.Sle2c1 and B6 mice at 5 months of age (5 mice per strain and per B cell type). CD43− sB cells were isolated with the Myltenyi B cell kit, leading to a population of > 90% B220hi cells. Pc macrophages were removed by a 2 h culture at 37° C in PBS supplemented with 10% FBS. Non-adherent Pc cells were first submitted to negative selection with a cocktail of biotinylated anti-Thy1.2 and anti-CD3ε Abs, then to positive selection with biotynylated-anti-CD5 Ab. Cells bound to biotynylated Abs were removed with streptavidin-conjugated magnetic beads (Myltenyi). This protocol led to a B220int CD5+ cell population with a 80% purity. RNA was extracted with the Qiagen RNeasy Micro Kit. cDNA was synthesized and labeled with the Ovation Biotin RNA Amplification and Labeling System (NuGEN Technologies, Inc.) before hybridization to Affymetrix Mouse Genome 430 2.0 arrays. All data analyses were based on the use of “internal standards” and generalization of the “Error Model” (19) as presented elsewhere (20). For RT-PCR analysis, cDNAs from total splenocytes, Pc B1a cells and sB cells purified as above were prepared using the Promega reverse transcription kit. RT-PCR was performed with the following primers: Cdkn2c forward: 5′-CAGTCCTTCTGTCAGCCTCC-3′ and reverse: 5′-CTCCGGATTTCCAAGTTTCA-3′; β-actin forward: 5′-ACCACACCTTCTACAATGAG-3′ and reverse: 5′-GGTACGACCAGAGGCATACA-3′. Band density was measured with the Quantity One software, and Cdkn2c mRNA expression was normalized to that of β-actin. Quantitative (q) RT-PCR was performed with TaqMan probes (ABI) for Cdkn2c, Cdkn1a, Cdkn1b, Cdkn2d and Gapdh using the comparative Ct method normalized to one B6 control value (such as 2[(Cdkn2 CT − Gapdh CT for Sle2c1) − (B6 Cdkn2c CT −B6 Gapdh CT)]). Gapdh expression normalized to cyclophylin did not vary between strains, B cell types, or experimental conditions (data not shown).
Antibody measurements
Anti-dsDNA IgG was measured as previously described (2) in sera from the NZB crosses diluted 1:100. Relative units were standardized using a B6.TC serum, arbitrarily setting the reactivity of a 1:100 dilution of this control serum to 100 units. Mice were immunized with 100 μg of NP-KLH (Biosearch Tech.) precipitated in Alum (Pierce Biotech.) at a 1:1 ratio as previously described (21). Primary immune responses were measured 7 d later. For secondary responses, mice were boosted with the same dose of NP-KLH and Alum three wks after the primary immunization. Mice were sacrificed 10 d after the secondary immunization. Anti-NP IgM and IgG were quantified by ELISA as previously described (21). Briefly, plates were coated with NP3-BSA (Biosearch Tech.) at 10 μg/ml. After blocking, serially diluted sera were incubated in duplicate, and the NP-bound Abs were detected by alkaline phosphatase (AP)-conjugated goat anti-mouse IgG (Chemicon) or IgM (Southern Biotech). Each immunization was performed in duplicate with three to seven mice per strain. For in vitro measurement of Ab production, sB cells were stimulated with 8 μg/ml of either LPS (Sigma) or anti-IgM Fab2 (Abcam) for up to 72 h, and Pc B1a cells were stimulated with 5 μg/ml of LPS for 48 h. Total IgM in supernatants diluted up to 1:250 was measured by sandwich ELISA on plates coated with goat-anti mouse IgM (ICN/Cappel) at 1 μg/ml and revealed with AP-conjugated goat-anti mouse IgM (Southern Biotech).
p18INK4c immunoprecipitation and Western blot
Cells were lysed in RIPA-lysis buffer (0.1% Nonidet P-40, 50 mM NaF, 2 mM sodium orthovanadate, 1 μg/ml aprotinin, 2 mM PMSF, 10 μg/ml leupeptin, and 2 μg/ml pepsatin A in PBS; all reagents from Sigma) on ice for 60 min. After centrifugation, the supernatants were assayed for protein concentration (Bradford assay; Bio-rad). Total proteins (250-500 μg) were immunoprecipitated (IP) with p18 mouse monoclonal Ab (sc-56453 Santa Cruz Biotech.) and protein-G beads (Amersham) at 4°C overnight. The beads were washed four times with cold RIPA buffer containing protease inhibitors, then boiled for 10 min in the presence of 2× sample buffer (Biorad), and the released proteins fractionated by SDS-PAGE in 10-15 % gels. Proteins were electroblotted onto 0.45-μm pore size nitrocellulose PVDF membranes, which were blocked at room temperature with 5% nonfat milk in TBS and 0.1% Tween-20. Membranes were then incubated overnight with goat anti-p18 (sc-864), or anti-IgG (Southern Biotech.). The blots were washed three times with 0.1% Tween-20 in TBS and incubated with Clean Blot IP detection reagent-HRP (Thermo Scientific). Blots were developed using an enhanced chemiluminescence detection system (Amersham).
Cdkn2c sequencing and promoter analysis
The Cdkn2c promoter, 5′UTR, exons and 3′ UTR were sequenced from genomic DNA extracted from B6 and B6.Sle2c1 mice. The PCR primers used for sequencing are listed in Table 1. Genomic DNAs from the Sle2c1 recombinant strains and parental strains were genotyped for the −74 C>T SNP with the following primers: 5′-AGCCTCTAAGGGCCTCCGCC-3′ and 5′-GCAACTGCTGCTACGGTTGCC-3′. Reporter constructs were generated by cloning into the firefly luciferase reporter plasmid pGL4-basic (Promega, Madison, WI) promoter segments of different lengths upstream from the putative translation initiation site of the mouse Cdkn2c gene. Two constructs starting at the −279 and −1209 position contained the −74 C>T SNP, and another construct starting at position −52 served as a negative control (numbering of the constructs is from the transcriptional start site). Promoter segments were amplified from either B6 or B6.Sle2c1 genomic DNA. The following primers were used: -1209 construct forward: 5′-TCCTCGAGCCCTTTGTTGTTACCTGATTC-3′ (XhoI site), and reverse: 5′;-ACTGAGATCTTGCCGGTTTCTTATCCCT-3′ (BglII site); −279 construct forward: 5′-AGGGCTCGAGGAGACTAGCGAAGCGAGA-3′ and reverse primer same as for the −1209 construct; −52 construct forward: 5′-TTCTCGAGTCAACTCAAAAAGCGCTCAAT-3′ and reverse: 5′-GACAAGATCTCTTGCGCGTCTTTCCTTTA-3′. All constructs were confirmed by sequencing. Transient transfections of NIH3T3 cells were performed using Lipofectamine 2000 (Invitrogen) in serum-free medium. Cells at 80-90% confluence were transfected with 1.6 μg of Cdkn2c reporter plasmids or PGL-4.10 empty vector, plus 160 ng of Renilla luciferase plasmid (Promega) as a control for transfection efficiency. In some experiments, 750 ng of the pCDNA3-E2F1 expression vector (a kind gift from Dr. Shiwu Li, University of Florida) was also transfected into the cells. After 6 h of incubation with the Lipofectamine:DNA complexes, the medium was changed to DMEM supplemented with 10% FBS, and incubated for an additional 48 h. Raji cells were transfected with 5 μg of Cdkn2c reporter plasmids and 0.5 μg of Renilla luciferase expression plasmid using DEAE-dextran (Millipore). Plasmid DNA mixed with DEAE-dextran in Tris-saline (pH 7.4) was added to 6 x 106 cells and incubated for 12-14 h at 37° C in serum-free media, when chloroquine diphosphate was added to the cells and incubated for an additional 2.5 h. After washing, the cells were re-plated and incubated for an additional 48 h at 37°C in complete Raji medium. Cell extracts were prepared from transfected cells with the Dual Luciferase Assay System (Promega), and luciferase activity was measured with a Lumat LB 9507 luminometer (Berthold Tech.). Firefly luciferase activity was normalized to Renilla luciferase activity. These experiments were repeated with three independent DNA preparations.
