

Abstract
The U3-LTR region of leukemia viruses transactivates cancer-related signaling pathways through the production of a non-coding RNA transcript although the role of this transcript in virus infection remains unknown. In this study we demonstrate for the first time that an LTR-specific small non-coding RNA is produced from a FeLV-infected feline cell line. RNA cloning identified this as a 104 base transcript that originates from the U3-LTR region. We also demonstrate that in in vitro assays this LTR RNA transcript activates NFκB signaling. Taken together, our findings suggest a possible role for this LTR transcript in FeLV pathogenesis.
Keywords: FeLV, LTR, non-coding RNA
1. Introduction
Long terminal repeat (LTR) regions of oncogene-deficient leukemia viruses such as murine leukemia virus (MuLV) or feline leukemia virus (FeLV), play an important role in their pathogenesis. Proto-oncogene activation by means of its strong promoter activity at the site of integration of the provirus is the most well understood function of the LTR [1–4]. Tissue-tropism, disease-specificity and pathogenic potential of leukemia viruses are also determined by the LTR [5–7]. Transcriptional enhancers present within the U3 region of the LTRs of MuLVs and FeLVs and selective expression of transcription factors in different tissues, impart these functionalities to the LTR. LTRs have also been implicated in the development of the preleukemic hematopoietic hyperplasia seen in leukemia virus-infected animals. Insertion of unrelated sequences such as the enhancer element of polyomavirus into the U3-LTR of Mo-MuLV has been shown to abrogate such hyperplasia and reduce the tumorigenic potential of the virus [8,9].
Several other studies demonstrated that LTR region of Mo-MuLV and FeLV independently activate AP-1 and NFκB signaling pathways, which are intimately associated with the cancer development. [10–14]. It was suggested that induction of these pathways could provide an actively growing target cell population for secondary infection and thereby presumably increases the chance of insertional activation of proto-oncogenes. A search for the minimum sequence necessary for LTR-mediated cellular gene transactivation demonstrated that the U5 and R regions were not necessary. In case of Mo-MuLV, the first 300 bases of the U3 region was sufficient for this activity [15]. A more detailed study on FeLV demonstrated that the length of the fragment could be even smaller [16]. In this study, a 210 base pair fragment of the U3-LTR (from −248 to −39) was found to be sufficient for activation of collagenase IV gene expression.
Analysis of the necessary FeLV U3-LTR sequence for gene transactivation revealed that this region does not have any valid protein-coding frame. It was demonstrated previously that leukemogenic viruses such as Mo-MuLV and FeLV generate LTR specific RNA transcripts [11,17]. Other studies on non-oncogenic endogenous FeLV LTRs demonstrated that they neither make specific transcripts like their exogenous counterpart, nor do they transactivate cellular gene expression [18]. Furthermore, an LTR-specific transcript was not detected from total RNA preparation from feline embryo fibroblast cell line AH927 although it contains endogenous FeLV LTRs. The functional implication of LTR transcript-mediated gene transactivation in virus pathogenesis is not yet clear, in part because isolated LTRs were used in all earlier studies. Whether such a transcript is indeed made in FeLV-infected cells has not been tested. In the present study we investigated production of the LTR-specific transcript in feline cells that actively produce FeLV. We show that two LTR-specific small RNAs are generated in FeLV infected cells. We cloned the smaller transcript of these RNAs and found that it originates in the U3 region of the LTR and lies within the minimum sequence necessary for the cellular gene transactivational activity that identified previously. We also demonstrate that the LTR transcript independently activates cellular gene transcription.
2. Materials and Methods
2.1. Cell culture and transfection
Feline embryo fibroblast cell line AH927 (a gift from Pradip Roy-Burman) and HEK293 cells stably expressing mouse-TLR3 (from Invivogen) were maintained in DMEM with 10% fetal calf serum with 100 U penicillin per ml and 100 μg streptomycin per ml. Mouse fibroblast line Balb-3T3 was purchased from ATCC and maintained in the same medium, but with 10% donor calf serum. AH927 and Balb-3T3 cells were transfected with lipofectamine plus reagent whereas HEK293-mTLR3 cells were transfected with lipofectamine 2000 reagent (from Invitrogen), according to manufacturer's protocol. Origin of U3-LTR construct 61E-LTR with wild-type cellular gene transactivational activity, smaller U3-LTR constructs clone H (minimal essential region required for cellular gene transactivation) and Clone G (activity negative) have been described earlier [16].
