

Abstract
Major depression, affecting an estimated 350 million people worldwide, poses a serious social and economic threat to modern societies. There are currently two major problems calling for innovative research approaches, namely, the absence of biomarkers predicting antidepressant response and the lack of conceptually novel antidepressant compounds. Both, biomarker predicting a priori whether an individual patient will respond to the treatment of choice as well as an early distinction of responders and nonresponders during antidepressant therapy can have a significant impact on improving this situation. Biosignatures predicting antidepressant response a priori or early in treatment would enable an evidence-based decision making on available treatment options. However, research to date does not identify any biologic or genetic predictors of sufficient clinical utility to inform the selection of specific antidepressant compound for an individual patient. In this review, we propose an optimized translational research strategy to overcome some of the major limitations in biomarker discovery. We are confident that early transfer and integration of data between both species, ideally leading to mutual supportive evidence from both preclinical and clinical studies, are most suitable to address some of the obstacles of current depression research.
1. Introduction
Major depressive disorder is a prevalent, severe, and life-threatening disorder with an enormous impact not only on all aspects of everyday life of the affected individual and their families, but also on secondary costs to society [1, 2] as it accounts for more lost productivity compared with any other disorder. Depression affects an estimated 350 million people worldwide and is considered by the WHO to be the leading cause of disability. Onset of the disorder is typically in the twenties and the course commonly recurrent or chronic, with depressive episodes occupying 20% of postdiagnosis life. Although the currently available treatments are safe, there is significant variability in antidepressant treatment outcome. Almost 60% of patients do not recover following a single antidepressant trial [3] and 20% of these patients fail to respond to any intervention.
In this review, we will put a spotlight on and critically discuss selected approaches for biomarker discovery in the treatment of depression. The purpose of this work will be to selectively focus on areas where there have been promising findings, as opposed to conducting an exhaustive literature review of studies which have failed to yield any significant breakthrough in our knowledge. There are excellent reviews providing a detailed summary on what has been published during the last years in the field of depression biomarkers [4, 5]. Finally, we will present an outlook on future approaches and discuss and submit an optimized translational strategy to improve the biomarker discovery process in depression research.
2. Current Problems and Unmet Needs in the Treatment of Depression: The Lack of Biomarkers/Biosignatures Predicting an Individual Patient's Response
Treating depression is not a one-size-fits-all approach. Although it would be ideal to better target available treatments to individual patients (i.e., a personalized treatment approach [6]) there are no clinically useful assessments that can predict with a reasonable high degree of certainty—a priori or early in treatment—whether a particular depressed patient will respond to a particular antidepressant. Among factors that have been shown to modulate antidepressant treatment response in major depression are disease severity, longer duration and frequency of the episodes, comorbid anxiety disorders, and an older age of onset [7]. However, due to their low sensitivity and specificity, research to date does not identify any biologic or genetic predictors of sufficient clinical utility to inform the selection of specific antidepressant compound for an individual patient.
Thus, the most effective antidepressant medication for each patient can presently only be identified through trial and error. During such a trial and error treatment sequence, each compound must be used for a sufficient length of time to determine whether or not the patients responds, an approach that may result in a prolonged sequence of several trials [8, 9]. If early on we could predict with a reasonable high degree of certainty that a medication will likely be ineffective for an individual patient, we could increase treatment efficacy and dramatically reduce costs. The latter becomes increasingly important as the average length of stay in hospital due to depressive disorder is high (>30 days), with considerable cross-national variation ranging from 5 to more than 40 days [10].
Therefore, the identification of individual factors predicting treatment response is one of the most pressing needs in depression treatment. Both, biomarkers predicting a priori whether an individual patient will respond to the treatment of choice as well as an early distinction of responders and nonresponders during antidepressant therapy can have a significant impact on improving this situation. Biomarkers/biosignatures would not only allow to monitor treatment response in clinical practice but also be assist in the evaluation of drug actions at an early stage in clinical trials which are frequently marred by late attrition [11]. The latter could be of particular interest considering that central nervous system (CNS) drugs entering clinical development have a considerably lower probability of reaching the marketplace (7%) compared with the industry average across other therapeutic areas (15%) [12].
Many definitions of “biomarker” exist, one of which is “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological response(s) to a therapeutic intervention” [13]. In the case of treatment response, as elaborated by Kraemer et al. [14], treatment moderators are factors that “specify for whom and under which conditions the treatment works… They also suggest to clinicians which of their patients might be most responsive to the treatment and for which patients other, more appropriate, treatments might be sought”.
Despite decades of research, the neurobiology of depressive disorder is largely unknown. Therefore, researchers have focused on the identification of neurobiological measurements mirroring a so-called “endophenotype” [15], such as the stress hormone system (hypothalamic pituitary adrenocortical system, HPA system), neuroimaging, pharmacogenetics, and genomics as well as biosignatures on the protein level (proteomics). Particulary in the context of polygenic diseases with a highly heterogeneous phenotype, a single genetic variant in one gene intuitively is likely to have a small impact, and combinations of specific biomarker, a so-called biosignature or biomarker panel, are considered to be more promising and informative [16]. This is in contrast to other disease areas (e.g., oncology), where, for example, overexpression of a single gene (HER2) in tumor tissue is an example of both a prognostic and predictive biomarker [17].