Table 1.
Cdkn2c primers.
| Forward primer | Reverse primer | amplicon* | |
|---|---|---|---|
| Promoter and Exon 1 | 5′-AGCCTCTAAGGGCCTCCGCC-3′ | 5′-GCAACTGCTGCTACGGTTGCC-3′ | −423 to 45 |
| Promoter and Exon 1 | 5′- TGGGGGCGGGTTTTTCAACTCA-3′ | 5′- GCGTCCGATGATGGGAAGGGT-3′ | −81 to +417 |
| Exon 1 | 5′-GCCGGAGGACCGCCAAGAAC-3′ | 5′- TCTCCGGAGGCTGGTGGGTG-3′ | +271 to +707 |
| Exon 1 | 5′- TCGCCGTGCACCGTCTTCAG-3′ | 5′- GTCAGCTCAGACAACACCGCGA-3′ | +607 to +1075 |
| Exon 1 | 5′- TCTGGACTACCCCCTTCGGCT-3′ | 5′- CCCGCCCCTCGATTCACACG-3′ | +920 to +1327 |
| Exon 2 | 5′- CAGTCCTTCTGTCAGCCTCC-3′ | 5′- CTCCGGATTTCCAAGTTTCA-3′ | +1386 to +1652 |
| Exon 3 | 5- TTGTTGTGGCTCAAGAGCTG-3′ | 5′- GCGGTGTTAGCCAATGAAAT-3′ | +1627 to +2377 |
| 3′UTR | 5′- TCGACTTGGCCAGGTTCTAT-3′ | 5′- CACACTACACCAGGCTTCCA-3′ | +1928 to +2510 |
All the locations were calculated relative to +1 transcription start site
Statistical analysis
Data was analyzed with Graph pad Prism 4.0 software with the statistical tests indicated in the text. Non-parametric tests were used when data was not distributed normally. Means, the standard errors of the mean (SEM), and the levels of statistical significance are shown in the figures.
Results
Fine-mapping of the expansion of the Pc B1a cell subset
The Sle2c1 interval is located in the NZB-derived terminal region of Sle2 (Fig. 1A). To fine-map the location of the gene responsible for expanding the Pc B1a cell subset, we generated three B6.Sle2c1 recombinant strains (Fig. 1B). The resolution was limited because most of the few SNPs in this region that are published as polymorphic between NZB and B6 have in fact the B6 allele (data not shown). The percentage and absolute number of Pc B1a cells, as well as the Pc B1a/B2 cell ratio in B6.Sle2c1.Rec1 and B6.Sle2c1.Rec1a mice were similar to that of B6.Sle2c1 mice, while the B6.Sle2c1.Rec1b values were similar to that of B6 (Fig. 2). These results map Sle2c1 to a ∼ 6 Mb critical interval located between rs28132547 and D4MIT278 (Fig. 1B). This interval contains 19 named genes, 14 of which are expressed in B cells (supplemental Table 1), as well as 14 anonymous protein-coding transcripts.
Figure 2.

Mapping the Pc B1a cell expansion in the Sle2c1 recombinants. A, Representative FACS plots of B220 and CD5 staining of Pc lymphocytes in B6, B6.Sle2c1 and its recombinants showing the B1a (solid line) and B2 (dashed line) gates. B, Percentages and absolute numbers of Pc B1a cells, and B1a/B2 cell ratios in the B6.Sle2c1 strain and its recombinants compared to B6. ***: p < 0.001; statistical significance of Dunnett' multiple comparison tests with B6 values. The graphs show means and SEM for 10-25 mice per strain at 5-6 months of age.
Sle2c1 enhances NZB autoimmune phenotypes
To determine the contribution of Sle2c1 to autoimmune pathogenesis, we compared cohorts of NxSle2 and NxSle2c1 females, in which the entire Sle2, or only Sle2c1 was expressed as a homozygous locus (NxSle2 and NxSle2c1, respectively) to NxB mice in which these loci were heterozygous, all on a NZB x B6 heterozygous background. This complementation analysis previously determined that Sle1 (22) and Sle1c (23) significantly enhanced the autoimmune phenotypes of the NZW and NZB heterozygous genomes, respectively. At 12 months of age, both NxSle2 and NxSle2c1 mice showed a significantly increased splenomegaly (Fig. 3A). In addition, NxSle2c1 mice showed an increased percentage of B1a cells in the spleen (Fig. 3B), and an expansion of the transitional T1 B cell subset at the expense of follicular B cells (Fig. 3C) as compared to NxB mice. These two phenotypes were even more accentuated in NxSle2 mice. However, the Pc B1a cell compartment was not expanded in NxSle2c1 mice, as it was in NxSle2 mice (Fig. 3D). Both NxSle2 and NxSle2c1 mice produced significantly more anti-dsDNA IgG than NxB mice at 7 months of age (Fig. 3E), but this difference was not maintained at either 9 or 12 months of age (data not shown). The expression of the whole Sle2 locus on the NZB background significantly enhanced renal pathology as compared to NxB (comparison of the distribution of GN scores, χ2 = 26.84, p < 0.001), but there was no difference between the renal pathology of NxSle2c1 and NxB mice (Fig. 3F). Furthermore, the amount of glomerular immune complex deposits was not different between these two latter stains (data not shown). Overall, these results show that Sle2c1 homozygosity enhanced NZB cellular phenotypes that are associated with autoimmunity, namely splenomegaly, and expanded transitional B cells and splenic B1a cells, and resulted in an accelerated production of anti-dsDNA Ab. But Sle2c1 expression was not sufficient to induce strong autoimmune phenotypes, including renal pathology, as was observed for the entire Sle2 locus. This suggests that Sle2c1 contribution to autoimmune pathogenesis requires the presence of other susceptibility loci.
