

Abstract
Mithramycin A (Mith) is an aureolic acid-type polyketide produced by various soil bacteria of the genus Streptomyces. Mith inhibits myeloid cell leukemia-1 (Mcl-1) to induce apoptosis in prostate cancer, but the molecular mechanism underlying this process has not been fully elucidated. The aim of this study was therefore to investigate the detailed molecular mechanism related to Mith-induced apoptosis in prostate cancer cells. Mith decreased the phosphorylation of mammalian target of rapamycin (mTOR) in both cell lines overexpressing phospho-mTOR compared to RWPE-1 human normal prostate epithelial cells. Mith significantly induced truncated Bid (tBid) and siRNA-mediated knock-down of Mcl-1 increased tBid protein levels. Moreover, Mith also inhibited the phosphorylation of mTOR on serine 2448 and Mcl-1, and increased tBid protein in prostate tumors in athymic nude mice bearing DU145 cells as xenografts. Thus, Mith acts as an effective tumor growth inhibitor in prostate cancer cells through the mTOR/Mcl-1/tBid signaling pathway.
Keywords: prostate cancer, Mithramycin A, myeloid cell leukemia-1, mTOR, truncated Bid
Introduction
Prostate cancer is the most commonly diagnosed malignancy and the second leading cause of cancer-related death among American men.(1,2) Current prostate cancer therapeutic strategies are no longer effective because cancerous cells have evolved the ability to grow in the absence of androgens, and most patients develop androgen-independent prostate cancer. Although androgen independence occurs through a gradual process, most cancers will eventually become androgen-independent because cancer cells transform themselves in a manner that promotes their growth in the absence of key survival factors such as androgens.(3,4) Therefore, new therapeutic approaches need to be implemented to manage androgen independent prostate cancer.
Mithramycin A (Mith), a polyauroleic acid isolated from Streptomyces, was initially evaluated as a chemotherapeutic agent in cancer patients during the 1960s and 70s.(5,6) Recently there has been renewed interest in clinical development of Mith.(7,8) It is known to be a GC-rich DNA binding agentthat inhibits the binding of specificity protein 1 (Sp1) and down-regulates numerous genes which mediate proliferation, invasion, and metastasis of cancer cells.(9,10) Myeloid cell leukemia-1 (Mcl-1) is normally up- and down-modulated in response to environmental signals and conditions in normal cells, but is constitutively expressed in cancer where it promotes cell survival and drug resistance.(11,12) Aicheberger et al.(13) found that exposure of cells to Mcl-1-specific siRNA resulted in reduced cell survival and increased apoptosis compared with untreated cells.(12) Lian et al.(14) also demonstrated that inhibition of Mcl-1 by the multikinase inhibitor, sorafenib suppressed cell growth and induced apoptosis in androgen-independent prostate cancer cells. In addition, we recently studies demonstrated that knock-down of Sp1 by siRNA or Mith inhibits Mcl-1 protein, thus inducing apoptosis and inhibiting tumors in prostate cancer.(15,16) These findings suggest that Mcl-1 plays a critical role in the survival of prostate cancer patients, and that is could be a potential new therapeutic target. Although our previous study clearly demonstrated that Mith inhibits Mcl-1 to induce apoptosis,(15) the molecular mechanism underlying this effect in prostate cancer remains poorly understood.
The mammalian target of rapamycin (mTOR) is a highly conserved serine/threonine kinase that regulates cell growth, cell cycle progression and metabolism.(17) Several studies have established that the activation of Akt/mTOR signaling is strongly associated with advanced prostate tumors.(18–20) Kremer et al.(21) also reported a marked increased in the expression level of mTOR signaling pathway in prostate cancer tissues. Although the precise mechanisms are unknown, mTOR-mediated alterations may play an important role in protein synthesis, aberrant cell cycle signaling, and inhibition of apoptosis.(22–24) Interestingly, other studies have shown that synthesis of Mcl-1 is regulated by the control of protein translation through mTOR.(25,26)
In this study, we investigated how Mith regulated Mcl-1 protein to induce apoptosis and what the downstream targets of Mcl-1 were in PC3 and DU145 human androgen-independent prostate cancer cell lines and tumor tissues from athymic nude mice xenografts.