3. Promising Findings in Biomarker Discovery: Will They Make It to Clinical Use?
3.1. Stress Hormone Regulation: Neuroendocrine Function Test as Biomarker of Treatment Response?
The cumulative evidence makes a strong case implicating dysregulation of the HPA system in the pathogenesis of affective disorders ( for review: [18, 19]). The reason for HPA hyperactivity in depression is not yet clear. Genetic and experience-related factors may interact to induce manifold changes in corticosteroid receptor signaling, finally resulting in hypersecretion of both corticotropin releasing hormone (CRH), the major activator of the system, and vasopressin [20–23]. Moreover, a considerable amount of evidence has been accumulated suggesting that normalization of the HPA system might be the final step necessary for stable remission of the disease [24]. On the basis of these findings, it was further hypothesized that antidepressants may act through normalization of HPA system function [19]. Prominent neuroendocrine abnormalities among patients with major depression are, among others, a pathological outcome in the combined dexamethasone-CRH-challenge test (dex-CRH test). Interestingly, this combined dex-CRH test proved particularly useful as a predictor of increased risk for relapse: in those patients where the neuroendocrine abnormality persisted, the risk of relapse or resistance to treatment was much higher [25–27]. It has been suggested that changes in HPA system regulation assessed with repeated dex/CRH tests could be a potential biomarker that may predict clinical outcome at followup. Such a biomarker could support decision processes in antidepressant drug treatment even earlier than the full clinical improvement has developed and could also be assist in reducing attrition rates in the development of novel compounds [28]. However, it still needs to be shown that HPA system restoration as a biomarker of treatment response does not only work for “classical” antidepressants for which modulation of the HPA system was consistently shown [24], but also for conceptually novel drugs that do not target the monoaminergic system.
3.2. Neuroimaging Add
Different neuroimaging techniques, such as positron emission tomography (PET), magnetic resonance spectroscopy (MRS), and functional magnetic resonance imaging (fMRI) have all been used to study whether baseline, pretreatment characteristics or changes in brain functioning and metabolism correlate with symptom improvement following antidepressant treatment [29–31]. If structural or functional magnetic resonance imaging (fMRI) studies could provide an accurate probability of a patient's chances of responding to a specific treatment modality such as, for example, antidepressant or psychotherapy, then there could be either clinically important utility to the information obtained by the MRI scan or could this information be of critical value in clinical trials investigating the efficacy of novel antidepressant compounds or other innovative treatment modalities. Indeed, several findings suggest that the likelihood of response may be predicted by imaging findings. In 2002, Mayberg [32] reviewed studies published to date examining the correlation between changes in brain metabolism patterns and symptom reduction in major depressive disorder following treatment with standard antidepressants. Frontal hypometabolism before and normalization of this reduced metabolic activity during treatment turned out to be a prognostic marker for response to both antidepressant treatment and cognitive behavioral therapy. An inverse pattern of serial changes in regional brain metabolism between responders and nonresponders to the treatment suggested that failure to induce these adaptive changes in brain metabolism may underlie treatment nonresponse [29]. It was concluded that normalization of frontal lobe hypometabolism as a correlate of clinical symptom improvement was the most consistent finding. Studies involving the use of fMRI have been instrumental in linking changes in brain metabolism during the performance of cognitive tasks and treatment outcome in depressive disorder. In addition, studies in patients with depression have shown an increased subcallosal cingulated gyrus activity that may be reversed by several antidepressants therapies. A recent study investigating the robustness of pre-treatment subgenual anterior cingulated cortex activity as predictive parameter for outcome in cognitive therapy concluded that neuroimaging might provide a clinically applicable way of assessing neural systems associated with treatment response (for recent review: [33]).
3.3. Pharmacogenetics and Genomics
Pharmacogenetics investigates how individual genetic differences modulate the response to drugs, both in terms of efficacy, that is, the therapeutic effect, as well as side effects. It has been shown that response to pharmacological treatment segregates in families, supporting the idea that the individual genetic endowment modulates, at least partially, the response to antidepressant treatment [34]. Only a few studies, however, have investigated familial patterns of response to antidepressants [35]. As the global drug response is the final result of a number of potentially interacting biological events, such as drug absorption, distribution, interaction with the putative target, biotransformation, and excretion, it is likely that a considerably large number of genes is involved in shaping these complex processes. Under this condition, a single genetic variant in one gene intuitively is likely to have a small impact, and combinations of specific mutations are considered to be more informative. This is in contrast to other disease areas, such as, oncology, where, for example, overexpression of a single gene (HER2 oncogene) in tumor tissue which occurs in 15–20% of invasive breast cancers is an example of both a prognostic and predictive biomarker. Her2 overexpression is associated with a diminished prognosis (e.g., higher risk of recurrence); however, it also predicts that a patient will more likely benefit from directed therapies that target Her2 (Trastuzumab, [17]). One source accounting for the variation in response to antidepressant treatment is genetic differences as currently analyzed by single nucleotide polymorphisms (SNP) mapping. So far, they mostly focused on metabolic enzymes of the cytochrome P450 (CYP) families and genes within the monoaminergic system with compelling evidence for an effect of CYP2D6 polymorphisms on antidepressant drug plasma levels [36] and of a serotonin transporter promoter polymorphism on clinical response to a specific class of antidepressants, the selective serotonin reuptake inhibitors (SSRI). In major depression, more than 75% of the patients worldwide are treated with SSRI. Among the many putative and still unknown targets of SSRIs is the serotonine transporter which clears the intrasynaptic neuronal space by reabsorbing serotonine into presynaptic neurons. One of the most replicated findings in the field of pharmacogenetics during the last years is the association between the short/long allele at the promoter of the serotonine transporter and the response to antidepressant treatment [37]. It has been shown that the long allele of the promoter is characterized by an increased expression rate and might be a predictor of antidepressant response and remission in Caucasians.