Figure 3.

Sle2c1 homozygosity enhanced splenic B1a and T1 B cell expansion but not renal pathology on an NZB heterozygous background. Comparisons between NxB, NxSle2c1 and NxSle2 spleen weight (A), percentage of splenic B1a cells (B), AA4.1+ IgM+ CD21lo CD23lo T1 over AA4.1− IgM+ CD21int CD23+ follicular B cell (T1/FOB) ratio (C), Pc B1a/B2 cell ratio (D), serum anti-dsDNA IgG at 7 months of age (E), and GN score distribution (F). Graphs A-E show individual mice and means plus SEM. *: P < 0.05, **: P < 0.01, and ***: P < 0.001. Graph E shows the distribution of GN scores in ten 12 mo old mice from each strain. Scores were grouped according to the type of lesion (H: hyaline, MM: mesangial matrix, MC: mesangial cellular, and P: proliferative) and the severity score (all scores were either 3 or 4).
Cdkn2c expression is significantly decreased in B6.Sle2c1 B cells
To gain insight into the mechanisms by which Sle2c1 expands the Pc B1a cell compartment, we performed microarray analyses on Pc B1a cells and sB cells obtained from B6.Sle2c1 and B6 mice. 534 genes for Pc B1a cells and 120 genes for sB cells were differentially expressed between the two strains (p ≤ 0.001). Among these genes, the Cyclins and Cell Cycle Regulation pathway was differentially expressed with LS permutation p values of 8 × 10−4 for Pc B1a cells and 10−3 for sB cells (Fig. 4A and B). The specific genes within this pathway that were differentially expressed between the two strains were not identical between the two cell types, which corroborates the existence of a B1a-specific cell cycle regulation (24). However, the expression of one gene, Cdkn2c, was about 4 folds lower in B6.Sle2c1 than in B6 for both Pc B1a and sB cells (Fig. 4A and B). Cdkn2c is located within the Sle2c1 interval (Fig. 1B). RT-PCR confirmed a significantly lower Cdkn2c expression in B6.Sle2c1 and Sle2c1-expressing B6.TC sB cells as compared to B6 sB cells (Fig. 4C). qRT-PCR showed that B6.Sle2c1, B6.Sle2c1.Rec1a and B6.TC sB cells expressed significantly less Cdkn2c than B6 while B6.Sle2c1.Rec1b sB cells expressed a similar level as B6 (Fig. 4D). Correspondingly, p18 protein was expressed at a low level in B6.Sle2c1.Rec1a sB cells comparatively to B6 and B6.Sle2c1.Rec1b (Fig. 4E). Cdkn2c message expression was also low in B6.Sle2c1.Rec1a and B6.TC Pc B1a cells, and high in B6.Sle2c1.Rec1b and B6 Pc B1a cells (Fig. 4F). A decreased expression of p18 protein was found in both B6.Sle2c1 and B6.TC Pc B1a cells as compared to B6 (Fig. 4I). Expression of the other cyclin-dependent kinase inhibitors was also examined by qRT-PCR in sB and Pc B1a cells (suppl. Fig 1). While the results were inconclusive for Cdkn1a, this analysis confirmed the microarray data for Cdkn1b expression, which was decreased in Sle2c1-expressing B1a cells, and for Cdkn2d, which was increased in sB cells and decreased in Pc B1a cells expressing Sle2c1. Overall, these results showed an association between a low level of Cdkn2c expression in B cells and elevated numbers of Pc B1a in the Sle2c1 congenic recombinants (this report), and in B6.TC mice that carry the Sle2c1 locus (2).
Figure 4.

Cdkn2c expression in B cells from B6.Sle2c1 recombinant mice. Cyclins and Cell Cycle Regulation pathway genes differentially expressed (p < 0.05) in Pc B1a (black) or sB (white) cells between B6 and B6.Sle2c1 mice, showing the p values (A) and the ratios of geometric means (5 mice per strain), in which negative values indicate genes under-expressed in B6.Sle2c1 B cells (B). The double arrow between A and B points to Cdkn2c. C, Representative Cdkn2c and β-actin (Actb) expression in B6.Sle2c1, B6.TC and B6 sB cells. D, Cdkn2c message expression in sB cells from B6, B6.Sle2c1, B6.Sle2c1.Rec1a, B6.Sle2c1.Rec1b and B6.TC mice. E, Representative p18 expression in B6.Sle2c1.Rec1a, B6.Sle2c1.Rec1b and B6 sB cells. IgG expression was used as control. F, Cdkn2c message expression in Pc B1a cells from B6, B6.Sle2c1, B6.Sle2c1.Rec1a, B6.Sle2c1.Rec1b and B6.TC mice. I, Representative p18 expression in Pc B1a cells from B6.Sle2c1 and B6 mice immunized with NP-KLH (right), and unimmunized B6 and B6.TC mice (left). The densitometry analysis normalized to IgG expression is shown on the graph on the right. In graphs D and F, qRT-PCR data was normalized to Gapdh and expressed as fold difference with one B6 value. Individual mouse values and means plus SEM are represented.
