

Abstract
Chromophore-assisted light inactivation (CALI) is a powerful technique for acute perturbation of biomolecules in a spatio-temporally defined manner in living specimen with reactive oxygen species (ROS). Whereas a chemical photosensitizer including fluorescein must be added to specimens exogenously and cannot be restricted to particular cells or sub-cellular compartments, a genetically-encoded photosensitizer, KillerRed, can be controlled in its expression by tissue specific promoters or subcellular localization tags. Despite of this superiority, KillerRed hasn't yet become a versatile tool because its dimerization tendency prevents fusion with proteins of interest. Here, we report the development of monomeric variant of KillerRed (SuperNova) by direct evolution using random mutagenesis. In contrast to KillerRed, SuperNova in fusion with target proteins shows proper localization. Furthermore, unlike KillerRed, SuperNova expression alone doesn't perturb mitotic cell division. Supernova retains the ability to generate ROS, and hence promote CALI-based functional analysis of target proteins overcoming the major drawbacks of KillerRed.
Chromophore-assisted light inactivation (CALI) is a technique for inactivating target proteins1. CALI uses a chromophore molecule as a photosensitizer. Stimulation with intense light irradiation yields short-lived reactive oxygen species (ROS) such as singlet oxygen. ROS damages proteins close to the chromophore through methionine oxidation and cross-linking2. Since the half-radius of photodamage of singlet oxygen (3–4 nm) is smaller than the mean protein-protein interaction distance inside a cell (8 nm) (ref. 2) specific proteins can be inactivated by this method, whereas neighboring molecules remain intact. Thus far, small synthetic dye-based photosensitizers such as malachite green and fluorescein have been used in CALI applications1,3,4. Although these synthetic photosensitizers can be combined with genetically encoded targeting technologies5,6 e.g. HaloTag, they must be added exogenously to a living specimen, which limits the versatility of the CALI approach.
KillerRed is the first reported example of a genetically encoded photosensitizer that has been used for CALI; its phototoxicity exceeds that of other fluorescent proteins at least 1,000-fold, sufficient for protein inactivation. KillerRed was developed via the molecular engineering of a green fluorescent protein (GFP)-like hydrozoan chromoprotein, anm2CP7. Theoretically, the creation of fusion proteins between KillerRed and other proteins of interest should overcome the difficulties associated with exogenous synthetic photosensitizer delivery and distribution. However, when fused to a protein of interest, the tendancy of KillerRed to homodimerize7 hampers the normal function of the target protein. This drawback motivated us to develop a fully monomeric KillerRed.
Results
In the absence of a crystal structure for KillerRed, to reduce dimerisation we began by substituting amino acid residues located in the possible dimer interface of the original KillerRed predicted by homology modeling with the 3D-JIGSAW8 server using DsRed as a template. We chose DsRed because it forms a homotetramer with two different interfaces called AB and AC respectively9, the structures of which are typical of hydrozoan fluorescent proteins. We aligned the putative KillerRed monomer with each component molecule in the DsRed tetramer structure to create a tetrameric model structure of KillerRed, which we then used to deduce the dimer interface. By side chain analysis we found a hydrophobic patch (similar to that of the AC interface in DsRed) in the predicted KillerRed oligomer. Subsequently the 3D structure of KillerRed has been determined10,11 and the predicted surface is in close agreement with that obtained crystallographically.
Initial attempts to break the dimeric state of KillerRed by introduction of positively charged side chain lysines at L160/F162 to introduce intermolecular electrostatic repulsion by the K160/K162 pair at the AC interface were made. However, KillerRed with L160K or F162K showed no absorbance and fluorescence, suggesting that the positive charge of the lysine side chain perturbed either protein or chromophore structure. Considering this we subsequently introduced threonine, which has an uncharged side chain, in the same position (L160 and F162) by site-directed mutagenesis. The resulting protein, KillerRed-L160T/F162T, exhibited faster migration in native PAGE analysis than the original KillerRed, suggesting its possible monomerization (Figure 1a). However, the absorption and fluorescence intensity of KillerRed-L160T/F162T were dramatically diminished. To recover the optical properties, in particular absorption and fluorescence intensity, we next performed random mutagenesis of the entire KillerRed-L160T/F162T gene by error-prone PCR. One substitution (N145S) yielded a brighter protein, but with an undesired green shifted absorbance peak at 512 nm. To restore red fluorescence to the monomer, we continued the directed evolution of the mutant protein by error-prone PCR for 4 generations. Eventually, we obtained our best variant, which contains a total of 6 mutations (G3V/N145S/L160T/F162T/L172K/M204T) compared with the original KillerRed sequence. We named this protein SuperNova.