Keeping the importance of alterations in the HPA system associated with depression and the robust effects of antidepressant treatment on restoration of the HPA system in mind, the search for polymorphisms in genes regulating the HPA axis which could have important impact on response to antidepressants is a straightforward candidate-driven pharmacogenetic approach [24]. Indeed, one of the most replicable findings in the pharmacogenetics of depression is that genetic variations in the 51 kDa immunophilin FKBP5 shape antidepressant treatment response [38–40]. FKBP5 is a cochaperone of hsp90 which regulates glucocorticoid receptor (GR) sensitivity. In particular, polymorphisms associated with enhanced expression of FKBP5 following glucocorticoid receptor activation have also been shown to respond faster to antidepressant treatment than patients with other FKBP5 genotypes and can be considered predictive of treatment outcome, and this effect appears independent of the class of antidepressant drug used. The latter observation might suggest that the mechanisms in which FKBP5 is involved in treatment response are downstream of the primary binding profile of antidepressant drugs.
As one important determinant of antidepressant treatment outcome is the concentration in which the compound of choice reaches the organ of interest, that is, the brain, factors modulating the penetration of antidepressant drugs into the central nervous system could play a critical role in determining response to pharmacological treatment. More than ten years ago, Uhr et al. have started to investigate the influence of the P-glycoprotein (P-gp), a drug efflux pump at the blood-brain barrier which is encoded by the ABCB1 gene, on the penetration of different classes of antidepressant into the brain in a transgenic mouse model. Using those animals deficient of P-gp, they could show that many antidepressants are substrates of the P-gp, among them, for example, widely prescribed compounds such as paroxetine and venlafaxine as well as tricyclic compounds like amitriptyline [41, 42]. They later translated their preclinical findings into the clinic and could finally show that polymorphisms in the ABCB1 gene predict the response to antidepressant treatment in those depressed patients receiving drugs that had previously been identified as substrates of ABCB1 using double-knockout mice. They concluded that the combined consideration of both the medication's capacity to act as an ABCB1-transporter substrate and the patient's ABCB1 genotype are strong predictors for achieving a remission [43]. The validity of ABCB1 polymorphism analysis on predicting treatment response to specific antidepressants is currently under investigation in larger clinical trials.
With the availability of genome-wide association studies (GWAS) and the rapid growth of available data repositories unbiased, that is, genome-wide screening to identify genetic factors that could assist in the prediction of an individual's drug response, has been a major focus in psychiatry research [44]. However, despite tremendous efforts in identifying predictive genes in large GWAS, the results are fairly modest [45]. None of the genetic polymorphisms identified has achieved genome-wide significance or was consistently replicated across studies. One possible explanation was that if antidepressant response is a polygenic phenotype associated with common variation, individual studies had been underpowered to detect all but the largest effects. However, despite increased statistical power in a very recent meta-analysis, again no reliable predictors of antidepressant treatment outcome could be identified [46], suggesting that alternative strategies looking beyond DNA need to be explored. With respect to disease vulnerability genes, a very recent approach from the Psychiatric Genomics Consortium moved forward in that they combined the analyses of genome-wide single nucleotide polymorphism (SNP) data for more than 33000 cases and more than 27000 controls distributed among the five major psychiatric disorders (major depressive disorder, bipolar disorder, schizophrenia, autism spectrum disorder, and attention deficit disorder) [47, 48]. The interesting and innovative aspect of this approach is the underlying assumption that there might be a shared genetic makeup for these diseases, which is in contrast to the hypothesis that some of the major limitations of GWAS could be overcome by improved patient stratification. However, with respect to treatment response, an approach focusing on the disease phenotype (i.e., symptoms, which very likely share common neurobiological mechanisms across disease categories) rather than on the artificial psychiatric diagnosis is complicated by the fact that still treatments are classically chosen according to the patient's diagnosis. To improve the power of pharmacogenomics studies investigating antidepressant treatment response, therefore, strategies focussing on a more narrowly defined set of core symptoms or focussing on extreme phenotypes (i.e., good/early and poor responder) has recently been suggested for future pharmacogenetics approaches [46].
3.4. Transcriptomics, Proteomics, and Metabolomics: Adding Additional Levels of Complexity to Potentially Predictive Biosignatures
Stimulated by the disappointing results of the GWAS for antidepressant response, researchers have started to explore the potential of gene expression and proteomics as sources of predictive biosignatures most recently. As the downstream effects of different predictors of antidepressant response—be they genetic and/or environmental—should ultimately be mediated by changes in gene function, investigating predictors and underlying biological factors associated with response by means of large-scale “omics” approaches (gene expression, i.e., transcriptomics, proteomics, and metabolomics) may provide valuable information [49, 50]. The regulation of gene expression has been proposed as one molecular mechanism that could mediate stable adaptations and maladaptations in the brain, as supposed to be involved in both the pathophysiology of depressive disorder as well as in the mechanism of action by which antidepressant treatment works. Therefore, researchers have started to explore the potential of gene expression and proteomics as sources of predictive biosignatures, but this has not been done until most recently [51–53]. Moreover, with the exception of small proof-of-concept studies providing first evidence that, for example, metabolomics could add a biochemical level of information to the panel of markers predicting response to a particular antidepressant in patients [54], there is no in-depth neither an appropriate translational animal experimental approach addressing this question systematically so far. In fact, animal studies for the identification of signature gene expression profiles or biosignatures predicting and shaping treatment response have been hampered by the fact that no appropriate animal model addressing the issue of heterogeneity in response to antidepressant treatment has yet been described.