2.2. Virus propagation
AH927 cells were transfected with replication-competent wild-type FeLV-A molecular clone (61E) or with a transactivational activity-negative but replication-competent LTR-mutant virus 61E-Mut as above. Transfected cells were passaged at 48 hrs post-transfection and then every 3 days until virus production reached a peak. Virus production was estimated by measuring reverse transcriptase activity in cell free culture supernatant, as described previously [16]. Virus antigen level in the culture supernatant was measured by p27 FeLV core antigen ELISA (Synbiotics Corporation).
2.3. Northern Blot
Total cellular RNA from AH927 cells expressing 61E or mutant virus 61E-Mut at their peak production level was used for northern blot analysis. Typically four to five passages were necessary for all cells to become infected and actively produce virus. Total cellular RNA from these cells was extracted by 4M guanidine thiocyanate lysis followed by ultracentrifugation through a 5.7 M CsCl cushion. Twenty-five microgram of each RNA sample were separated in 1% agarose-6.3% formaldehyde gel in 20 mM morpholine propanesulphonic acid (MOPS) buffer and transferred on to Duralon-UV membrane (Stratagene) by capillary action [19]. Membranes were hybridized with appropriate 32P-labelled probe using Express-Hyb hybridization solution (Clontech) according to manufacturer's protocol. DNA templates used for probe preparation were either the 341 bp U3-LTR insert in the 61E-LTR plasmid for LTR probe [11], or a PCR-amplified product of the envelope region of FeLV 61E using specific primers (FeLV EnvA and FeLV EnvB, Table 1).
Table 1.
Oligonucleotides used in the study.
| Namea | Sequence (5' to 3')b | Locationc |
|---|---|---|
| 3'-end RNA ligation linker | rAppTTTAACCGCGAATTCCAG/ddC | |
| 5'-end RNA ligation linker | TGGAATrUrCrUrCrGrGrGrCrArCrCrArArGrGrU | |
| miRNA-RT | GCTGGAATTCGCGGTTAA | |
| miRNA-Forward | TGGAATTCTCGGGCACCA | |
| F | CGGCTTGAGGCCAAGAAC | 61E U3LTR −197 to −180 |
| R | TCGAACTCTGGTCAACTG | 61E U3LTR −94 to −111 |
| M6 | ACTGGGGAGCtcGGAGACTGC | 61E U3LTR −108 to −128 |
| M7 | ATAGCAGAAttCGCGCGTACA | 61E U3LTR −28 to −48 |
| FeLV EnvA | GTCAGGACAATAACTGTGAGG | 61E gp70 6167 to 6187 |
| FeLV EnvB | CGGTCCCAATCCGTTTGGGAC | 61E gp70 6499 to 6479 |
| T7-F | TAATACGACTCACTATAGGCGGCTTGA--GGCCAAGAAC | T7 promoter at the 5' of `F' |
Ligation linkers were purchased from Integrated DNA Technologies.
bases with a prefix `r' denote ribonucleotides, dd is dideoxynucleotide, lower case letters in M6 and M7 denote deviation from original sequence introduced to create restriction enzyme sites. T-7 promoter binding site is underlined in oligonucleotide primer T7-F.
Locations of the oligonucleotide primers are based on FeLV-A 61E sequence (Genbank accession number M18247) where +1 is the beginning of the genomic RNA transcript at R.
2.4. Small RNA cloning
Total cellular RNA from 61E infected AH927 cells were separated on a 1% low melting point agarose gel using RNase-free reagents. Approximately 50–250 base size RNAs were purified and ligated with preadenylated 3'-end ligation linker (Table 1) using truncated T4 RNA-ligase 2 (New England Biolabs). The 3'-end ligated RNA was then further ligated with 5'-end ligation linker (table 1) using T4 RNA ligase 1 followed by phenol-chloroform extraction and ethanol precipitation. The RNA linkers were purchased from Integrated DNA Technologies. The doubly end-ligated RNA was then reverse transcribed using miRNA-RT primer and PCR amplified with miRNA-F primer (Table 1). PCR products of approximately 50–300 bp sizes were purified from agarose gel and cloned in pGEMT-easy T-A cloning system (Promega). Recombinant colonies were directly screened for LTR specific sequences by colony hybridization using the same 32P-labeled LTR probe used for northern blot described earlier. Inserts of the recombinant clones were analyzed by automated DNA sequencing.