p18, the cyclin-dependent kinase inhibitor encoded by Cdkn2c, prevents the activation of cyclin D2 and D3 by CDK4 and CDK6, and leads to early G1 cell cycle arrest (25). p18-mediated G1 arrest is necessary for differentiation into functional Ab-secreting plasma cells, and p18-deficient mice display a defective Ab production in response to immunization (26). We hypothesized that a functional consequence of a low p18 expression in the Sle2c1 recombinants should be a defective Ab response to immunization with NP-KLH. Cdkn2c message expression was significantly lower in the spleens of immunized B6.Sle2c1 and B6.Sle2c1.Rec1a than B6 and B6.Sle2c1.Rec1b mice (Fig. 5A). As previously reported (26), Cdkn2c expression increased in B6 mice upon secondary immunization, but it remained unchanged in B6.Sle2c1 mice. This result was confirmed at the protein level, with p18 being barely detectable in the spleen of B6.Sle2c1 mice after secondary immunization (Fig. 5B). Similar data was obtained in the primary immunization, although the overall level of p18 expression was much lower than in the secondary immunization (data not shown). Serum anti-NP IgM (Fig. 5C-D) and IgG (Fig. 5E-F) were significantly lower in B6.Sle2c1 and B6.Sle2c1.Rec1a than in B6 and B6.Sle2c1.Rec1b mice seven days after immunization, indicating that p18 expression affects functional plasma cell differentiation in the Sle2c1 recombinants as it did in the p18-deficient mice (26). As expected, B6.Sle2c1 mice also produced less anti-NP Ab than B6 mice after secondary immunization (Fig. 5G). The number and frequency of NP+ CD138+ plasma cells was however not affected by Sle2c1 expression (data not shown). Interestingly, B6.Sle2c1 heterozygous mice showed an intermediate level of Cdkn2c and p18 after secondary immunization (sup. Fig. 2A and Fig. 5B). The anti-NP IgM and IgG production was however similar between B6.Sle2c1 homozygous and heterozygous mice (sup. Fig. 2B and C). This suggests that the Sle2c1 allele controls p18 expression in a dose-dependent manner, but that a 50% reduction of p18 expression is sufficient to significantly impair plasma cell differentiation. Overall, these results show that mice expressing the NZB allele of Sle2c1 mount a lower Ab response to TD immunization as compared to mice expressing the B6 allele of Sle2c1, which is consistent with the respective levels of Cdkn2c expression found in the sB cells of these mice.
Figure 5.

Decreased Cdkn2c expression in B6.Sle2c1 mice is associated with an impaired Ab response to NP-KLH immunization. A, qRT-PCR of Cdkn2c expression in splenocytes of unimmunized and NP-KLH immunized mice (1st: primary, 2nd: secondary immunization). Data were normalized to Gapdh and to one of the unimmunized B6 samples. B, Western blot analysis of p18 expression in the spleens of B6, B6.Sle2c1 heterozygotes and B6.Sle2c1 mice after secondary NP-KHL immunization. IgG expression was probed as control. Quantitation of the bands by densitometry is shown on the right. C - D, Anti-NP IgM from B6, B6.Sle2c1.Rec1a, B6.Sle2c1.Rec1b and B6.Sle2c1 mice after primary NP-KLH immunization in sera diluted 1:100 (C), and in serially diluted sera (D). E - F, Anti-NP IgG from B6, B6.Sle2c1.Rec1a, B6.Sle2c1.Rec1b and B6.Sle2c1 mice after primary NP-KLH immunization in sera diluted 1:100 (E), and in serially diluted sera (F). G, Anti-NP IgM and IgG in 1:100 diluted sera from B6 and B6.Sle2c1 mice after secondary NP-KLH immunization. The graphs show means and SEM of at least 5 mice per group. *: p < 0.05, **: p < 0.01, and **: p < 0.001: statistical significance of non-parametric tests with B6 values with multiple test correction when appropriate. In E, the difference was not significant between B6 and B6.Sle2c1 values with a multiple comparison test, but was different (p = 0.02) with a Mann-Whitney test. In D and F, the tests were performed between the B6.Sle2c1.Rec1a and B6 values for each dilution.
To compare the effect of low Cdkn2c expression between sB and Pc B1a cells, purified cells were stimulated in vitro. Cdkn2c expression increased overtime in sB cells stimulated with LPS in all strains, but it remained significantly lower in B6.Sle2c1 and B6.Sle2c1.Rec1a than in B6 and B6.Sle2c1.Rec1b mice (Fig. 6A). Accordingly, a significantly lower amount of IgM was obtained from LPS-stimulated B6.Sle2c1 and B6.Sle2c1.Rec1a sB cells than from B6 and B6.Sle2c1.Rec1b sB cells (Fig. 6B). Similar results were obtained with sB cells stimulated with anti-IgM (Fig. 6C and data not shown). These results are consistent with those obtained with the TD-immunization showing an impaired Ab production from sB cells expressing low levels of Cdkn2c. Pc B1a cells from B6.Sle2c1 and B6.Sle2c1.Rec1a mice expressed a lower level of Cdkn2c than Pc B1a cells from B6 or B6.Sle2c1.Rec1b mice without significant change in the presence or absence of LPS stimulation (Fig. 6D). However, LPS-stimulated B6.Sle2c1 and B6.Sle2c1.Rec1a B1a cells secreted significantly more IgM than B6 or B6.Sle2c1.Rec1b B1a cells (Fig. 6E). This data corroborate earlier studies that have shown that B1a cells do not undergo G1 arrest for Ab secretion (27). Therefore high IgM secretion occurs in B1a cells expressing the NZB allele of Sle2c1, in spite of low Cdkn2c expression. Finally, we have previously shown that B6.Sle2 Pc B1a cells proliferated more than B6 Pc B1a cells, either spontaneously or in response to LPS, while no difference was observed for CD5-negative B cells (8). In vitro LPS stimulation indicated that this phenotype mapped to Sle2c1: In both spleen and Pc, more B6.Sle2c1 CD5+ B cells proliferated than B6 CD5+ B cells, but there was no difference for CD5− cells (Fig. 6F). Assessment of the spontaneous B cell proliferation status ex-vivo found a significantly higher number of Ki67+ proliferating Pc B1a cells in the strains expressing the NZB allele of Sle2c1 (B6.Sle2c1 and B6.Sle2c1.Rec1a) as compared to the B6 allele (B6 and B6.Sle2c1.Rec1b) (Fig. 6G). Overall, these results show that Cdkn2c, a gene located within the Sle2c1 critical interval, is expressed at lower levels in B6.Sle2c1 B cells. This low level expression impaired functional plasma cell differentiation in sB cells but not in Pc B1a cells, while it increased B1a but not B2 cell proliferation.
Figure 6.

Decreased Cdkn2c expression has opposite effects in B6.Sle2c1 sB cells and Pc B1a cells. A, Cdkn2c expression in sB cells stimulated with LPS. Values for B6.Sle2c1.Rec1a (square) and B6.Sle2c1.Rec1b (star) were obtained only at 72 h. B, IgM secretion by sB cells after 72 h stimulation with LPS. ELISA were performed on supernatants diluted 1:100. C, Cdkn2c expression in sB cells stimulated with anti-IgM. D, Cdkn2c expression in Pc B1a cells cultured for 72 h with or without LPS. E, IgM secretion by Pc B1a cells after 72 h stimulation with LPS. The X axis shows the supernatant dilutions at which the ELISA assay was performed. For A and C, Cdkn2c was normalized to unstimulated sB and Pc B1a cell B6 values. F. Representative CFSE dilution in splenic or Pc B220+ CD5+ (B1a) and B220+ CD5− (B2) cells after 72 h LPS stimulation performed in 5 mice per strain. The filled grey histograms indicate B6 and the open black histograms indicate B6.Sle2c1 values. G, Absolute numbers of proliferating Ki67+ sB cells and Pc B1a cells in B6, B6.Sle2c1, and B6.Sle2c1.Rec1a and B6.Sle2c1.Rec1b mice. In B, D, and G, graphs show individual mice and means plus SEM. Graphs in A, C, and E show means plus SEM, and the differences between B6 and B6.Sle2c1 values were significant at p < 0.05 at all time points or dilutions. Statistical significance in B, D and G was obtained with Dunnett's multiple comparison tests to B6 values. *: p < 0.05, and **: p < 0.01.