4. Micro-RNAs: Small RNAs with a Big Regulatory Impact
In recent years, micro-RNAs have emerged as key protagonists in regulating many physiological processes including those fundamental to the functioning of the central nervous system. MicroRNAs are highly conserved small regulatory molecules that cause posttranscriptional gene silencing by base pairing with target mRNA [55]. They have diverse functions in the brain, including the regulation of neuronal development and differentiation, synapse formation and modulation of synaptic plasticity. Interestingly, miRNA expression levels seem to be dynamic in the mammalian brain since they are altered by environmental stimuli. Evidence collected to date has already demonstrated that miRNA expression levels might be altered in patients suffering from depression [56] and could be considered an additional level of complexity for response biosignatures [57]. Furthermore, increasing evidence suggests that antidepressants utilize miRNAs as downstream effectors [58]. Because one miRNA has hundreds of target mRNAs, each miRNA has wide-reaching effects on gene expression, and together with their target mRNAs they form nonlinear gene networks. The discovery of microRNAs has added a new dimension to our understanding of complex gene regulatory networks. Recently, Juhila et al. described the miRNA expression patterns in the two regions of interest for antidepressant treatment response, namely, the prefrontal cortex and the hippocampus, of the mouse brain. Prefrontal cortex and hippocampus were shown to have distinct miRNA expression patterns which were reflected in the predicted gene regulatory pathways [59].
5. Looking Beyond DNA: Epigenetics, the Impact of Environmental Factors on Individual Antidepressant Response
Besides genetic variation, gene expression can be modulated by epigenetic factors. These include DNA methylation, histone modification, and RNA interference. In this review, we will focus on DNA methylation which involves the methylation of cytosines in cytosine-guanine (CpG) dinucleotides. By DNA methylation, access of transcription factors is reduced and while DNA methylation is most often associated with transcriptional repression, it can also lead to increased transcription by reducing the binding of specific transcriptional repressors. The discovery of those epigenetic modifications as a mechanism regulating long-term neurobiological adaptations and controlling over gene expression without altering the genetic code has added an additional level of complexity to depression research [60, 61]. It is now generally accepted that susceptibility to major depression is determined by a combined effect of genes and environment, with heritability estimates ranging from 30% to 40%, complemented by a major impact of stressful or aversive life events. It has been suggested that the combination of certain environmental factors with genetic predispositions would result in an epigenetic and persistent dysregulation of central nervous system transcriptional programs, leading to phenotypic manifestation of the disease [62].
Considering that modification of the epigenetic profile of neuronal DNA provides a mechanism for activity-dependent epigenetic regulation in the adult nervous system, epigenetic modifications could also be of importance as modulators of individual response to antidepressant treatment. In particular, changes in epigenetic modifications during lifespan and as a consequence of a plethora of environmental influences provides an attractive model to explain why an individual patient does not respond to the antidepressant drug which was convincingly effective in former episodes of the disease. The same holds true for differences in antidepressant response in monozygotic twins. In schizophrenia, it was shown most recently that histone deacetylase (HDAC) inhibitors prevented the repressive histone modifications induced at the metabotropic glutamate 2 receptor promotor by atypical antipsychotics and augmented their therapeutic effects [63]. However, the impact of individual epigenetic modifications on modulating treatment response to antidepressants is a neglected field so far. Necessarily, studies in humans often focus on epigenetic changes in peripheral tissues, which may or may not be representative of epigenetic changes in neuronal cells.
6. How Can We Advance the Field? The Future of Biomarker Discovery in Antidepressant Response
Considering the limitations of research for CNS disorders, that is, the sheer complexity of the brain and the fact that despite decades of research, the causality of depression is still largely unknown, the fact that current pharmacological treatment modalities are far from being sufficient is not very surprising [12]. Although the need for innovative research strategies including translational approaches is more than obvious, in fact only very little approaches finally can be considered to be innovative. Transferring research questions form the clinical situation to preclinical models, for example from bed to bench and back, still remains a challenge. With the exception of the GENDEP investigation (http://gendep.iop.kcl.ac.uk/index.php and [64]), there is no systematic translational approach to study the neurobiology of response to antidepressant treatment so far.
One of the major constraints in depression research is the lack of appropriate animal experimental approaches. Medical research relies extensively on the use of animal models to study pathology accounting for clinical conditions, and complex psychiatric diseases such as mood disorders are no exception [65]. Ideally, such an animal model should mimic the human condition of interest with respect to its etiology, symptomatology, treatment, and biological basis. With respect to complex psychiatric disorders, however, meeting such requirements seems to be impossible. These models can, however, be successfully employed if specific questions related to specific key symptoms prevalent in human depression are addressed. Of central importance to our approach is the availability of valid behavioural paradigms for evaluating the potential efficacy of antidepressant drugs.