Figure 1. Characterization of the molecular size of KillerRed variants.
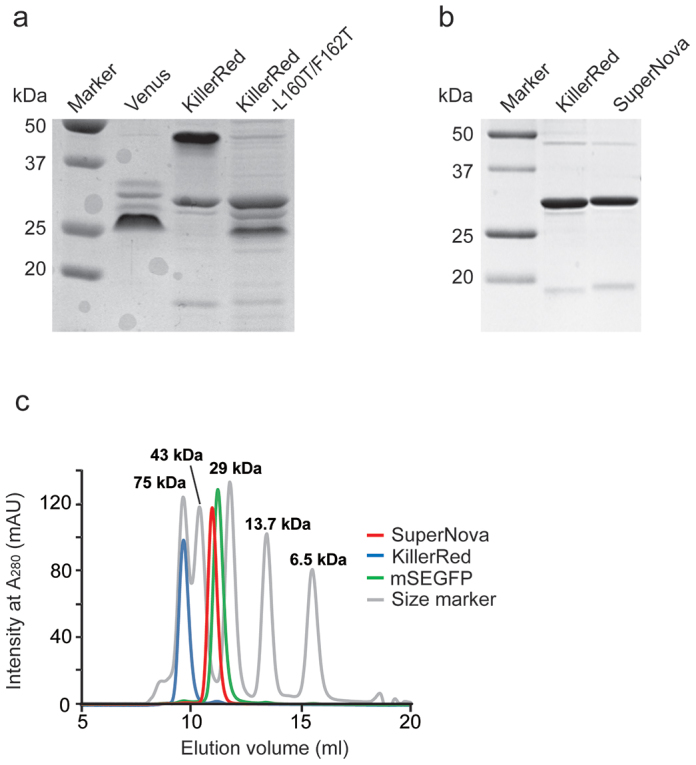
(a) Native PAGE analysis of KillerRed-L160T/F162T, KillerRed, and the YFP variant Venus. (b) SDS-PAGE analysis of KillerRed and SuperNova. (c) Gel filtration chromatography of SuperNova (red line), KillerRed (blue line), mSEGFP (green line) and molecular weight marker proteins (gray line) through a Superdex75 100/300GL column. Proteins were detected by their absorption at 280 nm.
Spectroscopic analyses revealed that SuperNova had fluorescence excitation/emission maxima at 579/610 nm, with an extinction coefficient of 33,600 M−1 cm−1 at 579 nm, and a quantum yield of 0.30 (Supplementary Figure 1a, b). KillerRed by comparison has a molar extinction coefficient of 45,000 M−1 cm−1 at 585 nm, and a quantum yield of 0.25 (ref. 7). These results suggest that SuperNova retains equivalent fluorescence properties to KillerRed in spite of the mutations introduced. In other analyses we determined that SuperNova has higher folding and maturation efficiency in vitro, with similar structural stability but lower photostability in living cells when compared with KillerRed (Supplementary Figure 1c–e). Overall these results convinced us that SuperNova would be a useful variant for further characterisation.
To further prove the oligomerisation state of Supernova, gel filtration analysis of this variant with an N-terminal His6-tag was performed; with an elution peak identified at a similar position to monomeric type GFP mSEGFP (Figure 1c). However, KillerRed, which has the same molecular size by SDS-PAGE, eluted earlier with an estimated molecular weight double that of the monomer (Figure 1b & c). In addition analytical ultracentrifugation sedimentation velocity analysis (AUC-SV) of 100 μM SuperNova solutions yielded a molecular mass estimate of 29.8 kDa in agreement with molecular weight predicted by the primary amino acid sequence (29.4 kDa). The sedimentation coefficient (C(s)) was equivalent to that for the monomeric fluorescent protein mCherry and smaller than that of the parental KillerRed protein (Figure 2a–d). Analytical ultracentrifugation sedimentation equilibrium analysis (AUC-SE) at different starting protein concentrations produced similar results (Supplementary Figure 2, Supplementary Results). Together these results strongly indicate that SuperNova, behaves as a monomeric protein under physiological conditions.
Figure 2. Results of analytical ultracentrifugation (AUS) analyses for SuperNova, mCherry and KillerRed.