A novel promoter SNP regulates Cdkn2c transcription
The sequence of the Cdkn2c exons, 5′ UTR, and 3′UTR regions was identical between B6 and B6.Sle2c1 mice (data not shown). We identified however a novel SNP at position −74 in the promoter in which the B6 allele is C and the Sle2c1 allele is T (suppl.Fig. 3A). This region is highly conserved in the Cdkn2c promoter in mammals (suppl. Fig. 3B). An in silico analysis (28) predicted that the −74 C → T transition results in the loss of binding sites for the NRF2 and Hunchback transcription factors, while it creates a binding site for YY1 that is adjacent to the already existing YY1 site. YY1 can either negatively or positively regulate transcription depending on the context of the adjacent transcription factors (29). Therefore a detailed analysis of the contribution of each transcription factor binding at this site will be necessary to understand how they impact Cdkn2c expression. No other mutation was found in the Sle2c1 allele of the Cdkn2c promoter. We genotyped the Sle2c1 recombinant strains as well as the parental strains for this SNP (Table 2) and found a perfect concordance between 1) the imputed genomic origin of the region (B6.Sle2, NZM2410, NZB, all carrying the T allele), and 2) the expansion of the Pc B1a cell compartment. Indeed, B6.Sle2c1.Rec1b mice carry the C allele and show B6-like levels of Pc B1a cells, while all the strains with T allele have been shown to expand this compartment ((3,9,30) and this report). Interestingly, NZW mice have elevated numbers of Pc B1a cells ((31) and Morel, unpublished), but they carry the −74 C allele.
Table 2. Genotype at the −74 C>T SNP in the B6.Sle2c1 recombinant and parental strains as well as the relative expansion of the Pc B1a cell compartment relative to B6.
| Strain | ↑Pc B1a | −74 C>T |
|---|---|---|
| B6 | − | C |
| B6.Sle2 | + | T |
| B6.Sle2c1 | + | T |
| B6.Rec1 | + | T |
| B6.Rec1a | + | T |
| B6.Rec1b | − | C |
| NZB | ++ | T |
| NZW | +/− | C |
| NZM2410 | ++ | T |
The impact of the −74 C/T SNP on Cdkn2c transcription was determined by comparing the expression of a luciferase reporter gene driven by each allele. Three promoter-luciferase constructs were produced within the region that has been shown to induce maximal transcription (32), with two constructs (−1209 and −279) containing the SNP, and one construct (-52) corresponding to the minimal promoter (33) not containing the SNP. As expected, the -52 construct induced a low luciferase expression that was similar between the two alleles (Fig. 7). For both constructs containing the SNP, however, the C allele induced a significantly higher level of transcription than the T allele. Cdkn2c expression is regulated by the E2F1 transcription factor (30), and the -1209 and -279 constructs contain two sites that loosely fit the consensus DNA sequence of E2F-binding sites (34) (suppl. Fig. 3A). As a preliminary to a detailed analysis of the Cdkn2c promoter, we extended these experiments to determine whether the difference of expression between the Sle2c1 and B6 allele is maintained in the presence of E2F1. E2F1 increased the activity of the Cdkn2c promoter constructs −1209 and −279 by 190 and 225 folds, respectively (Fig. 7B). Interestingly, even in the presence of this strong transcriptional induction, the C allele induced a significantly higher level of transcription than the T allele for both constructs. A decreased transcriptional activity of the −279 Sle2c1 construct was also observed in Raji cells (Fig. 7C). These results strongly suggest that the −74 C →T transition is responsible for the low level of P18INK4c found in the B cells expressing the NZB allele of Cdkn2c.
Figure 7.

The −74 C/T promoter SNP regulates Cdkn2c transcription. Luciferase assays in NIH3T3 (A-B) and Raji (C) cells transfected with three Cdkn2c promoter-luciferase constructs with serial 5′ deletions. In B, an E2F1 expression vector was co-transfected with the Cdkn2c promoter constructs. Data was expressed as the relative induction of luciferase activity induced by each promoter construct over empty vector. *: p < 0.05, **: p < 0.01, and ***: p < 0.001.
Discussion
Sle2c1 is the major locus contributing to the expansion of Pc B1a cells in the NZM2410 mouse model of lupus. Genetic analysis mapped Sle2c1 to a 6 Mb NZB-derived region of chromosome 4 based on the analysis of two new recombinant intervals, Sle2c1.Rec1a and Sle2c1.Rec1b. An expansion of both Pc B1a cells and splenic NKT cells was mapped by another group to a larger interval on the NZB chromosome 4 that includes Sle2c1 (35). The phenotype of the two recombinant intervals analyzed in that study are consistent with our results, and we predict that the NZBc4S interval that has a B6-like phenotype is telomeric to Sle2c1. In addition, B6.Sle2c1 and B6.Sle2 mice have a normal NKT cell level (data not shown), suggesting that the increased NKT cell number is an NZB phenotype that maps to the NZW-derived part of Sle2. The Sle2c1 critical interval contained too many genes, most of them with known expression in B cells, for a direct candidate gene analysis. Gene expression profiling of both sB and Pc B1a cells provided the leading cues by identifying among the many genes differentially expressed a significant involvement of the Cell Cycle regulation pathway. Furthermore, the expression of one of its member, Cdkn2c, was four folds lower in Sle2c1 B cells of both types, and mapped to the Sle2c1 interval. We have confirmed the difference in Cdkn2c expression with qRT-PCR. Moreover, we have identified a unique promoter polymorphism for which there is a complete concordance between, on one hand, the C allele and B6-like Pc B1a levels, and on the other hand, the T allele and an enlarged Pc B1a compartment. The only exception is the NZW strain, indicating that the accumulation of B1a cells in that strain is sustained through a different mechanism, which is not surprising since a large number of genetic alterations have already been found to affect the size of the B1a cell compartment (10).