2.5. In vitro transcription of LTR transcript and RNA transfection
To synthesize the 104 base LTR-RNA transcript that we cloned, we PCR amplified this sequence with forward (T7-F) and reverse primer (R) described in Table 1 using 61E-LTR as template. The primer T7-F in this PCR reaction had additional T7 promoter-binding sequence. The amplified PCR product with a T7 promoter-binding site was then used as template for RNA synthesis in vitro in standard T7-RNA polymerase reaction followed by DNase digestion, ethanol precipitation and spectrophotometric quantitation. To synthesize fluorescein-labeled LTR transcript, similar in vitro synthesis protocol was used using fluorescein RNA labeling mixture (Roche Applied Bioscience). Quality and size of the product was verified in 4% polyacrylamide gel using RNA molecular weight standards from Invitrogen. For RNA transfection in HEK293-mTLR3 cells, LTR transcript RNA or yeast t-RNA and NFκB-dependent luciferase reporter plasmid (3xKB-Luc) were mixed together before complex formation was initiated with lipofectamine 2000 reagent. Cells were harvested from luciferase activity 24 hrs post-transfection. Transfection of fluorescein-labeled RNA transcript to HEK293-mTLR3 cells was also done by lipofectamine 2000 reagent. Cells were fixed with 4% paraformaldehyde solution in PBS and mounted with Ultracruz Mounting media (Santa Cruz Biotechnology) containing 4',6-diamidino-2-phenylindole (DAPI).
3. Results
3.1. LTR specific small RNAs are made in FeLV-infected feline AH927 cells
Our previous work with isolated FeLV LTRs suggested that U3-LTR specific transcripts are generated from the LTR. To determine whether such transcripts are made naturally in virus-infected cells, we analyzed total cellular RNA from feline embryo fibroblast AH927 cells that were actively-producing FeLV-A virus 61E. To strengthen our analysis, we also included RNA from AH927 cells infected with a LTR mutant virus (61Mut), which replicates with similar kinetics as of 61E but does not support cellular gene transactivation as reported earlier [16], and also RNA from uninfected AH927 cells. We measured p27 core antigen level and reverse transcriptase activity in the culture supernatants for both viruses and confirmed that they both had comparable level of virus production (Figure 1A and 1B). Northern blot analysis with a LTR-specific probe detected one band of less than 200 base in both 61E and 61E-Mut RNA preparations, albeit in lesser amount in the mutant virus (Figure 2C). Although full-length genomic RNA (8.0 kb) and singly-spliced env RNA (3.0 kb) were readily detected, visualization of LTR-specific small RNA transcripts required probing as much as 25 microgram of total cellular RNA per lane. To verify further the LTR origin of this band we used a different probe corresponding to only FeLV envelope gp70 region in separate northern hybridization experiment. Both the genomic RNA and spliced env RNA were detected with this probe, as with LTR probe, but not the small RNA transcript (Figure 2C). As expected, the RNA from uninfected AH927 cells did not react to either probe. In some experiments we also found another transcript (approximately 350 bases) that hybridized with LTR probe only. The discrete and reproducible appearance and size of the small LTR-specific transcripts suggested that the small RNA species detected were not degradation product of the larger RNA species. These data thus demonstrated that LTR-specific small RNAs are made during FeLV infection.
Figure 1.

Detection of LTR-specific small RNAs in FeLV infected AH927 cells.
AH927 cells transfected with replication competent full-length FeLV molecular clone 61E or the LTR mutant 61E-Mut were passaged 5 times before analysis to allow spread of virus infection. Virus titer in the culture supernatant was measured by reverse transcriptase activity (A) and p27 antigen ELISA (B). Positive and negative controls for the ELISA were supplied in the kit. Standard deviation was calculated with data from three separate assays. Northern blotting was performed with either FeLV LTR-specific or Env-specific 32P-labelled probes (C). Molecular weight of the primary transcripts was estimated from the relative location of 28S and 18S ribosomal RNAs on a photograph fo the ethidium bromide-stained gel (D). The ethidium bromide-staining pattern was also used to demonstrate equal loading and quality of RNA preparations.
Figure 2.