Materials and Methods
Reagents
The antibodies for Mcl-1, phospho-mTOR, mTOR, tBid, Bim, Bak, Bax, Bcl-xL, Bcl-2 and cleaved-caspase 3 were obtained from Cell Signaling Technology, Inc. (Danvers, MA) and poly (ADP-ribose) polymerase (PARP) antibody was purchased from BD Biosciences (San Diego, CA). Actin antibody was acquired from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). 4'-6-Diamidino-2-phenylindole (DAPI) was purchased from Sigma-Aldrich Chemical Co. (St Louis, MO). The Mcl-1 antibodies used for immunocytochemistry were purchased from Abcam (Cambridge, MA). Mithramycin A (Mith) was acquired from Sigma-Aldrich Chemical Co. (St Louis, MO).
Cell culture and chemical treatment
The DU145 and PC3 cells were kindly provided by Prof. Hwan-Mook Kim (Gacheon University, Incheon, Korea). RWPE-1 normal prostate epithelial cells were purchased from American Type Culture Collection (Manassas, VA). DU145 and PC3 cells were grown in RPMI1640 medium containing 10% FBS and 100 U/mL of penicillin and streptomycin for DU145 and PC3 cells and keratinocyte-SFM containing bovine pituitary extract and human recombinant epidermal growth factor for RWPE-1 in an atmosphere containing 5% CO2. Cells were seeded and allowed to attach. When the cells reached 50–60% confluence, they were treated with DMSO or Mith for 72 h. Mith was dissolved in 0.1% DMSO (vehicle control).
Western blot analysis
DU145 and PC3 cells were seeded in 60 mm2 dishes and treated with DMSO or Mith for 72 h. Whole cell lysates were extracted with lysis buffer and protein concentrations were measured using a DC Protein Assay (Bio-Rad Laboratories. Hercules, CA). Samples containing equal amounts of protein were separated by SDS-PAGE and then transferred to an Immun-BlotTM PVDF membranes (Bio-Rad Laboratories). Membranes were blocked with 5% skim milk in TBST at room temperature for 1 h, incubated with primary antibodies overnight at 4°C and then with HRP-conjugated secondary antibodies for 90 min at room temperature. Antibody-bound proteins were detected using an ECL Western Blotting Luminol reagent (Santa Cruz Biotechnology, Inc.) and exposed to film.
Mcl-1 small interfering RNA (siRNA)
On TARGET plus SMART-pool siRNA sequences targeting Mcl-1 and a non-targeting control were purchased from Dharmacon Research, Inc. (Lafayette, CO). Transfection was performed according to the manufacturer’s instructions. DU145 and PC3 cells were seeded in six-well plates and transfected transiently with 50 nM siRNA using DharmaFECT2 transfection reagent (Thermo Scientific, Lafayette, CO). After 72 h, DU145 and PC3 cells were analyzed for apoptosis using Western blot analysis and DAPI staining.
Statistical analysis
Statistical analyses of the experimental data were performed using a two-sided Student’s t test. Significance was set at p<0.05.
Results
Mith down-regulates the phosphorylation of mTOR which is overexpressed in prostate cancer cells
Previously, our group demonstrated that Mith down-regulates Mcl-1 to induce apoptotic cell death in human prostate cancer cells,(15) so we intend to investigate how Mith regulates Mcl-1 protein and induces apoptosis. Other previous reports have suggested that over-expression of mTOR and Mcl-1 in prostate cancer cells may be strongly regulated by the mTOR signaling pathway.(26,27) We thus investigated the total and phosphrylated forms of mTOR in RWPE-1, DU145 and PC3 cells. Our results confirmed that the phosphorylation of mTOR was over-expressed in the prostate cancer cell lines compared to RWPE-1 cells (Fig. 1A). As shown in Fig. 1B and 1C, Mith also significantly decreased phosphorylation of mTOR in a concentration-dependent manner in DU145 and PC3 cells. To investigate the mTOR activation status, we examined the phosphorylation of S6K1 and eIF4E and the results showed that Mith clearly decreased their phosphorylation levels (Supplemental Fig. 1*). These findings suggest that mTOR may be a molecular target for Mith-induced apoptosis and that mTOR may modulate Mcl-1 protein levels in prostate cancer cells.
Fig. 1.
