

Abstract
Receptor for Advanced Glycation End-products (RAGE) is a multi-ligand receptor ubiquitous present on epithelial, neuronal, vascular and inflammatory cells, usually expressed at low levels in homeostasis and to increased degrees at sites of stress or injury. The aim of the present study was to evaluate sRAGE plasma levels in patients with Acute Coronary Syndrome (ACS) and to assess its diagnostic efficacy in identification of patients with acute events. Plasma levels of sRAGE were determined in 860 patients with Coronary Artery Disease (CAD): 530 patients presented stable angina and 330 were observed during acute ischemic event (147 with unstable angina and 183 with myocardial infarction). sRAGE plasma levels were significantly lower in patients with ACS than in patients with stable angina: [median 584 pg/mL (IQR: 266–851 pg/mL) in MI patients, median 769 pg/mL (IQR: 394–987 pg/mL) in patients with unstable angina, median 834 pg/mL (IQR 630–1005 pg/mL) in patients with stable angina; P < 0.001]. sRAGE levels did not differ among ACS patients stratified by the extent of coronary artery disease. In conclusion, this study confirm the role of sRAGE in activation and progression of inflammatory process and suggests the possibility that sRAGE can be considered an indicator of destabilization of vulnerable plaque.
1. Introduction
The inflammatory process is actively involved in atherosclerosis and underlies all phases of atherosclerotic plaque development: the beginning, the progression, and the plaque rupture [1]. In the last years, mediators and effectors of this cascade are deeply studied in order to better define the mechanism that leads to acute clinical events, and systemic approaches are pursued to discover serum biomarkers useful to identify patients with plaque at risk of future vascular events [2, 3].
Advanced glycation end products (AGEs) are a heterogeneous and complex group of biochemical compounds, resulting from nonenzymatic glycation and oxidation of protein, nucleic acids, and lipids [4, 5]. The receptor for advanced glycation end-products (RAGE) is a multiligand member of the immunoglobulin superfamily ubiquitous present on epithelial, neuronal, vascular, and inflammatory cells, usually expressed at low levels in homeostasis and to increased degrees at sites of stress or injury [6].
RAGE-mediated mechanism plays a role in ischemic myocardial injury through triggering RAGE-dependent cellular activation, inducing oxidative stress, and promoting inflammatory proliferative responses leading to vascular dysfunction [7, 8]. Via activation of signal transduction cascades and transcription factors such as nuclear factor (NF)-kB, AGE-RAGE interaction yields oxidative stress, and increased expression of inflammatory and prothrombotic species in atherosclerosis-prone vessels [9].
In addition to AGEs, RAGE binds certain members of the amyloid-b (Ab) peptide cleavage product of b-amyloid precursor protein [10], the S100/calgranulin family [11], and the high mobility group 1 DNA binding protein (HMGB1) amphoterin [12, 13]. Numerous studies suggested that the interaction between S100/calgranulin or HMGB1 and RAGE may activate inflammatory and stress signaling pathways of inflammatory cells, vascular cells, epithelial cells, terminally differentiated cells such as neurons and cardiomyocytes, and transformed cells [11, 12, 14]. These data supported the hypothesis that S100/calgranulin and HMGB1 ligands may exert profound effects on cellular phenotype [15].
Besides the full-length RAGE protein in humans, nearly 20 natural occurring RAGE splicing variants were described on mRNA and protein level. It is thought that these isoforms, characterized by either N-terminally or C-terminally truncations, are possible regulators of the full-length RAGE receptor either by competitive ligand binding or by displacing the full-length protein in the membrane. The soluble C-truncated RAGE isoforms are the most focused isoforms in research and clinics; the deregulations of the circulating levels of soluble RAGE forms were reported in several RAGE-associated pathological disorders [16]. These soluble isoforms may be formed by two processes: alternative splicing forming the endogenous secretory receptor for AGE (esRAGE) or proteolytic cleavage mediated by disintegrins and metalloproteinases that produce sRAGE [17].
The aim of the present study was to evaluate sRAGE plasma levels in patients with acute coronary syndrome (ACS) and to assess its diagnostic efficacy in the identification of patients with acute events.