(a) AUC sedimentation velocity (AUC-SV) analysis at loading a concentration of 100 μM. Plot of the distribution of sedimentation coefficients (C (s) versus s, where s is plotted in Svedberg units, (S) calculated from AUC sedimentation velocity experiments. Majority of SuperNova, mCherry and KillerRed were 3.1 S, 3.0 S and 4.7 S, respectively. (b–d) Concentration gradient of AUC sedimentation equilibrium experiments at 20,000 r.p.m. Data sets (black) were fit to the monomeric (green) and dimeric (red) molecular weights. Determined average molecular weights (Mw) were 29.8 kDa, 31.5 kDa and 60.2 kDa for (b) SuperNova, (c) mCherry and (d) KillerRed, respectively.
To explain why these mutations resulted in monomerization, we analyzed the crystal structure of the mutant at 2.3 Å resolution (Supplementary Figure 3 and Table 1). Although the mutant is found as a dimer due to crystal packing, the interface area of 1131.3 Å2 is clearly much smaller than that of KillerRed (1671.6 Å2). In particular mutations introduced to KillerRed reduced the central hydrophobic interaction (between Phe162 of each monomer; between Leu160 and Phe162) and broke the peripheral salt bridge (between Glu99 and Arg158) at the interface (Supplemental Results), suggesting that these residues are essential for KillerRed multimerisation.
It is well known that the obligate multimerisation of naturally occurring fluorescent proteins limits their usefulness as fusion protein partners. The same is true for KillerRed which when fused with fibrillarin, keratin, or Cx43, showed abnormal subcellular localization (Figure 3a, d and g) compared with previously reported monomeric fluorophores in living HeLa cells12,13. For example, Cx43-KillerRed fusions form aggregates (arrows in Figure 3g), and fibrillarin fusions leak into the cytosol. By contrast, the same proteins could successfully be fused with SuperNova (Figure 3b, e and h) without disruption of subcellular localisation. Moreover, in quantitative analysis of fibrillarin localization, by fluorescence intensity ratio measurement between nucleolus and cytosol, e.g. Figure 3 a–c, we found that targeting of SuperNova was much better than KillerRed (p < 0.001), and almost equal to EGFP (p > 0.1) (Figure 3j). The usefulness of SuperNova as a fusion partner, in particular the ability not to perturb localization, was extended by analysis of other constructs (Supplementary Figure 4).
Figure 3. Expression patterns of SuperNova, KillerRed or EGFP fusion proteins in HeLa cells.
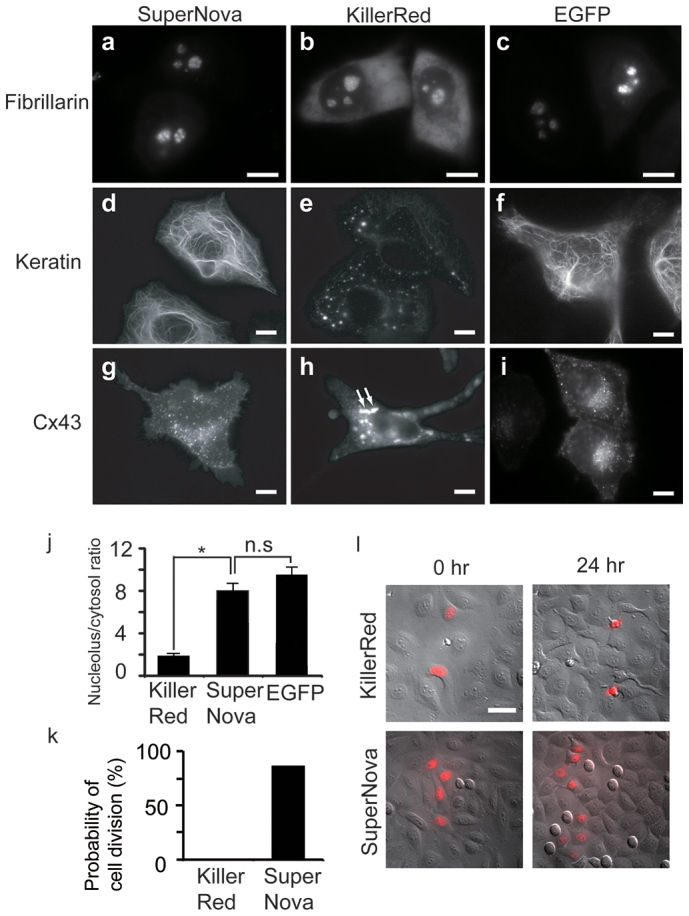
(a–c) fibrillarin, (d–f), keratin or (g–i) Cx43 fused with KillerRed (a, d and g), SuperNova (b, e and h) or EGFP (c, f and i), respectively. Scale bar, 10 μm. (j) Quantitative analysis of localization of fibrillarin fused with KillerRed, SuperNova or EGFP. Fluorescence intensity ratios between nucleolus and cytosol are indicated. (k) The probability of cell division during 24 hours is shown for HeLa cells expressing KillerRed-H2B or SuperNova-H2B following time lapse imaging. (l) Merged images of the differential interference contrast and the fluorescence analyzed in (k). Scale bar, 50 μm.