We have initially reported that Sle2c did not increase clinical disease in B6.Sle1.Sle3 mice (9). This experiment was however conducted with a long Sle2c interval, in which we have identified, in addition to Sle2c1, a strong Sle2c2 suppressor locus (16). Such “gene masking” resulting from the proximity of susceptibility and suppressor loci has been reported in the NOD mouse model of diabetes (36), and is likely to represent a frequent occurrence in the genetic architecture of complex traits. When Sle2c1 contribution was analyzed separately from Sle2c2, we found that it promoted a severe renal and skin pathology in B6.lpr mice by favoring Th17 differentiation (15). Here we showed an allele-dose effect of Sle2c1 expression on a heterozygous (NZB X B6)F1 background, with the homozygous expression of Sle2c1 inducing lymphocyte expansion and an increased number and percentage of splenic B1a and T1 B cells, as well as an accelerated production of anti-dsDNA IgG Abs. These phenotypes have all been implicated in lupus pathogenesis, but were not sufficient to induce renal pathology as we observed when the entire Sle2 locus was expressed on an NZB heterozygous background. Therefore the Sle2c1 locus is associated not only with B1a cell homeostasis but also with a subset of phenotypes leading to lupus pathogenesis, the severity of which depends on the nature of the other susceptibility loci it interacts with.
It is well-established that cell cycle regulation differs between B1a and conventional B cells (24). Cyclins D2 and D3 are both expressed in B cells with overlapping functions that are not totally defined. Cyclin D2 is required for B1a but not B2 cell development (37), while cyclin D3 deficiency reduces the number of follicular B cells (38) and severely impairs the development of germinal center B cells (39), but does not affect B1a cell numbers or functions (38). However, temporal inactivation of cyclin D3 complexes in late G1 phase blocks B1a cell proliferation (38). In addition, phorbol ester (PMA) stimulation is sufficient to induce proliferation in B1a but not in B2 cells. This difference has been attributed to the activation of cyclin D3-CDK4 complexes that phosphorylate the retinoblastoma gene product (pRb) in PMA-stimulated B1a but not B2 cells (40). Overall these results emphasize the crucial role of cyclin D2 and D3 complexes in B1a cell proliferation, and predict that gene products such as p18, which fine-tune the amount of activated D2 and D3 complexes, would impact the accumulation of the self-renewing B1a cells. It is not surprising that a decreased level of p18 has a different outcome on B1a and B2 cells due to the differences in cell cycle regulation between these two cell types. The reduced p18 level in Sle2c1 sB cells reproduced the impaired T-dependent humoral response of p18-deficient B cells due to the p18-dependent G1-arrest in plasma cells (26). The impaired response to T-dependent immunization that we have previously reported in B6.TC mice (21) could be due at least to their expression of Sle2c1 and p18-deficiency. Other genetic factors positively regulating the development of plasma cells in the NZM2410 model (41) could compensate for this defect and allow the robust differentiation of autoreactive plasma cells that we have observed in B6.TC mice (21,41). It should be noted that according to recent results obtained in the BWF1 mice, some of these anti-dsDNA plasma cells should be of B1a origin (11), and may therefore bypass the G1-arrest requirement. The proliferation of Sle2c1 B2 cells was not affected, suggesting that p18 plays a non-essential role in the cell cycle regulation of this cell type until terminal differentiation, which is corroborated by the normal number of splenic B cells in B6.Sle2c1 mice. In contrast, Ab production was not impaired in Sle2c1 B1a cells, but their proliferation was increased. This latter phenotype provides an explanation for the age-dependent accumulation of B1a cells observed in these mice.
The identification of p18 as a key regulator of B1a cell homeostasis is novel, and it should open new venues to understand the specificity of cell cycle regulation in the various lymphocyte subsets. In addition, the NZB mouse has long been recognized as a model for chronic lymphocytic leukemia (CLL) (42). Defects in p18 expression have been associated with multiple tumors, including multiple myeloma (43), but not CLL so far. As Sle2c1 represents a major locus responsible for NZB B1a cell accumulation, a better analysis of cell cycle regulation in Sle2c1 B1 a cells may shed light on CCL induction.
An association between a dysregulated cell cycle due to a deficiency in a cyclin kinase inhibitor and lupus has been established for Cdkn1a/p21CIP1/WAF1. p21-deficient mice develop a lupus-like disease through the accumulation of activated/memory T cells (44,45), and human CDKN1A polymorphisms leading to decreased p21 levels have been associated with SLE (46). Contrary to Cdkn2c, Cdkn1a expression does not seem to be significantly affected in B cells expressing Sle2c1. In addition, it was very recently shown that B cell homeostasis is regulated by the RAPL-mediated translocation of Cdkn1b/p27Kip1 to the nucleus, and that the sequestration of p27 in the cytoplasm leads to a lupus-like phenotype (47). Cdkn2c/p18 has never been associated with lupus or any autoimmune phenotype, and to our knowledge, the −74 C → T SNP is the first naturally occurring polymorphism that has been identified to regulate the size of the B1a cell compartment. The recent developments that have uncovered new mechanisms by which B1a cells can contribute to autoimmunity, either directly through the production of pathogenic antibodies, or indirectly thought their promotion of Th17 differentiation, identify Cdkn2c/p18 as a novel type of lupus susceptibility gene, whose characterization is likely to unravel emerging functional pathways contributing to this complex disease.
Supplementary Material
Acknowledgments
We thank Dr. Thomas Chiles for critical review of the manuscript, Cecilia Lopez for microarray processing, Leilani Zeumer for excellent technical help, and Xuekun Su for outstanding animal care.
This work was supported by a National Institutes of Health grant RO1 AI068965 to LM and K01AR056725 to ZX.