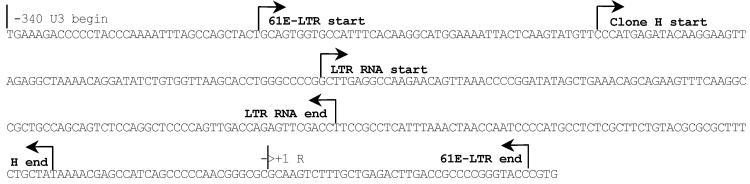
Mapping of the LTR transcript.
Coordinates of the LTR transcript are shown on the FeLV 61E LTR sequence. Clone 61E-LTR and clone H are the transactivational activity-positive U3-LTR clones used in our earlier studies. Location and orientation of the small LTR-transcript is shown. `R' is the beginning of normal genomic transcript.
3.2. Coordinates of the FeLV LTR transcript lie within the U3 region
To begin characterization of the small LTR transcript generated from FeLV, we molecularly cloned the RNA using an approach typically used for cloning small non-coding RNAs. As we had previously demonstrated that a 210 bp U3-LTR region (clone H) was sufficient for gene transactivation and produced RNA transcripts [16], we focused our attention on characterizing the smaller of the two bands identified in our hybridization analyses. We purified the smaller RNA transcript from low melting agarose gel and ligated at both the 5'- and 3'-ends using a specific RNA cloning linker. Ligated product was then reversed transcribed and subjected to PCR amplification using primers that were homologous to the RNA cloning linker. PCR products up to 300 bp were gel-purified and cloned into pGEMT-Easy T-A cloning vector and screened by colony hybridization. We identified one colony that strongly hybridized with the LTR probe, purified the clone, and the plasmid was sequenced in its entirety. The cloned transcript was identified as a 104 base positive-sense RNA fragment, which exactly matched the FeLV 61E proviral sequence from −197 to −93 (+1 being the start of R). The orientation of the cloned RNA was determined from the 5'-end and 3'-end RNA linker sequences in the clone. This sequence does not have any open protein-coding frame. The sequence coordinates of this LTR RNA transcript as well as the two other U3-LTR clones, 61E-LTR and clone H, are shown in Figure 2. The relative position of this transcript in comparison to the other LTR clones used in our study is shown in Figure 3A. Interestingly, the coordinates of the LTR RNA transcript lie fully within the minimum sequence necessary for gene transactivation activity (Clone H) that we identified previously [16].
Figure 3.

Transactivational activity of the U3-LTR transcript RNA.
A. Location of the LTR transcript clone F-R and other U3-LTR constructs used in this study. Locations of the primers used to amplify specific regions of the LTR are indicated by open and closed boxes (forward and reverse primers, respectively). B. Transactivational activity of the plasmid clones. Balb-3T3 cells were co-transfected with various LTR plasmid clones and NFκB dependent reporter plasmid 3xKB-Luc as described previously [14]. Cells were harvested for luciferase assay 48 hrs after transfection. C. Transactivational activity of the LTR transcript RNA. HEK293-mTLR3 cells were co-transfected with in vitro synthesized LTR transcript RNA or yeast tRNA (200 or 400 ng) and reporter plasmid 3xKB-Luc. Two hundred nanogram of pTZ19U plasmid vector DNA was included as control in all RNA transfections. Error bars were calculated from three independent transfections.
3.3. Small LTR transcript transactivates gene expression
We next wished to determine whether the LTR RNA transcript is biologically active. To address this question we subcloned the 104 bp DNA sequence that encodes the LTR transcript, into the pGEMT vector and tested its transactivation potential in a NFκB-dependent luciferase reporter assay. We also tested the in vitro synthesized LTR transcript RNA in similar NFκB reporter assays. We found that the plasmid clone of the LTR transcript (F-R) was unable to transactivate the NFκB-dependent reporter (Figure 3B) in Balb-3T3 cells. The 61E-LTR and clone H, but not clone G, induced reporter expression as we have reported previously [16]. To verify that in vitro synthesized RNA can be introduced into the cells, we transfected fluorescein-labeled LTR RNA in HEK293-mTLR3 cells and analyzed the presence of the LTR transcript in cells by immunofluorescence microscopy. We chose HEK293-mTLR3 cells over Balb-3T3 cells for this purpose because of their higher transfection efficiency and suitability for RNA transfection. We also have shown previously that these cells support LTR-mediated NFκB transactivation. As shown in Figure 4, transfected LTR transcript was readily detected inside the cells. In reporter assays, the LTR RNA transcript strongly activated expression of NFκB-dependent luciferase gene in a dose dependent manner (Figure 3C). Transfected yeast tRNA (small RNAs of comparable size to the LTR transcript) however, failed to activate the NFκB reporter in the same assay. Our data thus demonstrated that the LTR transcript we identified and cloned in this study is biologically active. The failure of the DNA equivalent of the LTR transcript to transactivate gene expression on the other hand, suggested the need for additional sequences for its activity.