Yet even when localization appears equivalent between SuperNova and KillerRed fusions, in whole animals or single cells we found significant physiological differences between the cells expressing the two constructs. For example behavioral assays of neural function14 are abnormal in KillerRed, but preserved in SuperNova expressing Caenorhabditis elegans animals with selective expression of the fluorescent proteins in mechanosensory neurons even in the absence of light irradiation (Supplementary Figure 5). In single cells we found that the probability of cell division within 24 hrs when H2B is fused with SuperNova or KillerRed was dramatically different. As shown in Fig. 3k and l, cells expressing KillerRed-H2B frequently died (83%) and never completed mitosis (n = 80 cells, 0%) in timelapse analysis. In contrast, most of the cells expressing SuperNova-H2B underwent normal division (n = 105 cells, 87%). This microscopic studies show the evidence that SuperNova may not interfere with normal cell division rate. Multimerisation may account for this observation as cell cycle could be suppressed and cell death promoted by the abnormal oligomerisation of histones via the KillerRed dimer tendency as cell division is sensitive to the monomerisation state of histones15.
An alternative explanation is that ROS generation by KillerRed exceeds that of SuperNova, and thus differential outcomes may be attributable to direct photoxicity of the fluorescent protein alone in simple timelapse analysis. Indeed Asn145 in KillerRed is required for its phototoxic activity7, and this residue is mutated (to Ser) in Supernova. Furthermore, as previously highlighted, we found that early intermediates during SuperNova production (KillerRed-L160T/F162T/N145S) have a green shifted absorption peak. The previously described green shifted KillerRed variant E68Q (514 nm absorption maxima) is practically non-phototoxic, supposedly due to inhibition of red chromophore formation11. Logically therefore while KillerRed-L160T/F162T/N145S also fails to form a red chromophore, the subsequent mutations introduced during the development of SuperNova which influence the chemical environment to restore red chromophore maturation and, red fluorescence may, or may not, restore the phototoxicity required for CALI.
In order to test whether the changes made in SuperNova altered phototoxic activity compared to the parental KillerRed we measured the generation of reactive oxygen species (ROS) from SuperNova. In addition, since KillerRed ROS generation is predominantly a superoxide (type I reaction) rather than singlet oxygen (type II)11 product we measured singlet oxygen and superoxide generation from SuperNova in comparison to equimolar amounts of KillerRed. Singlet oxygen generation was measured by photobleaching of ADPA (anthracene-9,10-dipropionic acid) after irradiation with intense orange light (1.4 W/cm2). As shown in Supplementary Figure 6a, SuperNova generated singlet oxygen slightly more (~ 5%) readily than KillerRed. Superoxide generation was measured by photobleaching of DHE (Dihydroethidium). Here the efficiency of KillerRed was slightly higher (~ 10%) than that of SuperNova (Supplementary Figure 6b). In order to test whether these small differences were relevant in biological systems we tested the relative phototoxicity of SuperNova and KillerRed in E. coli (Supplementary Figure 6c) and HeLa (Supplementary Figure 1d).
In the prokaryotic system, exposure of E.coli expressing KillerRed, SuperNova, mCherry or EGFP to 15 minutes of intense illumination from a filtered 200 W mercury light source produced equivalent, but impaired, cell survival in those expressing the anm2CP derivatives KillerRed and SuperNova. Eukaryotes have evolved more complex systems to limit the impact of reactive oxygen species, therefore a different assay system has been used previously to show phototoxicity of KillerRed in cultured animal cells7; we emulated this approach and targeted SuperNova to mitochondria in HeLa cells by fusing it with 2 tandem copies of a mitochondrial localization signal derived from cytochrome c oxidase (2MLS-SuperNova; Supplementary Figure 7a). Irradiation of the HeLa cells with intense orange light (8 W/cm2) for 90 s caused photobleaching of SuperNova in the mitochondria and then induced apoptotic cell death within several hours in 76.9% of the cells expressing 2MLS-SuperNova (n = 13; Supplementary Figure 7b & c) paralleling previous reports for 2MLS-KillerRed7 albeit at superior efficiency (n = 16; p < 0.0001 by Wilcoxon test). By contrast in identical experimental conditions, cells expressing conventional monomeric fluorescent proteins (2MLS-mSEGFP or 2MLS-mCherry) tended to survive (10%; n = 10 cells and 16%; n = 19 cells, respectively).