Abbreviations used in this paper
- B6
C57BL/6
- Pc
peritoneal cavity
- B6.TC
BcN/LmJ
- NxSle2
(NZB X B6.Sle2)F1
- NxSle2c1
(NZB X B6.Sle2c1)F1
- NxB
(NZB X B6)F1
- GN
glomerulonephritis
- sB cells
splenic B cells
- IP
immunoprecipitation
- p18
p18INK4c
- SEM
standard error of the mean
- CLL
chronic lymphocytic leukemia
References
- 1.Morel L, Rudofsky UH, Longmate JA, Schiffenbauer J, Wakeland EK. Polygenic control of susceptibility to murine systemic lupus erythematosus. Immunity. 1994;1:219–229. doi: 10.1016/1074-7613(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 2.Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, Wakeland EK. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci U S A. 2000;97:6670–6675. doi: 10.1073/pnas.97.12.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohan C, Morel L, Yang P, Wakeland EK. Genetic dissection of systemic lupus erythematosus pathogenesis - Sle2 on murine chromosome 4 leads to B cell hyperactivity. J Immunol. 1997;159:454–465. [PubMed] [Google Scholar]
- 4.Duan B, Morel L. Role of B-1a cells in autoimmunity. Autoimmun Rev. 2006;5:403–408. doi: 10.1016/j.autrev.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 5.Karras JG, Wang ZH, Huo L, Howard RG, Frank DA, Rothstein TL. Signal transducer and activator of transcription-3 (STAT3) is constitutively activated in normal, self-renewing B-1 cells but only inducibly expressed in conventional B lymphocytes. J Exp Med. 1997;185:1035–1042. doi: 10.1084/jem.185.6.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tumang JR, Frances R, Yeo SG, Rothstein TL. Cutting Edge: Spontaneously Ig-secreting B-1 cells violate the accepted paradigm for expression of differentiation-associated transcription factors. J Immunol. 2005;174:3173–3177. doi: 10.4049/jimmunol.174.6.3173. [DOI] [PubMed] [Google Scholar]
- 7.Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70−. J Exp Med. 2011;208:67–80. doi: 10.1084/jem.20101499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Z, Butfiloski EJ, Sobel ES, Morel L. Mechanisms of peritoneal B-1a cells accumulation induced by murine lupus susceptibility locus Sle2. J Immunol. 2004;173:6050–6058. doi: 10.4049/jimmunol.173.10.6050. [DOI] [PubMed] [Google Scholar]
- 9.Xu Z, Duan B, Croker BP, Wakeland EK, Morel L. Genetic dissection of the murine lupus susceptibility locus Sle2: contributions to increased peritoneal B-1a cells and lupus nephritis map to different loci. J Immunol. 2005;175:936–943. doi: 10.4049/jimmunol.175.2.936. [DOI] [PubMed] [Google Scholar]
- 10.Berland R, Wortis HH. Origins and functions of B-1 cells with notes on the role of CD5. Ann Rev Immunol. 2002;20:253–300. doi: 10.1146/annurev.immunol.20.100301.064833. [DOI] [PubMed] [Google Scholar]
- 11.Enghard P, Humrich JY, Chu VT, Grussie E, Hiepe F, Burmester GR, Radbruch A, Berek C, Riemekasten G. Class switching and consecutive loss of dsDNA reactive B1a B cells from the peritoneal cavity during murine lupus development. Eur J Immunol. 2010 doi: 10.1002/eji.200940050. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 12.Zhong X, Gao W, Degauque N, Bai C, Lu Y, Kenny J, Oukka M, Strom TB, Rothstein TL. Reciprocal generation of Th1/Th17 and T(reg) cells by B1 and B2 B cells. Eur J Immunol. 2007;9:2400–2404. doi: 10.1002/eji.200737296. [DOI] [PubMed] [Google Scholar]
- 13.Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL, Le Tv, Lorenz RG, Xu H, Kolls JK, Carter RH, Chaplin DD, Williams RW, Mountz JD. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 14.Doreau A, Belot A, Bastid J, Riche B, Trescol-Biemont MC, Ranchin B, Fabien N, Cochat P, Pouteil-Noble C, Trolliet P, Durieu I, Tebib J, Kassai B, Ansieau S, Puisieux A, Eliaou JF, Bonnefoy-Berard N. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. 2009;10:778–785. doi: 10.1038/ni.1741. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Kyttaris VC, Tsokos GC. The role of IL-23/IL-17 axis in lupus nephritis. J Immunol. 2009;183:3160–3169. doi: 10.4049/jimmunol.0900385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu Z, Vallurupalli A, Fuhrman C, Ostrov D, Morel L. An NZB-derived locus suppresses chronic graft versus host disease and autoantibody production through non-lymphoid bone-marrow derived cells. J Immunol. 2011 doi: 10.4049/jimmunol.1003512. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Z, Cuda CM, Croker BP, Morel L. The NZM2410-derived lupus susceptibility locus Sle2c1 increases TH17 polarization and induces nephritis in Fas-deficient mice. Arthritis Rheum. 2010 doi: 10.1002/art.30146. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morel L, Yu Y, Blenman KR, Caldwell RA, Wakeland EK. Production of congenic mouse strains carrying genomic intervals containing SLE-susceptibility genes derived from the SLE-prone NZM2410 strain. Mammalian Genome. 1996;7:335–339. doi: 10.1007/s003359900098. [DOI] [PubMed] [Google Scholar]
- 19.Rocke DM, Durbin B. A model for measurement error for gene expression arrays. J Comput Biol. 2001;8:557–569. doi: 10.1089/106652701753307485. [DOI] [PubMed] [Google Scholar]
- 20.Dozmorov I, Centola M. An associative analysis of gene expression array data. Bioinformatics. 2003;19:204–211. doi: 10.1093/bioinformatics/19.2.204. [DOI] [PubMed] [Google Scholar]
- 21.Niu H, Sobel ES, Morel L. Defective B-cell response to T-dependent immunization in lupus-prone mice. Eur J Immunol. 2008;38:3028–3040. doi: 10.1002/eji.200838417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morel L, Tian XH, Croker BP, Wakeland EK. Epistatic modifiers of autoimmunity in a murine model of lupus nephritis. Immunity. 1999;11:131–139. doi: 10.1016/s1074-7613(00)80088-6. [DOI] [PubMed] [Google Scholar]
- 23.Giles BM, Tchepeleva SN, Kachinski JJ, Ruff K, Croker BP, Morel L, Boackle SA. Augmentation of NZB autoimmune phenotypes by the Sle1c murine lupus susceptibility interval. J Immunol. 2007;178:4667–4675. doi: 10.4049/jimmunol.178.7.4667. [DOI] [PubMed] [Google Scholar]
- 24.Piatelli MJ, Tanguay D, Rothstein TL, Chiles TC. Cell cycle control mechanisms in B-1 and B-2 lymphoid subsets. Immunologic Research. 2003;27:31–51. doi: 10.1385/IR:27:1:31. [DOI] [PubMed] [Google Scholar]