2. Methods
2.1. Study Population
This study included 860 Caucasian subjects (681 males and 179 females) consecutively recruited among subjects with coronary artery disease (CAD) referred to the Department of Cardiology of University Hospital. The patients included in the study presented at least one angiographically documented coronary stenosis (≥75% stenosis). Patients underwent accurate examinations and a gathering of physiological, pathological, remote and proximal, familiar, and pharmacological anamnestic data. One hundred and eighty-three patients present acute myocardial infarction (MI), 147 present unstable angina, and 530 patients had a stable angina. Patients with heart failure, cardiomiopathy, infective diseases, or chronic inflammation and patients affected by renal failure, severe liver complications, cancer, or haematological disorders were excluded from the study.
All cardioactive assumed drugs were registered, with particular regard to β-blockers, calcium-antagonists, ACE-inhibitors, antiaggregant, and nitrates. The gathered data included age and mass body index (calculated dividing the weight measured in Kg by the height squared measured in meters). Cardiovascular risk factors were defined as follows: gender, hypertension (systolic blood pressure >140 mmHg or diastolic blood pressure >90 mmHg or antihypertension therapy), and family history of cardiovascular pathology (documented CAD in parents or siblings manifested before 60 years of age in men and 70 years of age in women); cigarettes smoking was dichotomized into ever versus never, with ever smoking defined as having smoked daily for 1 year or more; patients' lipid range (total cholesterol, LDL and HDL, and triglycerides, defining hypercholesterolemia and hypertriglyceridemia as cholesterol and triglycerides plasma concentrations, respectively, higher than 200 mg/dL and 150 mg/dL) and patients' glycaemia (normal values were considered if less than 100 mg/dL, reduced glycaemic tolerance if glycaemia was 100–110 mg/dL, and diagnosed diabetes if the plasma concentration of glucose was >126 mg/dL) were considered.
All participants gave written informed consent. The study protocol is conformed to the guidelines of the Helsinki Declaration for human research and was approved by our local ethics committee.
2.2. Coronary Angiography
The coronary angiography was delivered in all of the patients using the Sones technique. We considered as hemodynamically significant the presence of a ≥ 75% stenosis in the principle coronaries. The angiographic severity of the CAD was measured by the number of the coronaries with stenosis and assigning a number between 0 and 3 corresponding to the number of obstructed vases. A ≥ 75% lesion of the left main coronary was considered a two-vessel disease. The left ventriculography was affected before the coronary angiography. On the basis of the result of coronary angiography, the CAD patients were classified as having one, two, or three major epicardial coronary arteries with a ≥ 75% luminal obstruction.
2.3. Laboratory Methods
Venous blood samples were collected at time from symptom onset, before revascularization, in Vacutainer tubes containing ethylenediaminetetraacetic acid as anticoagulant for determination of plasma levels of sRAGE and lipid parameters. The samples were centrifuged at 1000 g for 30 and immediately divided into aliquots. All laboratory tests were performed in blind.
The serum total cholesterol and triglycerides were determined using a standard enzymatic procedure. HDL cholesterol was determined enzymatically after precipitation of other lipoproteins with dextran sulfate magnesium. Blood glucose was determined by the method of glucose oxidase. Plasma levels of sRAGE were determined using a kit for the immunoadsorption enzyme that is commercially available (Quantikine, R and D Systems) according to the manufacturer's protocol. Briefly, a monoclonal antibody against sRAGE has been used to capture sRAGE from plasma. sRAGE captured was marked with a polyclonal anti-human sRAGE. After washing, the plates were incubated with streptavidin-HRP, developed on an appropriate substrate, and OD450 was determined using a plate reading immunoadsorption enzyme. The measurements were made in duplicate and the results compared. The values of the coefficients of variation intra- and interdetermination were, respectively, less than 6% and <8%.
2.4. Statistical Analysis
Data were analysed with the statistics program 8.0 (StatSoft Software 8.0, Tulsa, OK, USA). We used the Kolmogorov-Smirnov analysis to test if the continuous variables presented a normal distribution. The continuous variables were expressed by an average ± standard deviation or by a median and interquartile range if data did not fall into a normal distribution pattern. The categorical variables were presented as frequencies and percentages. The differences between the groups were valued with the Student's t-test for normally distributed variables, with Mann-Whitney's U Test for the not normally distributed variables, and with the chi-squared test for the categorical variables.
The correlations among the study variables were evaluated calculating the correlation coefficient according to Spearman analysis. The correlations between the variables of our study were valued calculating the correlation coefficients. Values of P < 0.05 were considered statistically significant.