What could be an innovative approach to improve the discovery of predictive biomarker or biosignatures in depression treatment? Recently, a large randomized clinical trial designed to identify factors that moderate response to three treatments for patients with major depression among patients never treated previously for the condition has been initiated [66]. However, we still are confident that carefully designed, truly translational strategies which fully exploit the advantages of animal research, that is, the unlimited access to the central nervous system and the strictly standardized experimental conditions are best suited to advance the field. Of course, any animal experimental finding finally needs to be corroborated in the human being, should it be considered important in the context of depressive disorder, as shown in some very recent translational investigations [67, 68].
An optimized translational strategy which we would like to propose here (see Figure 1) should originate from questions that arise from daily clinical problems and translates those into a valid animal experimental approach modeling the clinical situation as close as possible. In case of the enigma of individual differences in antidepressant response, a mouse model focussing on extremes (i.e., good or early responder versus poor or non-responder) in response to treatment with the most commonly prescribed antidepressant compounds, that is, serotonin reuptake inhibitors and serotonine-norepinephrine reuptake inhibitors, could be considered a straightforward approach. Such an animal model enables us to identify potential candidates, for example genes, proteins, or biosignatures predicting antidepressant response in our mouse model a priori, that is, before treatment, or early in treatment. Already at this very early step animal data need to be integrated with patients' data to generate strong candidates. Only those candidates or biomarker panels which show up in both species are considered strong candidates which then can be investigated in detail with respect to their potential predicting antidepressant drug response. Further steps will be the development of a diagnostic kit based on the quantitative assessment of protein and/or metabolite levels or gene expression in patient blood prior to or early after the onset of treatment. The results of this assay will predict whether a particular treatment will be effective for an individual patient and enable the psychiatrist to make an educated and objective decision on what antidepressant to use for which patient.
Figure 1.
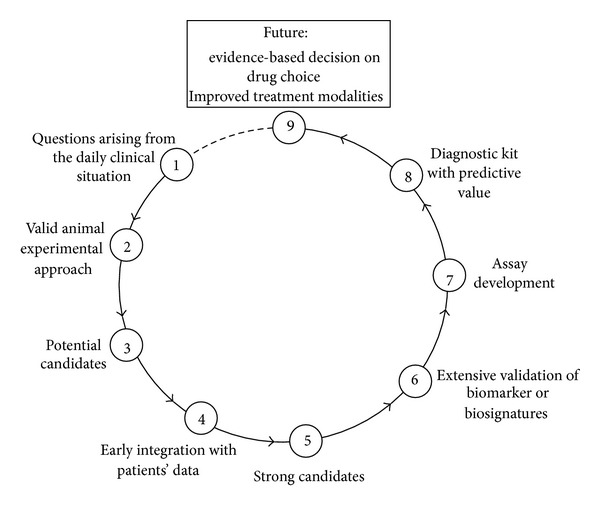
An optimized translational approach for the discovery of biosignatures predictive of antidepressant treatment response. To overcome some of the major constraints of current depression research, translational research needs to start with questions that arise from daily clinical problems and translates those into a valid animal experimental approach modelling the clinical situation as close as possible. This enables us to identify potential candidates, for example genes, proteins, or biosignatures predicting antidepressant response in our mouse model. Already at this very early step animal data need to be integrated with patients' data to generate strong candidates. Only those candidates or biomarker panels which show up in both species are considered strong candidates which then can be investigated in detail with respect to their potential predicting antidepressant drug response. Further steps will be the development of a diagnostic kit based on the quantitative assessment of protein and/or metabolite levels or gene expression in patient blood prior to or early after the onset of treatment. The results of this assay will predict whether a particular treatment will be effective for an individual patient and enable the psychiatrist to make an educated and objective decision on what antidepressant to use for which patient.
Steps which differentiate the proposed strategy from most ones in the field are (1) the conviction that the questions need to arise from the clinical situation and then be translated into an animal experimental approach and (2) the early integration of animal data with patients' data to allow for the early selection of strong candidates (which is particularly important for hypothesis-free omics approaches where the selection of the right candidates out of long lists of potential ones is a critical point).
We are confident that the proposed approach strongly supports the transfer and integration of data between both species, ideally leading to mutual supportive evidence from both preclinical and clinical studies, and is most suitable to address and finally overcome some of the obstacles of current depression research.
Authors' Contribution
Christiana Labermaier and Mercè Masana contributed equally and are listed in alphabetical order.