- 25.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 26.Tourigny MR, Ursini-Siegel J, Lee H, Toellner KM, Cunningham AF, Franklin DS, Ely S, Chen MH, Qin XF, Xiong Y, MacLennan ICM, Chen-Kiang S. CDK inhibitor p18(INK4c) is required for the generation of functional plasma cells. Immunity. 2002;17:179–189. doi: 10.1016/s1074-7613(02)00364-3. [DOI] [PubMed] [Google Scholar]
- 27.Tanguay DA, Colarusso TP, Pavlovic S, Irigoyen M, Howard RG, Bartek J, Chiles TC, Rothstein TL. Early induction of cyclin D2 expression in phorbol ester-responsive B-1 lymphocytes. J Exp Med. 1999;189:1685–1690. doi: 10.1084/jem.189.11.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marinescu V, Kohane I, Riva A. MAPPER: a search engine for the computational identification of putative transcription factor binding sites in multiple genomes. BMC Bioinformatics. 2005;6:79. doi: 10.1186/1471-2105-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gordon S, Akopyan G, Garban H, Bonavida B. Transcription factor YY1: structure, function, and therapeutic implications in cancer biology. Oncogene. 2005;25:1125–1142. doi: 10.1038/sj.onc.1209080. [DOI] [PubMed] [Google Scholar]
- 30.Mohan C, Morel L, Yang P, Wakeland EK. Accumulation of splenic B1a cells with potent antigen-presenting capability in NZM2410 lupus-prone mice. Arthritis Rheum. 1998;41:1652–1662. doi: 10.1002/1529-0131(199809)41:9<1652::AID-ART17>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 31.Kikuchi S, Fossati-Jimack L, Moll T, Amano H, Amano E, Ida A, Ibnou-Zekri N, Laporte C, Santiago-Raber ML, Rozzo SJ, Kotzin BL, Izui S. Differential role of three major New Zealand Black-derived loci linked with Yaa-induced murine lupus nephritis. J Immunol. 2005;174:1111–1117. doi: 10.4049/jimmunol.174.2.1111. [DOI] [PubMed] [Google Scholar]
- 32.Tallack MR, Keys JR, Perkins AC. Erythroid Kruppel-like factor regulates the G1 cyclin dependent kinase inhibitor p18INK4c. J Mol Biol. 2007;369:313–321. doi: 10.1016/j.jmb.2007.02.109. [DOI] [PubMed] [Google Scholar]
- 33.Blais A, Monté D, Pouliot F, Labrie C. Regulation of the human cyclin-dependent kinase inhibitor p18INK4c by the transcription factors E2F1 and Sp1. J Biol Chem. 2002;277:31679–31693. doi: 10.1074/jbc.M204554200. [DOI] [PubMed] [Google Scholar]
- 34.Slansky JE, Farnham PJ. Introduction to the E2F family: protein structure and gene regulation. Curr Top Microbiol Immunol. 1996;208:1–30. doi: 10.1007/978-3-642-79910-5_1. [DOI] [PubMed] [Google Scholar]
- 35.Loh C, Cai YC, Bonventi G, Lajoie G, MacLeod R, Wither JE. Dissociation of the genetic loci leading to B1a and NKT cell expansions from autoantibody production and renal disease in b6 mice with an introgressed new zealand black chromosome 4 interval. J Immunol. 2007;178:1608–1617. doi: 10.4049/jimmunol.178.3.1608. [DOI] [PubMed] [Google Scholar]
- 36.Ridgway WM, Peterson LB, Todd JA, Rainbow DB, Healy B, Burren OS, Wicker LS. Chapter 6 Gene-Gene Interactions in the NOD Mouse Model of Type 1 Diabetes. In: U Emil R, H, editors. Advances in Immunology Immunopathogenesis of Type 1 Diabetes Mellitus. Academic Press; 2008. p. 151. [DOI] [PubMed] [Google Scholar]
- 37.Solvason N, Wu WW, Parry D, Mahony D, Lam EW, Glassford J, Klaus GG, Sicinski P, Weinberg R, Liu YJ, Howard M, Lees E. Cyclin D2 is essential for BCR-mediated proliferation and CD5 B cell development. Int Immunol. 2000;12:631–638. doi: 10.1093/intimm/12.5.631. [DOI] [PubMed] [Google Scholar]
- 38.Mataraza JM, Tumang JR, Gumina MR, Gurdak SM, Rothstein TL, Chiles TC. Disruption of cyclin D3 blocks proliferation of normal B-1a cells, but loss of cyclin D3 is compensated by cyclin D2 in cyclin D3-deficient mice. J Immunol. 2006;177:787–795. doi: 10.4049/jimmunol.177.2.787. [DOI] [PubMed] [Google Scholar]
- 39.Peled JU, Yu JJ, Venkatesh J, Bi E, Ding BB, Krupski-Downs M, Shaknovich R, Sicinski P, Diamond B, Scharff MD, Ye BH. Requirement for cyclin D3 in germinal center formation and function. Cell Res. 2010 doi: 10.1038/cr.2010.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanguay DA, Colarusso TP, Doughty C, Pavlovic-Ewers S, Rothstein TL, Chiles TC. Cutting edge: Differential signaling requirements for activation of assembled cyclin D3-cdk4 complexes in B-1 and B-2 lymphocyte subsets. J Immunol. 2001;166:4273–4277. doi: 10.4049/jimmunol.166.7.4273. [DOI] [PubMed] [Google Scholar]
- 41.Erickson LD, Lin LL, Duan B, Morel L, Noelle RJ. A genetic lesion that arrests plasma cell homing to the bone marrow. Proc Natl Acad Sci U S A. 2003;100:12905–12910. doi: 10.1073/pnas.2131686100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phillips JA, Mehta K, Fernandez C, Raveche ES. The NZB mouse as a model for chronic lymphocytic leukemia. Cancer Res. 1992;52:437–443. [PubMed] [Google Scholar]
- 43.Leone PE, Walker BA, Jenner MW, Chiecchio L, Dagrada G, Protheroe RKM, Johnson DC, Dickens NJ, Brito JL, Else M, Gonzalez D, Ross FM, Chen-Kiang S, Davies FE, Morgan GJ. Deletions of CDKN2C in multiple myeloma: Biological and clinical implications. Clin Cancer Res. 2008;14:6033–6041. doi: 10.1158/1078-0432.CCR-08-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arias CF, Ballesteros-Tato A, Garcia MI, Martin-Caballero J, Flores JM, Martinez A, Balomenos D. p21CIP1/WAF1 controls proliferation of activated/memory T cells and affects homeostasis and memory T cell responses. J Immunol. 2007;178:2296–2306. doi: 10.4049/jimmunol.178.4.2296. [DOI] [PubMed] [Google Scholar]
- 45.Balomenos D, Martin-Caballero J, Garcia MI, Prieto I, Flores JM, Serrano M, Martinez A. The cell cycle inhibitor p21 controls T-cell proliferation and sex-linked lupus development. Nat Med. 2000;6:171–176. doi: 10.1038/72272. [DOI] [PubMed] [Google Scholar]
- 46.Kim K, Sung YK, Kang CP, Choi CB, Kang C, Bae SC. A regulatory SNP at position -899 in CDKN1A is associated with systemic lupus erythematosus and lupus nephritis. Genes Immun. 2009;10:482–486. doi: 10.1038/gene.2009.5. [DOI] [PubMed] [Google Scholar]
- 47.Katagiri K, Ueda Y, Tomiyama T, Yasuda K, Toda Y, Ikehara S, Nakayama KI, Kinashi T. Deficiency of Rap1-binding protein RAPL causes lymphoproliferative disorders through mislocalization of p27kip1. Immunity. 2011;34:24–38. doi: 10.1016/j.immuni.2010.12.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.