3. Results
3.1. Subject Characteristics
Our population is composed of 860 patients: demographic, clinical, echocardiographic, and biochemicals characteristics of the studied population are shown in Table 1. Five hundred and thirty of the 860 patients observed in our study presented a stable angina and three hundred thirty were observed during acute ischemic event (unstable angina and MI). The common cardiovascular risk factors are distributed equally in the three subpopulations, whilst patients with stable angina are more dyslipidemic and have a higher mean age than those with acute events (P < 0.01).
Table 1.
Demographic, clinical, and biochemical characteristics of the studied population.
| Myocardial infarction patients (n = 183) |
Unstable angina patients (n = 147) |
Stable angina patients (n = 530) |
P value | |
|---|---|---|---|---|
| Age, y | 59.9 ± 5.7 | 60.6 ± 7.1 | 64 ± 10 | P < 0.01 |
| Male, n (%) | 146 (80%) | 109 (74%) | 428 (81%) | Ns |
| BMI, kg/m2 | 27 ± 3 | 28 ± 3 | 27 ± 5 | Ns |
| Total cholesterol | 190 ± 38 | 192 ± 41 | 189 ± 40 | Ns |
| Triglycerides | 148 ± 51 | 155 ± 49 | 152 ± 54 | Ns |
| HDL cholesterol | 49 ± 19 | 45 ± 23 | 48 ± 22 | Ns |
| Hypertension, n (%) | 126 (69%) | 94 (64%) | 334 (63%) | Ns |
| Diabetes, n (%) | 37 (20%) | 37 (25%) | 151 (28%) | Ns |
| Dyslipidemia, n (%) | 123 (67%) | 91 (61%) | 434 (82%) | P < 0.01 |
| Ever smoking, n (%) | 106 (58%) | 82 (56%) | 314 (59%) | Ns |
| Creatinine, mg/dL | 0.9 ± 0.3 | 1 ± 0.1 | 1.1 ± 0.5 | Ns |
| Hemoglobin, g/L | 12.9 ± 1.7 | 13.5 ± 1.4 | 12.3 ± 2.5 | Ns |
| sRAGE, pg/mL | 584 (266–851) | 769 (394–987) | 834 (630–1005) | P < 0.001 |
3.2. sRAGE Plasma Levels
Plasma levels of sRAGE have a non-Gaussian distribution in CAD patients and were significantly lower in ACS patients (MI and unstable angina) (median 632 pg/mL (IQR: 335–947 pg/mL)) than in stable group median (834 pg/mL (IQR 630–1005 pg/mL); P < 0.001). In particular, patients with MI had lower value of sRAGE plasma levels than patients with unstable angina (median 584 pg/mL (IQR: 266–851 pg/mL) in MI patients, median 769 pg/mL (IQR: 394–987 pg/mL) in patients with unstable angina; P < 0.001).
The correlation between sRAGE plasma levels and the clinical and biochemical parameters was evaluated. Plasma concentrations of sRAGE were not associated with age, diabetes, smoke, hypertension, BMI, creatinine, eGFR and CRP. We observed an inverse correlation between sRAGE plasma levels and total cholesterol values (r = −0.75; P = 0.0004) and triglycerides values (r = −0.79; P = 0.0005) as reported in Table 2.
Table 2.
Correlation between sRAGE plasma levels and clinical and biochemical parameters in the studied population.
| R | P value | |
|---|---|---|
| Age | 0.09 | Ns |
| Diabetes | 0.14 | Ns |
| Smoke | −0.08 | Ns |
| Hypertension | 0.19 | Ns |
| BMI | 0.18 | Ns |
| Total cholesterol, mg/dL | −0.75 | 0.0004 |
| HDL cholesterol, mg/dL | 0.12 | Ns |
| Triglycerides, mg/dL | −0.79 | 0.0005 |
| Creatinine, mg/dL | −0.05 | Ns |
| eGFR, mL/min | 0.08 | Ns |
| CRP, mg/dL | 0.5 | Ns |
We have also evaluated the possible effect of statin therapy on sRAGE plasma levels separately. Eighty percent of patients with stable angina and 67% of ACS patients were on statins therapy. Similar levels of soluble RAGE were found between patients in statins therapy and patients not users of statins, and we found that patients with ACS had lower plasma levels of sRAGE in those with stable angina in both groups of subjects treated with statins (median 624 pg/mL (IQR: 315–938 pg/mL) versus 863 pg/mL (IQR: 618–1089 pg/mL), respectively, P value < 0.001) than in what they did not undertake this therapy (median 681 pg/mL (IQR: 491–942 pg/mL) versus 943 pg/mL (IQR: 740–1349 pg/mL), respectively, P < 0.001) (Figure 1). In particular, 21% of patients were taking rosuvastatin, 50% were taking atorvastatin, and 29% were taking simvastatin. Nevertheless, our data showed that patients under treatment with different types of statins have no significant different levels of sRAGE in plasma.