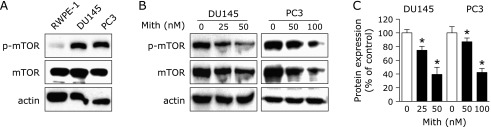
Mithramycin A down-regulates phosphorylation of mTOR in human prostate cancer cells. (A) Levels of total mTOR and phospho-mTOR (p-mTOR) were analyzed by Western blot analysis in RWPE1, DU145 and PC3 cells. (B) p-mTOR and total mTOR expression levels in DU145 and PC3 cells treated with dimethyl sulfoxide or different concentrations of Mithramycin A (Mith) were determined by Western blot analysis. (C) Results are expressed as mean ± SD of three independent experiments and statistical significance (p<0.05) compared with DMSO-treated cells is indicated (*).
Mith increases truncated Bid through down-regulation of Mcl-1
Given the fact that Mith plays a significant role in the inhibition of Mcl-1 protein and inactivation of the mTOR signaling pathway, we next investigated the proteins associated with this pathway. The expression of pro- and anti-apoptotic proteins, Bid, Bim, Bak, Bax, Bcl-2 and Bcl-xL, were analyzed using Western blot analysis. As shown in Fig. 2A, we observed that Mith increased truncated Bid (tBid) and decreased Bcl-xL protein in both cell lines. To determine whether the knock-down of Mcl-1 regulated the expression of tBid and Bcl-xL, we used a siRNA technology. Fig. 2B showed that the exposure to Mcl-1 siRNA caused an increase in tBid, but Bcl-xL was not affected by Mcl-1 siRNA. These results indicate that Mcl-1 has anti-proliferative and anti-apoptotic activities by inhibiting tBid.
Fig. 2.

Down-regulation of Mcl-1 by Mithramycin A or siRNA increases truncated Bid. (A) Bcl-2 family members [truncated Bid (tBid), Bim, Bak, Bax, Bcl-xL and Bcl-2] in DU145 and PC3 cells treated with dimethyl sulfoxide or various concentration of Mithramycin A (Mith) were evaluated by Western blot analysis. The results are representative of three independent experiments. (B) Cells were transiently transfected with either control siRNA (sicon) or siRNA specific to Mcl-1 (siMcl-1) for 72 h. Mcl-1, tBid and Bcl-XL proteins were analyzed by Western blot analysis.
Mith affects the mTOR/Mcl-1/tBid signaling pathway in a tumor xenograft of DU145 cells
As showed Fig. 3A, the expression phospho-mTOR and Mcl-1 protein levels were decreased in tumors and tBid protein was clearly increased in Mith-treated mice compared with controls. Tumor sections were obtained from animals from previous studies which showed that Mith inhibited prostate tumor growth in athymic nude mice bearing DU145 cells as xenografts.(15) A summary of the working model by which the treatment of Mith induces apoptosis in human prostate cancer through mTOR/Mcl-1/tBid signaling pathway is illustrated in Fig. 3B.
Fig. 3.

Mithramycin A regulates the mTOR/Mcl-1/truncated Bid signaling pathway. (A) Levels of phospho-mTOR (p-mTOR), total mTOR, Mcl-1 and truncated Bid (tBid) extracted from athymic nude mice xenografts bearing DU145 cells were determined by Western blot analysis. (B) Working model by which the treatment of Mithramycin A (Mith) induces apoptosis in human prostate cancer through the mTOR/Mcl-1/tBid signaling pathway.
Discussion
Several studies have indicated renewed interest in clinical development of Mith due to its inhibitory activities against tumor cell growth.(8,28–30) Because Mith acts as a gene selective Sp1 inhibitor by rapidly binding to GC-rich DNA sequences, mechanistic studies on Mith have mostly focused on targeting of the Sp1 protein. On the other hand, Mith can affect downstream targets such as Mcl-1 by displacing Sp1 transcription factor from its binding sites on the promoters of oncogenes to inhibit their expression.(16,31) Studies in this laboratory also previously demonstrated that Mith inhibits Mcl-1 protein to exhibit potent anti-tumorigenic activity in rodent models for prostate and oral cancers.(15,32)
Numerous reports have suggested that the aberrant activation of mTOR is closely linked with tumorigenesis,(33,34) and the activation of the mTOR pathway correlates with poor prognosis and reduced patient survival.(35) Thus, mTOR has been considered as a promising target for the treatment of human cancers. In this study, we also found that phosphorylation of mTOR is over-expressed in human prostate cancer cell lines compared to RWPE-1 normal prostate epithelial cells (Fig. 1A) indicating the potential relationship between mTOR and prostate cancer development. Pradelli et al.(25) and Mills et al.(26) have reported that the synthesis of Mcl-1 is regulated by control of protein translation through the mTOR signaling pathway.(36) Previous study in our laboratory showed that Mith regulates Mcl-1 protein through proteasome-dependent degradation and the inhibition of protein synthesis indicating that mTOR can modulate Mith-inhibited Mcl-1 protein expression levels in prostate cancer cells.(15) To validate whether Mith affected mTOR signaling, we conducted Western blot analysis and the results clearly showed that phosphor-mTOR was inhibited by Mith. Thus, the inhibition of Mcl-1 protein synthesis may be due to dephosphorylation of mTOR.