References
- 1.Murray CJ, Vos T, Lozano R, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the global burden of disease study. The Lancet. 2012;380(9859):2197–2223. doi: 10.1016/S0140-6736(12)61689-4. [DOI] [PubMed] [Google Scholar]
- 2.Olesen J, Gustavsson A, Svensson M, Wittchen HU, Jönsson B. The economic cost of brain disorders in Europe. European Journal of Neurology. 2012;19(1):155–162. doi: 10.1111/j.1468-1331.2011.03590.x. [DOI] [PubMed] [Google Scholar]
- 3.Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. The American Journal of Psychiatry. 2006;163(1):28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 4.Leuchter AF, Cook IA, Hamilton SP, et al. Biomarkers to predict antidepressant response. Current Psychiatry Reports. 2010;12(6):553–562. doi: 10.1007/s11920-010-0160-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papakostas GI. Surrogate markers of treatment outcome in major depressive disorder. International Journal of Neuropsychopharmacology. 2012;15(6):841–854. doi: 10.1017/S1461145711001246. [DOI] [PubMed] [Google Scholar]
- 6.Holsboer F. How can we realize the promise of personalized antidepressant medicines? Nature Reviews Neuroscience. 2008;9(8):638–646. doi: 10.1038/nrn2453. [DOI] [PubMed] [Google Scholar]
- 7.Kemp AH, Gordon E, Rush AJ, Williams LM. Improving the prediction of treatment response in depression: integration of clinical, cognitive, psychophysiological, neuroimaging, and genetic measures. CNS Spectrums. 2008;13(12):1066–1086. doi: 10.1017/s1092852900017120. [DOI] [PubMed] [Google Scholar]
- 8.Rush AJ, Wisniewski SR, Warden D, et al. Selecting among second-step antidepressant medication monotherapies: predictive value of clinical, demographic, or first-step treatment features. Archives of General Psychiatry. 2008;65(8):870–880. doi: 10.1001/archpsyc.65.8.870. [DOI] [PubMed] [Google Scholar]
- 9.Adli M, Rush AJ, Möller HJ, Bauer M. Algorithms for optimizing the treatment of depression: making the right decision at the right time. Pharmacopsychiatry. 2003;36(supplement 3):S222–S229. doi: 10.1055/s-2003-45134. [DOI] [PubMed] [Google Scholar]
- 10.Hölzel LP, Kriston L, Weiser AK, Härter M. Cross-national differences in inpatient depression treatment. European Psychiatric Review. 2011;4:32–34. [Google Scholar]
- 11.Pangalos MN, Schechter LE, Hurko O. Drug development for CNS disorders: strategies for balancing risk and reducing attrition. Nature Reviews Drug Discovery. 2007;6(7):521–532. doi: 10.1038/nrd2094. [DOI] [PubMed] [Google Scholar]
- 12.Agid Y, Buzsáki G, Diamond DM, et al. How can drug discovery for psychiatric disorders be improved? Nature Reviews Drug Discovery. 2007;6(3):189–201. doi: 10.1038/nrd2217. [DOI] [PubMed] [Google Scholar]
- 13.Wagner JA. Overview of biomarkers and surrogate endpoints in drug development. Disease Markers. 2002;18(2):41–46. doi: 10.1155/2002/929274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kraemer HC, Wilson GT, Fairburn CG, Agras WS. Mediators and moderators of treatment effects in randomized clinical trials. Archives of General Psychiatry. 2002;59(10):877–883. doi: 10.1001/archpsyc.59.10.877. [DOI] [PubMed] [Google Scholar]
- 15.Kendler KS, Neale MC. Endophenotype: a conceptual analysis. Molecular Psychiatry. 2010;15(8):789–797. doi: 10.1038/mp.2010.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon GE, Perlis RH. Personalized medicine for depression: can we match patients with treatments? The American Journal of Psychiatry. 2010;167(12):1445–1455. doi: 10.1176/appi.ajp.2010.09111680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. The New England journal of medicine. 2011;365(14):1273–1283. doi: 10.1056/NEJMoa0910383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holsboer F. The rationale for corticotropin-releasing hormone receptor (CRH-R) antagonists to treat depression and anxiety. Journal of Psychiatric Research. 1999;33(3):181–214. doi: 10.1016/s0022-3956(98)90056-5. [DOI] [PubMed] [Google Scholar]
- 19.Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23(5):477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 20.de Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. Brain corticosteroid receptor balance in health and disease. Endocrine Reviews. 1998;19(3):269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- 21.Raadsheer FC, Hoogendijk WJG, Stam FC, Tilders FJH, Swaab DF. Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology. 1994;60(4):436–444. doi: 10.1159/000126778. [DOI] [PubMed] [Google Scholar]
- 22.Purba JS, Hoogendijk WJG, Hofman MA, Swaab DF. Increased number of vasopressin- and oxytocin-expressing neurons in the paraventricular nucleus of the hypothalamus in depression. Archives of General Psychiatry. 1996;53(2):137–143. doi: 10.1001/archpsyc.1996.01830020055007. [DOI] [PubMed] [Google Scholar]
- 23.Müller MB, Landgraf R, Preil J, et al. Selective activation of the hypothalamic vasopressinergic system in mice deficient for the corticotropin-releasing hormone receptor 1 is dependent on glucocorticoids. Endocrinology. 2000;141(11):4262–4269. doi: 10.1210/endo.141.11.7767. [DOI] [PubMed] [Google Scholar]
- 24.Holsboer F, Barden N. Antidepressants and hypothalamic-pituitary-adrenocortical regulation. Endocrine Reviews. 1996;17(2):187–205. doi: 10.1210/edrv-17-2-187. [DOI] [PubMed] [Google Scholar]