Figure 1.
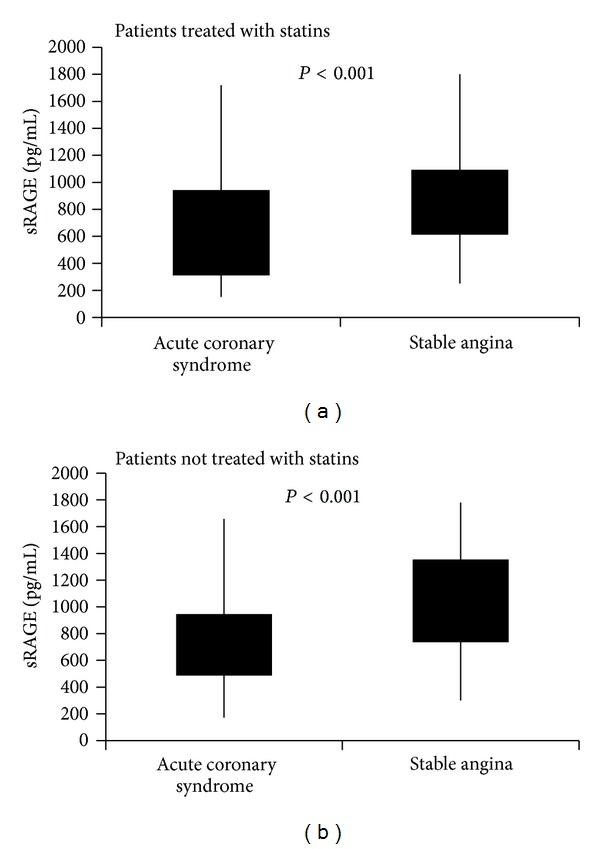
sRAGE plasma levels in ACS patients and patients with stable angina divided according to statins treatment: (a) patients treated with statins; (b) patients not treated with statins.
3.3. Extension of Coronary Artery Disease
Among the 330 patients with ACS, 139 (42%) had one-vessel disease, 120 (36%) had two-vessel disease, and 71 (22%) had three-vessel disease. The extent of CAD was different in the 530 patients with stable angina: 90 (17%) had one-vessel disease, 164 (31%) had two-vessel disease, and 276 (52%) had three-vessel disease.
Median sRAGE levels did not differ among ACS patients stratified by the extent of coronary artery disease: those with one epicardial coronary involved (672 pg/mL (IQR: 491–942 pg/mL)), those with two-vessel disease (667 pg/mL (IQR: 170–1005 pg/mL)), and those with significant stenosis involved three principle coronaries sRAGE (657 pg/mL (IQR: 375–874 pg/mL)). Age, gender, BMI, and the main cardiovascular risk factors and the clinical, biochemical, and demographic characteristics considered in the study are not statistically different in these groups.
4. Discussion
Our present study evidenced a strong correlation between plasma sRAGE levels and acute coronary events (MI and unstable angina) confirming clinical relevance to this biomarkers as indicator of inflammatory status. Plasma concentrations of sRAGE result inverse correlated with total cholesterol values and triglycerides values. In addition, sRAGE levels did not differ among ACS patients stratified by the number on coronary artery disease.
The role of sRAGE in systemic and coronary atherosclerotic disease has been amply demonstrated both in animal and human studies [18–20]. The present study confirms the role of sRAGE in humans and extends the analysis of the role of sRAGE in patients with ACS. Two recent researches investigating the role of RAGE and its ligands in ACS showed conflicting data. Cai et al. found higher sRAGE levels in ACS patients compared with controls; they also reported an association between three RAGE ligands (S100B, S100A6, and S100P) and ACS and demonstrated that, in patients and in rat models of myocardial infarction, the expression of these protein is related to myocardial injury [21]. On the contrary, McNair et al. showed lower serum levels of sRAGE in patients with NSTEMI compared to controls and demonstrated its negative correlation with those of hs-CRP [22].