Finally to prove the utility of SuperNova in CALI based interventions in living cells, we developed a simple assay based on actin turnover in lamellipodia in living mammalian cells. This process is known to be regulated by cofilin, which regulates actin dynamics at the leading edge of motile cells by stimulating actin filament disassembly and accelerating actin filament turnover16. We constructed a cofilin-SuperNova fusion protein and transiently expressed it with actin-EGFP in living COS7 cells (Figure 4a). During 10 minutes before CALI, actin filaments were freely moving (arrows in Figure 4b, upper panels). However following intense orange light irradiation, the motility of actin filaments in protrusive lamellipodia was suppressed qualitatively (arrowheads in Figure 4b, lower panels; supplemental movie), and quantitatively by temporal correlation coefficient analysis. In this assay higher temporal correlation equates to less active lamellipodia formation. The observed increase in temporal correlation after the CALI of cofilin is therefore consistent with its previously described function17. Importantly, the same effect was not observed in control experiments using cells lacking fluorescent protein tagged cofilin or cells expressing cofilin-CFP. These results suggest that the capacity to use SuperNova in CALI applications is comparable to that of KillerRed (Figure 4c) in terms of ROS generation, but significantly improved as a fusion partner by virtue of the engineered monomerisation
Figure 4. Molecular inactivation of cofilin with SuperNova.

(a) COS7 cells co-expressing actin-GFP and cofilin-SuperNova were made. The fluorescence image of actin-GFP and cofilin-SuperNova before and after CALI is shown. Bleaching of SuperNova's fluorescence was clearly observed after CALI. The dashed square in the actin-GFP image is the pseudopod region selected in time lapse imaging and analysis. Scale bar, 10 μm. (b) Still frames obtained during time lapse imaging of actin-GFP motion in lamellipodia before and after CALI by cofilin-SuperNova are shown. The numbers indicate time (min) after light irradiation (9.6 W/cm2, 1 min with through a 580AF20 from a mercury lamp). Scale bar, 10 μm. (c) Histograms of the temporal correlation in the cells expressing no fluorescent protein-tagged cofilin (NO, n = 14), cofilin-CFP (CFP, n = 20 cells), cofilin-KillerRed (n = 14 cells) and cofilin-SuperNova (n = 19 cells). Temporal correlation after CALI normalized by the pre-CALI value was represented as fold change in temporal correlation, which indicates the duration time of initial fluorescence pattern of actin-GFP. Therefore, a higher value reflects the slower movement. p < 0.001 for NO vs SuperNova, p > 0.10 for NO vs CFP, p = 0.083 for NO vs KillerRed, t-test.
Discussion
In this report, we obtained a monomeric photosensitizing protein SuperNova by introducing amino acids mutations into KillerRed. SuperNova fusion proteins localized normally within cells whereas KillerRed fusion proteins did not. In addition in vitro measurements showed a 5% increase of singlet oxygen generation and 10% decrease of superoxide generation in SuperNova when compared with those of KillerRed. These small in vitro differences do not impact on the controlled phototoxic effects of SuperNova in E.coli or apoptosis induction in HeLa cells when compared to KillerRed. By contrast KillerRed expression alone was harmful to cells and whole organisms even without illumination suggesting that its intrinsic multimerisation limits its biological compatibility. The N145 residue in KillerRed was previously considered to be indispensable for the pronounced phototoxic properties of this protein, indeed KillerRed-N145S has no phototoxicity7. However, SuperNova, which includes the N145S change has equivalent photosensitizing activity to KillerRed. We believe that the N145S mutation in SuperNova is compensated by other mutations affecting the hydrogen bond network around the chromophore. However, it is quite difficult to explain the small difference of ROS generation between KillerRed and SuperNova for two reasons. First, there is not much structural difference between the chromophores. Second, the other mutated residues in SuperNova are away from the chromophore. KillerRed's structure does not help us understand why KillerRed mainly produces superoxide10,11. Therefore, further physicochemical analyses should be performed to try and resolve the selective mechanism of ROS generation in genetically encoded photosensitizers. For example following the crystal structure analysis of KillerRed, it was suggested that one of the key structural features responsible for its CALI activity was a water-filled channel that connects the chromophore region to the end cap of the β-barrel10,11. Therefore, E70 and S121 in SuperNova, which are located adjacent to the chromophore, should be key amino acid residues required for CALI activity.