- 25.Heuser I, Yassouridis A, Holsboer F. The combined dexamethasone/CRH test: a refined laboratory test for psychiatric disorders. Journal of Psychiatric Research. 1994;28(4):341–356. doi: 10.1016/0022-3956(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 26.Holsboer F, Gerken A, Stalla GK, Müller OA. Blunted aldosterone and ACTH release after human CRH administration in depressed patients. The American Journal of Psychiatry. 1987;144(2):229–231. doi: 10.1176/ajp.144.2.229. [DOI] [PubMed] [Google Scholar]
- 27.Zobel AW, Yassouridis A, Frieboes R, Holsboer F. Prediction of medium-term outcome by cortisol response to the combined dexamethasone-CRH test in patients with remitted depression. The American Journal of Psychiatry. 1999;156(6):949–951. doi: 10.1176/ajp.156.6.949. [DOI] [PubMed] [Google Scholar]
- 28.Ising M, Horstmann S, Kloiber S, et al. Combined dexamethasone/corticotropin releasing hormone test predicts treatment response in major depression—a potential biomarker? Biological Psychiatry. 2007;62(1):47–54. doi: 10.1016/j.biopsych.2006.07.039. [DOI] [PubMed] [Google Scholar]
- 29.Mayberg HS, Brannan SK, Tekell JL, et al. Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biological Psychiatry. 2000;48(8):830–843. doi: 10.1016/s0006-3223(00)01036-2. [DOI] [PubMed] [Google Scholar]
- 30.Kennedy SH, Evans KR, Krüger S, et al. Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. The American Journal of Psychiatry. 2001;158(6):899–905. doi: 10.1176/appi.ajp.158.6.899. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy SH, Konarski JZ, Segal ZV, et al. Differences in brain glucose metabolism between responders to CBT and venlafaxine in a 16-week randomized controlled trial. The American Journal of Psychiatry. 2007;164(5):778–788. doi: 10.1176/ajp.2007.164.5.778. [DOI] [PubMed] [Google Scholar]
- 32.Mayberg HS. Modulating limbic-cortical circuits in depression: targets of antidepressant treatments. Seminars in Clinical Neuropsychiatry. 2002;7(4):255–268. doi: 10.1053/scnp.2002.35223. [DOI] [PubMed] [Google Scholar]
- 33.Siegle G, J G, Thompson WK, et al. Toward clinically useful neuroimaging in depression treatment: prognostic utility of subgenual cingulate activity for determining depression outcome in cognitive therapy across studies, scanners, and patient characteristics. Archives of General Psychiatry. 2012;69(9):913–924. doi: 10.1001/archgenpsychiatry.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Leon J. Pharmacogenomics: the promise of personalized medicine for CNS disorders. Neuropsychopharmacology. 2009;34(1):159–172. doi: 10.1038/npp.2008.147. [DOI] [PubMed] [Google Scholar]
- 35.Franchini L, Serretti A, Gasperini M, Smeraldi E. Familial concordance of fluvoxamine response as a tool for differentiating mood disorder pedigrees. Journal of Psychiatric Research. 1998;32(5):255–259. doi: 10.1016/S0022-3956(98)00004-1. [DOI] [PubMed] [Google Scholar]
- 36.Kirchheiner J, Seeringer A, Viviani R. Pharmacogenetics in psychiatry—a useful clinical tool or wishful thinking for the future? Current Pharmaceutical Design. 2010;16(2):136–144. doi: 10.2174/138161210790112728. [DOI] [PubMed] [Google Scholar]
- 37.Porcelli S, Fabbri C, Serretti A. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with antidepressant efficacy. European Neuropsychopharmacology. 2012;22(4):239–258. doi: 10.1016/j.euroneuro.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 38.Binder EB, Salyakina D, Lichtner P, et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nature Genetics. 2004;36(12):1319–1325. doi: 10.1038/ng1479. [DOI] [PubMed] [Google Scholar]
- 39.Ellsworth KA, Moon I, Eckloff BW, et al. FKBP5 genetic variation: association with selective serotonin reuptake inhibitor treatment outcomes in major depressive disorder. Pharmacogenetics and Genomics. 2013;23(3):156–166. doi: 10.1097/FPC.0b013e32835dc133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lekman M, Laje G, Charney D, et al. The FKBP5-gene in depression and treatment response-an association study in the sequenced treatment alternatives to relieve depression (STAR*D) cohort. Biological Psychiatry. 2008;63(12):1103–1110. doi: 10.1016/j.biopsych.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uhr M, Grauer MT, Holsboer F. Differential enhancement of antidepressant penetration into the brain in mice with abcb1ab (mdr1ab) P-glycoprotein gene disruption. Biological Psychiatry. 2003;54(8):840–846. doi: 10.1016/s0006-3223(03)00074-x. [DOI] [PubMed] [Google Scholar]
- 42.Uhr M, Steckler T, Yassouridis A, Holsboer F. Penetration of amitriptyline, but not of fluoxetine, into brain is enhanced in mice with blood-brain barrier deficiency due to Mdr1a P-glycoprotein gene disruption. Neuropsychopharmacology. 2000;22(4):380–387. doi: 10.1016/S0893-133X(99)00095-0. [DOI] [PubMed] [Google Scholar]
- 43.Uhr M, Tontsch A, Namendorf C, et al. Polymorphisms in the drug transporter gene ABCB1 Predict antidepressant treatment response in depression. Neuron. 2008;57(2):203–209. doi: 10.1016/j.neuron.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 44.Keers R, Aitchison KJ. Pharmacogenetics of antidepressant response. Expert Review of Neurotherapeutics. 2011;11(1):101–125. doi: 10.1586/ern.10.186. [DOI] [PubMed] [Google Scholar]