In the present study, we found lower sRAGE plasma levels in patients with acute event than patients with stable angina. Our data seems to support the hypothesis that increases in production of oxygen radicals are present in acute events, mediated by uninhibited increase interaction of RAGE to its ligands. RAGE-AGE axis may be involved in plaque rupture and endothelial erosion, two major causes of coronary thrombosis and hence ACS. Low levels of sRAGE would promote increase interaction AGEs-RAGE on the cells surface, resulting in increased production of cytokines [23] and reactive oxygen species [24]. Overexpression of matrix metalloproteinases, activated by reactive oxygen species, weakens the atherosclerotic plaque and produces its rupture [25]. In addition, the interaction between S100/calgranulin or HMGB1 and RAGE may activate inflammatory and stress signaling pathways causing profound effects on cellular phenotype [15].
In our population with ACS, we observed a higher prevalence of hypertension and diabetes. We previously demonstrated that plasma sRAGE levels are decreased in patients with essential hypertension and are inversely related to pulse pressure indicating the possibility that sRAGE may play a role in arterial stiffening and its complications [26]. In addition, several studies indicate a reduction in sRAGE plasma levels in diabetic patients [27]. Since there is a greater prevalence of hypertensive and diabetic patients in the population of ACS, the observed lower value of sRAGE in this population may be partly due to the preponderance of these two cardiovascular risk factors. We also observed an inverse correlation between sRAGE plasma levels and total cholesterol and triglycerides values. Lower plasma levels of sRAGE in hypercholesterolemic subject free of lipid-lowering treatment were demonstrated [28], and recent evidence shows that statin downregulated the expression of RAGE in human atherosclerotic plaques [29]. For this reason, the possible effect of statins therapy in sRAGE levels was taken into account separately, and we found that patients with ACS had lower plasma levels of sRAGE than patients with stable angina both in patients treated with statins and in patients that did not undertake lipid-lowering treatment.
Several studies have shown that the association between inflammatory markers and the severity of CAD are weak and are mostly explained by concomitant burden of cardiovascular risk factors. Our data show that, in our study population, the extent of coronary artery disease is different between acute and stable angina patients. In patients with acute event, there is prevalence of one-vessel disease, while patients with stable angina have higher rate of three-vessel coronary heart disease. sRAGE levels did not differ among ACS patients stratified by the number on coronary artery disease. The putative theory is that the clinical syndrome of unstable angina is caused by rupture of the atherosclerotic plaque with superimposed thrombus formation. Vulnerable (rupture prone) plaques contain a soft, lipid-rich core that is covered by a thin and inflamed cap of fibrous tissue and greater plaque burden than stable plaques [30]. Our data support the theory of “culprit lesions caused by ruptured”, eroded, or spot-calcified plaques and often containing thrombus, which is responsible for acute myocardial infarction event. The structure of culprit lesions in patients with ACS has been evaluated by different imaging techniques, and it was demonstrated that these lesions had significantly greater plaque area and a higher remodeling index than nonculprit lesions [31]. In addition, other studies have identified in culprit lesion area a greater activation of the inflammatory process, while the levels of inflammatory markers did not differ greatly between normal coronaries and nonculprit lesions [32].
Taken together, our data seems to confirm the role of sRAGE as a marker for inflammation and provides an interesting explanation for its prognostic value in the prediction of future cardiac events in patients with CAD. Our findings suggest that sRAGE is probably linked to the process transforming chronic atherosclerosis into an active state leading to unstable angina and acute myocardial infarction. So sRAGE, know maker of chronic activation as expression of a process that repeats over time in stable CAD, may be also considered an indicator of destabilization of vulnerable plaque in ACS.
In the interpretation of our data, is necessary to consider several important limitations. We measured in our population only the share of total sRAGE, having used a detection system that cannot discriminate between specific variants of sRAGE. We should therefore be aware that decreased levels of sRAGE measured by this method could be due to a reduction in circulating sRAGE isoforms. The results of our research will also share the limitations of observational compared studies. Indeed, we have evaluated an association and not a causal relationship or predictability.