Bid, a member of the BH3-only proteins, is activated via caspase-8-mediated cleavage in response to ligation of the death receptor, and its active form, tBid translocates to mitochondria from cytosol causing the activation of the mitochondrial death pathway.(37) Several studies have been devoted to analyses of the interactions between pro- and anti-apoptotic proteins, and in particular, between BH3-only and anti-apoptotic proteins as well as their putative relationships during the response to anti-cancer treatment.(37,38) In particular, Mcl-1 is known to antagonize the function of many pro-apoptotic BH and BH3-only proteins(39) and Clohessy et al.(40) identified Mcl-1 as a potent tBid-binding partner. Therefore, the BH3-only protein Bid may be a sensor of apoptotic stimuli in the mTOR and Mcl-1 network regulating life and death of prostate cancer cells. Here, we investigated whether tBid protein is regulated by the inhibition of Mcl-1 by Mith in DU145 and PC3 cells and the results showed that Mith significantly increased tBid and the knock-down of Mcl-1 by Mcl-1 specific siRNA also resulted in truncated Bid protein (Fig. 2), suggesting that Mcl-1 protein may act upstream to regulate Bid protein leels and ultimately bring about apoptotic cell death. To rule out the involvement of other Bcl-2 family members on Mith-induced apoptosis in prostate cancer, we evaluated whether Mith affected other Bcl-2 family members and the result showed that only Bcl-xL protein levels were altered by Mith in both cell lines. Knock-down of Mcl-1 by siRNA did not result in any changes in Bcl-xL protein levels in either cell line, indicating that Bcl-xL may not be a downstream target of Mcl-1 protein. Previous studies showed that Mith decreases tumor growth and weight in athymic nude mice bearing DU145 as xenografts.(15) Here we found that Mith inhibited phospho-mTOR and Mcl-1 protein levels and increased tBid protein levels in tumor sections from the same animals demonstrating comparable induction of these proteins in both in vivo and in vitro tumor models.
In summary, our study demonstrates that down-regulation of Mcl-1 protein by Mith may be due to the inactivation of mTOR signaling and tBid is a key downstream target of Mcl-1 protein for Mith-induced apoptosis. Therefore, mTOR/Mcl-1/tBid pathway might be a critical signaling pathway of Mith to induce apoptotic cell death in human prostate cancer.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2012001497 and 2012003731).