- 45.Ising M, Lucae S, Binder EB, et al. A genomewide association study points to multiple loci that predict antidepressant drug treatment outcome in depression. Archives of General Psychiatry. 2009;66(9):966–975. doi: 10.1001/archgenpsychiatry.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.GENDEP Investigators. Common genetic variation and antidepressant efficacy in major depressive disorders: a meta-analysis of three genome-wide pharmacogenetic studies. The American Journal of Psychiatry. 2013;170(2):207–217. doi: 10.1176/appi.ajp.2012.12020237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smoller JW, Craddock N, Kendler K, et al. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. The Lancet. 2013;381(9875):1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Serretti A, Fabbri C. Shared genetics among major psychiatric disorders. The Lancet. 2013;381(9875):1339–1341. doi: 10.1016/S0140-6736(13)60223-8. [DOI] [PubMed] [Google Scholar]
- 49.de Souza DM, Harris LW, Guest PC, Turck CW, Bahn S. The role of proteomics in depression research. European Archives of Psychiatry and Clinical Neuroscience. 2010;260(6):499–506. doi: 10.1007/s00406-009-0093-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Souza DM. Comprehending depression through proteomics. International Journal of Neuropsychopharmacology. 2012;15(10):1373–1374. doi: 10.1017/S146114571200034X. [DOI] [PubMed] [Google Scholar]
- 51.Mamdani F, Berlim MT, Beaulieu M-, Labbe A, Merette C, Turecki G. Gene expression biomarkers of response to citalopram treatment in major depressive disorder. Translational Psychiatry. 2011;1(article e13) doi: 10.1038/tp.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Webhofer C, Gormanns P, Reckow S, et al. Proteomic and metabolomic profiling reveals time-dependent changes in hippocampal metabolism upon paroxetine treatment and biomarker candidates. Journal of Psychiatric Research. 2013;47(3):289–298. doi: 10.1016/j.jpsychires.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 53.Malki K, Campbell J, Davies M, et al. Pharmacoproteomic investigation into antidepressant responsein two inbred mouse strains. Proteomics. 2012;12(14):2355–2365. doi: 10.1002/pmic.201100306. [DOI] [PubMed] [Google Scholar]
- 54.Kaddurah-Daouk R, Boyle SH, Matson W, et al. Pretreatment metabotype as a predictor of response to sertraline or placebo in depressed outpatients: a proof of concept. Translational Psychiatry. 2011;1(article e26) doi: 10.1038/tp.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nature Reviews Genetics. 2004;5(7):522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 56.Mouillet-Richard S, Baudry A, Launay J, Kellermann O. MicroRNAs and depression. Neurobiology of Disease. 2012;46(2):272–278. doi: 10.1016/j.nbd.2011.12.035. [DOI] [PubMed] [Google Scholar]
- 57.Oved K, Morag A, Pasmanik-Chor M, et al. Genome-wide miRNA expression profiling of human lymphoblastoid cell lines identifies tentative SSRI antidepressant response biomarkers. Pharmacogenomics. 2012;13(10):1129–1139. doi: 10.2217/pgs.12.93. [DOI] [PubMed] [Google Scholar]
- 58.O’Connor RM, Dinan TG, Cryan JF. Little things on which happiness depends: MicroRNAs as novel therapeutic targets for the treatment of anxiety and depression. Molecular Psychiatry. 2012;17(4):359–376. doi: 10.1038/mp.2011.162. [DOI] [PubMed] [Google Scholar]
- 59.Juhila J, Sipilä T, Icay K, et al. MicroRNA expression profiling reveals MiRNA families regulating specific biological pathways in mouse frontal cortex and hippocampus. PLoS ONE. 2011;6(6) doi: 10.1371/journal.pone.0021495.e21495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nature Reviews Neuroscience. 2007;8(5):355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- 61.Vialou V, Feng J, Robinson AJ, Nestler EJ. Epigenetic mechanisms of depression and antidepressant action. Annual Review of Pharmacology and Toxicology. 2013;53:59–87. doi: 10.1146/annurev-pharmtox-010611-134540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun H, Kennedy PJ, Nestler EJ. Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology. 2013;38(1):124–137. doi: 10.1038/npp.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kurita M, Holloway T, Garzia-Bea A, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promotor activity. Nature Neuroscience. 2012;15(9):1245–1254. doi: 10.1038/nn.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malki K, Uher R, Paya-Cano J, et al. Convergent animal and human evidence suggests a role of PPM1A gene in response to antidepressants. Biological Psychiatry. 2011;69(4):360–365. doi: 10.1016/j.biopsych.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 65.Berton O, Hahn CG, Thase ME. Are we getting closer to valid translational models for major depression? Science. 2012;338(6103):75–79. doi: 10.1126/science.1222940. [DOI] [PubMed] [Google Scholar]
- 66.Dunlop BW, Binder EB, Cubells JF, et al. Predictors of remission in depression to individual and combined treatments (PReDICT): study protocol for a randomized controlled trial. Trials. 2012;13(article 106) doi: 10.1186/1745-6215-13-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ganea K, Menke A, Schmidt MV, et al. Convergent animal and human evidence suggests the activin/inhibin pathway to be involved in antidepressant response. Translational Psychiatry. 2012;2(article e177) doi: 10.1038/tp.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kohli MA, Lucae S, Saemann PG, et al. The neuronal transporter gene SLC6A15 confers risk to major depression. Neuron. 2011;70(2):252–265. doi: 10.1016/j.neuron.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]