Figure 4.

Internalization of in vitro synthesized RNA by transfection
Fluorescein-labeled in vitro synthesized LTR transcript RNA was transfected in HEK293-mTLR3 cells by lipofectamine 2000 reagent and visualized 16 hr post-transfection by fluorescence microscopy. A. Fluorescence, B. Bright field, C. DAPI, D. Merge of A and C.
4. Discussion
Several earlier studies suggested that the cancer-related cellular gene transactivational activity of the U3-LTR region of leukemia viruses contributes to preleukemic hyperplasia induced by these viruses. Our previous studies showed that the gene transactivational activity of FeLV LTR is related to its ability to make a small non-coding RNA transcript [18]. It was not known however whether such a non-coding RNA is made during actual virus infection, and if it is, whether that RNA would have any role in cellular gene transactivation. In this study we demonstrated that small LTR-specific RNA transcripts are made from FeLV infected cells. Cloning and sequence analysis of the transcript confirmed that the coding region of this transcript is within the U3 region of the LTR. We also demonstrated that the LTR transcript RNA we identified in this study independently activates NFκB signaling. Therefore, our data suggests that production of LTR-specific small RNA plays a role in cellular gene transactivation.
Oncogene-deficient retroviruses such as Moloney MuLV and FeLV induce tumor development in all infected animals with a predictable time course. Although insertional activation of proto-oncogene is the most well understood mechanism of tumorigenesis, the non-specific nature of retrovirus integration suggests important role of other virus-directed mechanisms in pathogenesis. Our data demonstrates that LTR-specific small non-coding RNAs are generated in FeLV-infected cells, which then activates the NFκB signaling pathway. As chronic stimulation of NFκB signaling has been linked to the development of various cancers [20], our findings provide an alternate mechanism that may contribute in leukemia virus pathogenesis. Further characterization of the promoter elements for this LTR transcript will be necessary to understand the control of its expression. Our data suggests that these elements very likely exist outside the coding region of this transcript. Analysis of expression of this transcript in infected animals and its role in tumor development would be interesting future direction of this work.
Acknowledgements
We appreciate the technical assistance of Virginia Newman. This work was supported in part from a New Investigator Grant from Massachusetts division of American Cancer Society (S.K.G.), Institutional Research Grant IRG7200124 from the American Cancer Society (S.K.G.), a grant from the National Cancer Institute (CA112102) and the Karin Grunebaum Cancer Research Foundation (D.V.F.).
References
- [1].Fan H. Leukemogenesis by Moloney murine leukemia virus: a multi-step process. Trends Microbiol. 1997;5:74–82. doi: 10.1016/S0966-842X(96)10076-7. [DOI] [PubMed] [Google Scholar]
- [2].Selten G, Cuypers HT, Berns A. Proviral activation of the putative oncogene pim-1 in MuLV-induced T cell lymphomas. EMBO Journal. 1985;4:1793–1798. doi: 10.1002/j.1460-2075.1985.tb03852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Levy LS, Lobelle-Rich PA, Overbaugh J. flvi-2, a target of retroviral insertional mutagenesis in feline thymic lymphosarcomas, encodes bmi-1. Oncogene. 1993;8:1833–1838. [PubMed] [Google Scholar]