Abbreviations
- BSA
bovine serum albumin
- DAPI
4'-6-diamidino-2-phenylindole
- DMSO
dimethyl sulfoxide
- Mcl-1
myeloid cell leukemia-1
- Mith
mithramycin A
- mTOR
mammalian target of rapamycin
- PARP
poly (ADP-ribose) polymerase
- PBS
phosphate buffered saline
- RT
room temperature
- Sp1
specificity protein 1
- tBid
truncated Bid
Conflict of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Karnak D, Xu L. Chemosensitization of prostate cancer by modulating Bcl-2 family proteins. Curr Drug Targets. 2010;11:699–707. doi: 10.2174/138945010791170888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saraon P, Jarvi K, Diamandis EP. Molecular alterations during progression of prostate cancer to androgen independence. Clin Chem. 2011;57:1366–1375. doi: 10.1373/clinchem.2011.165977. [DOI] [PubMed] [Google Scholar]
- 4.Assikis VJ, Simons JW. Novel therapeutic strategies for androgen-independent prostate cancer: an update. Semin Oncol. 2004;31 (2 Suppl 4):26–32. doi: 10.1053/j.seminoncol.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Lombó F, Menéndez N, Salas JA, Méndez C. The aureolic acid family of antitumor compounds: structure, mode of action, biosynthesis, and novel derivatives. Appl Microbiol Biotechnol. 2006;73:1–14. doi: 10.1007/s00253-006-0511-6. [DOI] [PubMed] [Google Scholar]
- 6.Kofman S, Eisenstein R. Mithramycin in the Treatment of Disseminated Cancer. Cancer Chemother Rep. 1963;32:77–96. [PubMed] [Google Scholar]
- 7.Zhang M, Mathur A, Zhang Y, et al. Mithramycin represses basal and cigarette smoke-induced expression of ABCG2 and inhibits stem cell signaling in lung and esophageal cancer cells. Cancer Res. 2012;72:4178–4192. doi: 10.1158/0008-5472.CAN-11-3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malek A, Núñez LE, Magistri M, et al. Modulation of the activity of Sp transcription factors by mithramycin analogues as a new strategy for treatment of metastatic prostate cancer. PLoS One. 2012;7:e35130. doi: 10.1371/journal.pone.0035130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Previdi S, Malek A, Albertini V, et al. Inhibition of Sp1-dependent transcription and antitumor activity of the new aureolic acid analogues mithramycin SDK and SK in human ovarian cancer xenografts. Gynecol Oncol. 2010;118:182–188. doi: 10.1016/j.ygyno.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Venkov C, Plieth D, Ni T, et al. Transcriptional networks in epithelial-mesenchymal transition. PLoS One. 2011;6:e25354. doi: 10.1371/journal.pone.0025354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krajewski S, Bodrug S, Krajewska M, et al. Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programmed cell death in vivo. Am J Pathol. 1995;146:1309–1319. [PMC free article] [PubMed] [Google Scholar]
- 12.Kucukzeybek Y, Gul MK, Cengiz E, et al. Enhancement of docetaxel-induced cytotoxicity and apoptosis by all-trans retinoic acid (ATRA) through downregulation of survivin (BIRC5), MCL-1 and LTbeta-R in hormone- and drug resistant prostate cancer cell line, DU-145. J Exp Clin Cancer Res. 2008;27:37. doi: 10.1186/1756-9966-27-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aichberger KJ, Mayerhofer M, Gleixner KV, et al. Identification of MCL1 as a novel target in neoplastic mast cells in systemic mastocytosis: inhibition of mast cell survival by MCL1 antisense oligonucleotides and synergism with PKC412. Blood. 2007;109:3031–3041. doi: 10.1182/blood-2006-07-032714. [DOI] [PubMed] [Google Scholar]
- 14.Lian J, Ni Z, Dai X, et al. Sorafenib sensitizes (–)-gossypol-induced growth suppression in androgen-independent prostate cancer cells via Mcl-1 inhibition and Bak activation. Mol Cancer Ther. 2012;11:416–426. doi: 10.1158/1535-7163.MCT-11-0559. [DOI] [PubMed] [Google Scholar]
- 15.Choi ES, Jung JY, Lee JS, Park JH, Cho NP, Cho SD. Myeloid cell leukemia-1 is a key molecular target for mithramycin A-induced apoptosis in androgen-independent prostate cancer cells and a tumor xenograft animal model. Cancer Lett. 2013;328:65–72. doi: 10.1016/j.canlet.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 16.Choi ES, Shim JH, Jung JY, et al. Apoptotic effect of tolfenamic acid in androgen receptor-independent prostate cancer cell and xenograft tumor through specificity protein 1. Cancer Sci. 2011;102:742–748. doi: 10.1111/j.1349-7006.2011.01871.x. [DOI] [PubMed] [Google Scholar]