In conclusion, this study shows association between acute coronary event and lower value of soluble RAGE which confirms the role of sRAGE in the activation and progression of inflammatory process and suggests the possibility that sRAGE can be considered an indicator of destabilization of vulnerable plaque. Our study indicated that sRAGE plasma levels in patients with CAD were correlated to clinical manifestation of the disease and not to the extension of the disease.
Is sRAGE only a biomarker for coronary atherosclerosis or a new risk factor? Other studies are necessary to evaluate this hypothesis.
Conflict of Interests
The authors declare that they have no conflict of interests.
References
- 1.Libby P, Ridker PM, Hansson GK. Inflammation in Atherosclerosis. From Pathophysiology to Practice. Journal of the American College of Cardiology. 2009;54(23):2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uno K, Nicholls SJ. Biomarkers of inflammation and oxidative stress in atherosclerosis. Biomarkers in Medicine. 2010;4(3):361–373. doi: 10.2217/bmm.10.57. [DOI] [PubMed] [Google Scholar]
- 3.Drakopoulou M, Toutouzas K, Stefanadi E, Tsiamis E, Tousoulis D, Stefanadis C. Association of inflammatory markers with angiographic severity and extent of coronary artery disease. Atherosclerosis. 2009;206(2):335–339. doi: 10.1016/j.atherosclerosis.2009.01.041. [DOI] [PubMed] [Google Scholar]
- 4.Sourris KC, Forbes JM. Interactions between Advanced Glycation End-products (AGE) and their receptors in the development and progression of diabetic nephropathy—are these receptors valid therapeutic targets. Current Drug Targets. 2009;10(1):42–50. doi: 10.2174/138945009787122905. [DOI] [PubMed] [Google Scholar]
- 5.Bierhaus A, Hofmann MA, Ziegler R, Nawroth PP. AGEs and their interaction with AGE-receptors in vascular disease and diabetes mellitus. I. The AGE concept. Cardiovascular Research. 1998;37(3):586–600. doi: 10.1016/s0008-6363(97)00233-2. [DOI] [PubMed] [Google Scholar]
- 6.Ramasamy R, Yan SF, Schmidt AM. RAGE: therapeutic target and biomarker of the inflammatory response—the evidence mounts. Journal of Leukocyte Biology. 2009;86(3):505–512. doi: 10.1189/jlb.0409230. [DOI] [PubMed] [Google Scholar]
- 7.Chavakis T, Bierhaus A, Al-Fakhri N, et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. Journal of Experimental Medicine. 2003;198(10):1507–1515. doi: 10.1084/jem.20030800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalea AZ, Schmidt AM, Hudson BI. RAGE: a novel biological and genetic marker for vascular disease. Clinical Science. 2009;116(8):621–637. doi: 10.1042/CS20080494. [DOI] [PubMed] [Google Scholar]
- 9.Yan SF, Ramasamy R, Schmidt AM. The receptor for advanced glycation endproducts (RAGE) and cardiovascular disease. Expert reviews in molecular medicine. 2009;11:p. e9. doi: 10.1017/S146239940900101X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan SD, Chen X, Fu J, et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382(6593):685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 11.Hofmann MA, Drury S, Fu C, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97(7):889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 12.Hori O, Brett J, Slattery T, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of RAGE and amphoterin in the developing nervous system. Journal of Biological Chemistry. 1995;270(43):25752–25761. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 13.Taguchi A, Blood DC, del Toro G, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405(6784):354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 14.Dumitriu IE, Baruah P, Valentinis B, et al. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. Journal of Immunology. 2005;174(12):7506–7515. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 15.Mahajan N, Dhawan V. Receptor for advanced glycation end products (RAGE) in vascular and inflammatory diseases. International Journal of Cardiology. 2013;5273(13):00922–00924. doi: 10.1016/j.ijcard.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 16.Sterenczak KA, Nolte I, Murua Escobar H. RAGE splicing variants in mammals. Methods in Molecular Biology. 2013;963:265–276. doi: 10.1007/978-1-62703-230-8_16. [DOI] [PubMed] [Google Scholar]