- [4].Tsatsanis C, Fulton R, Nishigaki K, Tsujimoto H, Levy L, Terry A, Spandidos D, Onions D, Neil JC. Genetic determinants of feline leukemia virus-induced lymphoid tumors: patterns of proviral insertion and gene rearrangement. J. Virol. 1994;68:8296–8303. doi: 10.1128/jvi.68.12.8296-8303.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Short MK, Okenquist SA, Lenz J. Correlation of leukemogenic potential of murine retroviruses with transcriptional tissue preference of the viral long terminal repeats. J. Virol. 1987;61:1067–1072. doi: 10.1128/jvi.61.4.1067-1072.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Golemis E, Li Y, Fredrickson TN, Hartley JW, Hopkins N. Distinct segments within the enhancer region collaborate to specify the type of leukemia induced by nondefective Friend and Moloney viruses. J. Virol. 1989;63:328–337. doi: 10.1128/jvi.63.1.328-337.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chatis PA, Holland CA, Silver JE, Frederickson TN, Hopkins N, Hartley JW. A 3' end fragment encompassing the transcriptional enhancers of nondefective Friend virus confers erythroleukemogenicity on Moloney Leukemia virus. J. Virol. 1984;52:248–254. doi: 10.1128/jvi.52.1.248-254.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Davis B, Linney E, Fan H. Suppression of leukaemia virus pathogenicity by polyoma virus enhancers. Nature. 1985;314:550–553. doi: 10.1038/314550a0. [DOI] [PubMed] [Google Scholar]
- [9].Davis BR, Brightman BK, Chandy KG, Fan H. Characterization of a preleukemic state induced by Moloney murine leukemia virus: evidence for two infection events during leukemogenesis. Proc. Natl. Acad. Sci. USA. 1987;84:4875–4879. doi: 10.1073/pnas.84.14.4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Faller DV, Weng H, Choi SY. Activation of collagenase IV gene expression and enzymatic activity by the Moloney murine leukemia virus long terminal repeat. Virology. 1997;227:331–342. doi: 10.1006/viro.1996.8345. [DOI] [PubMed] [Google Scholar]
- [11].Ghosh SK, Faller DV. Feline leukemia virus long terminal repeat activates collagenase IV expression through AP-1. J. Virol. 1999;73:4931–4940. doi: 10.1128/jvi.73.6.4931-4940.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Weng H, Choi SY, Faller DV. The Moloney leukemia retroviral long terminal repeat trans-activates AP-1-inducible genes and AP-1 transcription factor binding. J. Biol. Chem. 1995;270:13637–13644. doi: 10.1074/jbc.270.23.13637. [DOI] [PubMed] [Google Scholar]
- [13].Faller DV, Weng H, Graves DT, Choi SY. Moloney murine leukemia virus long terminal repeat activates monocyte chemotactic protein-1 protein expression and chemotactic activity. J. Cell. Physiol. 1997;172:240–252. doi: 10.1002/(SICI)1097-4652(199708)172:2<240::AID-JCP11>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- [14].Abujamra AL, Spanjaard RA, Akinsheye I, Zhao X, Faller DV, Ghosh SK. Leukemia virus long terminal repeat activates NFkappaB pathway by a TLR3-dependent mechanism. Virology. 2006;345:390–403. doi: 10.1016/j.virol.2005.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Choi SY, Faller DV. The long terminal repeats of a murine retrovirus encode a trans-activator for cellular genes. J. Biol. Chem. 1994;269:19691–19694. [PubMed] [Google Scholar]
- [16].Abujamra AL, Faller DV, Ghosh SK. Mutations that abrogate transactivational activity of the feline leukemia virus long terminal repeat do not affect virus replication. Virology. 2003;309:294–305. doi: 10.1016/s0042-6822(03)00069-2. [DOI] [PubMed] [Google Scholar]
- [17].Choi SY, Faller DV. A transcript from the long terminal repeats of a murine retrovirus associated with trans activation of cellular genes. J. Virol. 1995;69:7054–7060. doi: 10.1128/jvi.69.11.7054-7060.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ghosh SK, Roy-Burman P, Faller DV. Long Terminal Repeat Regions from Exogenous but not Endogenous Feline Leukemia Viruses Transactivate Cellular Gene Expression. J. Virol. 2000;74:9742–9748. doi: 10.1128/jvi.74.20.9742-9748.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pal-Ghosh R, Morrow CD. A poliovirus minireplicon containing an inactive 2A proteinase is expressed in vaccinia virus-infected cells. J. Virol. 1993;67:4621–4629. doi: 10.1128/jvi.67.8.4621-4629.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Okamoto T, Sanda T, Asamitsu K. NF-kappa B signaling and carcinogenesis. Curr Pharm Des. 2007;13:447–462. doi: 10.2174/138161207780162944. [DOI] [PubMed] [Google Scholar]