- 17.Jin HO, Hong SE, Woo SH, et al. Silencing of Twist1 sensitizes NSCLC cells to cisplatin via AMPK-activated mTOR inhibition. Cell Death Dis. 2012;3:e319. doi: 10.1038/cddis.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Floc’h N, Kinkade CW, Kobayashi T, et al. Dual targeting of the Akt/mTOR signaling pathway inhibits castration-resistant prostate cancer in a genetically engineered mouse model. Cancer Res. 2012;72:4483–4493. doi: 10.1158/0008-5472.CAN-12-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kinkade CW, Castillo-Martin M, Puzio-Kuter A, et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008;118:3051–3064. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao H, Ouyang X, Banach-Petrosky WA, Shen MM, Abate-Shen C. Emergence of androgen independence at early stages of prostate cancer progression in Nkx3.1; Pten mice. Cancer Res. 2006;66:7929–7933. doi: 10.1158/0008-5472.CAN-06-1637. [DOI] [PubMed] [Google Scholar]
- 21.Kremer CL, Klein RR, Mendelson J, et al. Expression of mTOR signaling pathway markers in prostate cancer progression. Prostate. 2006;66:1203–1212. doi: 10.1002/pros.20410. [DOI] [PubMed] [Google Scholar]
- 22.Cao C, Subhawong T, Albert JM, et al. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 2006;66:10040–10047. doi: 10.1158/0008-5472.CAN-06-0802. [DOI] [PubMed] [Google Scholar]
- 23.Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- 24.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 25.Pradelli LA, Bénéteau M, Chauvin C, et al. Glycolysis inhibition sensitizes tumor cells to death receptors-induced apoptosis by AMP kinase activation leading to Mcl-1 block in translation. Oncogene. 2010;29:1641–1652. doi: 10.1038/onc.2009.448. [DOI] [PubMed] [Google Scholar]
- 26.Mills JR, Hippo Y, Robert F, et al. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci U S A. 2008;105:10853–10858. doi: 10.1073/pnas.0804821105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai B, Kong YY, Ye DW, Ma CG, Zhou X, Yao XD. Activation of the mammalian target of rapamycin signalling pathway in prostate cancer and its association with patient clinicopathological characteristics. BJU Int. 2009;104:1009–1016. doi: 10.1111/j.1464-410X.2009.08538.x. [DOI] [PubMed] [Google Scholar]
- 28.Torrance CJ, Agrawal V, Vogelstein B, Kinzler KW. Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat Biotechnol. 2011;19:940–945. doi: 10.1038/nbt1001-940. [DOI] [PubMed] [Google Scholar]
- 29.Sleiman SF, Berlin J, Basso M, Karuppagounder SS, Rohr J, Ratan RR. Histone deacetylase inhibitors and Mithramycin A impact a similar neuroprotective pathway at a crossroad between cancer and neurodegeneration. Pharmaceuticals (Basel) 2011;4:1183–1195. doi: 10.3390/ph4081183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia Z, Gao Y, Wang L, et al. Combined treatment of pancreatic cancer with mithramycin A and tolfenamic acid promotes Sp1 degradation and synergistic antitumor activity. Cancer Res. 2010;70:1111–1119. doi: 10.1158/0008-5472.CAN-09-3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee KA, Chae JI, Shim JH. Natural diterpenes from coffee, cafestol and kahweol induce apoptosis through regulation of specificity protein 1 expression in human malignant pleural mesothelioma. J Biomed Sci. 2012;19:60. doi: 10.1186/1423-0127-19-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin JA, Jung JY, Ryu MH, Safe S, Cho SD. Mithramycin A inhibits myeloid cell leukemia-1 to induce apoptosis in oral squamous cell carcinomas and tumor xenograft through activation of Bax and oligomerization. Mol Pharmacol. 2013;83:33–41. doi: 10.1124/mol.112.081364. [DOI] [PubMed] [Google Scholar]
- 33.Strimpakos AS, Karapanagiotou EM, Saif MW, Syrigos KN. The role of mTOR in the management of solid tumors: an overview. Cancer Treat Rev. 2009;35:148–159. doi: 10.1016/j.ctrv.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 34.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 35.Campone M, Levy V, Bourbouloux E, et al. Safety and pharmacokinetics of paclitaxel and the oral mTOR inhibitor everolimus in advanced solid tumours. Br J Cancer. 2009;100:315–321. doi: 10.1038/sj.bjc.6604851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coloff JL, Macintyre AN, Nichols AG, et al. Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res. 2011;71:5204–5213. doi: 10.1158/0008-5472.CAN-10-4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 38.Hikita H, Takehara T, Kodama T, et al. BH3-only protein bid participates in the Bcl-2 network in healthy liver cells. Hepatology. 2009;50:1972–1980. doi: 10.1002/hep.23207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010;584:2981–2989. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 40.Clohessy JG, Zhuang J, de Boer J, Gil-Gómez G, Brady HJ. Mcl-1 interacts with truncated Bid and inhibits its induction of cytochrome c release and its role in receptor-mediated apoptosis. J Biol Chem. 2006;281:5750–5759. doi: 10.1074/jbc.M505688200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.