- 17.Tam XHL, Shiu SWM, Leng L, Bucala R, Betteridge DJ, Tan KCB. Enhanced expression of receptor for advanced glycation end-products is associated with low circulating soluble isoforms of the receptor in Type 2 diabetes. Clinical Science. 2011;120(2):81–89. doi: 10.1042/CS20100256. [DOI] [PubMed] [Google Scholar]
- 18.Wautier J-L, Zoukourian C, Chappey O, et al. Receptor-mediated endothelial cell dysfunction in diabetic vasculopathy: soluble receptor for advanced glycation end products blocks hyperpermeability in diabetic rats. Journal of Clinical Investigation. 1996;97(1):238–243. doi: 10.1172/JCI118397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park L, Raman KG, Lee KJ, et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nature Medicine. 1998;4(9):1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 20.Falcone C, Emanuele E, D’Angelo A, et al. Plasma levels of soluble receptor for advanced glycation end products and coronary artery disease in nondiabetic men. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(5):1032–1037. doi: 10.1161/01.ATV.0000160342.20342.00. [DOI] [PubMed] [Google Scholar]
- 21.Cai XY, Lu L, Wang YN, et al. Association of increased S100B, S100A6 and S100P in serum levels with acute coronary syndrome and also with the severity of myocardial infarction in cardiac tissue of rat models with ischemia-reperfusion injury. Atherosclerosis. 2011;217(2):536–542. doi: 10.1016/j.atherosclerosis.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 22.McNair ED, Wells CR, Mabood Qureshi A, et al. Modulation of high sensitivity C-reactive protein by soluble receptor for advanced glycation end products. Molecular and Cellular Biochemistry. 2010;341(1-2):135–138. doi: 10.1007/s11010-010-0444-3. [DOI] [PubMed] [Google Scholar]
- 23.Hofmann MA, Drury S, Fu C, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97(7):889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 24.Reznikov LL, Waksman J, Azam T, et al. Effect of advanced glycation end products on endotoxin-induced TNF-α, IL-1β and IL-8 in human peripheral blood mononuclear cells. Clinical Nephrology. 2004;61(5):324–336. doi: 10.5414/cnp61324. [DOI] [PubMed] [Google Scholar]
- 25.Shah PK, Falk E, Badimon JJ, et al. Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques: potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation. 1995;92(6):1565–1569. [PubMed] [Google Scholar]
- 26.Geroldi D, Falcone C, Emanuele E, et al. Decreased plasma levels of soluble receptor for advanced glycation end-products in patients with essential hypertension. Journal of Hypertension. 2005;23(9):1725–1729. doi: 10.1097/01.hjh.0000177535.45785.64. [DOI] [PubMed] [Google Scholar]
- 27.Skrha J, Jr., Kalousová M, Svarcová J, et al. Relationship of soluble RAGE and RAGE ligands HMGB1 and EN-RAGE to endothelial dysfunction in type 1 and type 2 diabetes mellitus. Experimental and Clinical Endocrinology & Diabetes. 2012;120(5):277–281. doi: 10.1055/s-0031-1283161. [DOI] [PubMed] [Google Scholar]
- 28.Santilli F, Bucciarelli L, Noto D, et al. Decreased plasma soluble RAGE in patients with hypercholesterolemia: effects of statins. Free Radical Biology and Medicine. 2007;43(9):1255–1262. doi: 10.1016/j.freeradbiomed.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 29.Cuccurullo C, Iezzi A, Fazia ML, et al. Suppression of rage as a basis of simvastatin-dependent plaque stabilization in type 2 diabetes. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(12):2716–2723. doi: 10.1161/01.ATV.0000249630.02085.12. [DOI] [PubMed] [Google Scholar]
- 30.Ridker PM. High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation. 2001;103(13):1813–1818. doi: 10.1161/01.cir.103.13.1813. [DOI] [PubMed] [Google Scholar]
- 31.Hoffmann U, Moselewski F, Nieman K, et al. Noninvasive assessment of plaque morphology and composition in culprit and stable lesions in acute coronary syndrome and stable lesions in stable angina by multidetector computed tomography. Journal of the American College of Cardiology. 2006;47(8):1655–1662. doi: 10.1016/j.jacc.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 32.Kirbiš S, Breskvar UD, Šabovič M, Zupan I, Sinkovič A. Inflammation markers in patients with coronary artery disease—comparison of intracoronary and systemic levels. Wiener Klinische Wochenschrift. 2010;122(2):31–34. doi: 10.1007/s00508-010-1343-z. [DOI] [PubMed] [Google Scholar]