Recently a smaller genetically encoded singlet oxygen generating protein, miniSOG, was developed and used for photo-inducible cell ablation in C. elegans18. The chromophore in miniSOG requires a flavin mononucleotide (FMN) that is ubiquitously present in all cells. Thus even if FMN is not bound to miniSOG, it has intrinsic photosensitizing activity so that nonspecific effects in biological systems cannot be excluded. SuperNova does not have the same limitation because of its intrinsic photosensitizing chromophore. In the future it may be possible to evolve color variants of this protein by introducing mutations into the chromophore itself or vicinal residues for multicolor CALI. Thus by incorporating these changes with further improvements in light delivery and image analysis, SuperNova may become a useful reagent for spatiotemporal analysis of protein function in living cells due to the exquisite precision afforded by the CALI technique.
Methods
Random mutagenesis
The KillerRed gene was subjected to error-prone PCR-based random mutagenesis using the Diversify PCR random mutagenesis kit (Clontech) with a T7 primer and a pRSETB reverse primer. The mutation rate was 1.4–4.8 per 1,000 bp. Mutated DNA fragments were double-digested with BamHI and EcoRI and inserted into the BamHI/EcoRI site of the pRSETB vector (Invitrogen). Escherichia coli JM109 (DE3) was transformed with the vectors, and the bacteria were grown on LB plates for 1–3 days until pigmented colonies appeared. The pigmented colonies were then cultured overnight in 1.5 mL of LB medium containing 0.1 mg/mL carbenicillin and processed for plasmid purification. The cDNA sequences of the mutated KillerRed genes were verified by dye terminator cycle sequencing using Big Dye (Applied Biosystems).
Gene construction
Site-directed mutagenesis was performed to introduce L160T/F162T mutations into KillerRed as previously described19. The digested PCR product was cloned into the BamHI/EcoRI site of pRSETB. For SuperNova expression in mammalian cells SuperNova/pRSETB was digested with BamHI and EcoRI, and subcloned between BamHI and EcoRI site of pcDNA3 yielding SuperNova/pcDNA3. Eukaryotic expression vectors of the various fusion proteins described in this work were derived by substituting the FP component of mTFP-keratin; Cx43-mRFP113; 2MLS-KillerRed7; Phamret-fibrillarin/pcDNA320; Phamret-H2B/pcDNA320; pVimentin-mTFP112; pEB3-mTFP1 vector12; Lyn-SECFP/pcDNA3; YFP-RhoV14-C1; FPC-1myc LK1 WT vector21; cofilin-CFP vector21 was replaced with SuperNova or KillerRed respectively.
Protein purification
Recombinant fluorescent proteins with N-terminal polyhistidine tags were expressed in JM109 (DE3) at 23°C, with gentle shaking at 80 rpm for at least 4 days without IPTG induction. The polyhistidine-tagged proteins were purified by Ni-NTA agarose chromatography (Qiagen) and ion exchange chromatography (HiTrapQ) followed by buffer exchange to 50 mM HEPES-KOH (pH 7.4) with a PD-10 column (GE Healthcare).
Molecular size analysis
Size-exclusion chromatography was performed with a Superdex75 100/300GL column on the ÄKTA explorer 10S (GE Healthcare). Analytical ultracentrifugation analyses were performed in 10 mM HEPES, pH 7.2 containing 100 mM NaCl, using Proteomelab XL-1 Analytical Ultracentrifuge (Beckman-Coulter, Fullerton, CA). In sedimentation velocity experiments, 400 μL aliquots of KillerRed, SuperNova and mCherry dissolved in the same buffer were respectively sedimented at 60,000 rpm at 20°C using 12-mm aluminum double sector centerpieces with sapphire windows. The sedimentation behavior was monitored with Rayleigh interference optics. All sedimentation velocity data were analyzed by the continuous C(s) distribution model included in the software program SEDFIT ver. 14.1 (ref. 22). The parameters used for analysis, i.e., partial specific volume of the protein calculated from the amino acid composition, KillerRed: 0.7201 cm3/g, SuperNova: 0.7193 cm3/g, and mCherry: 0.7328 cm3/g), buffer density (ρ = 1.00314 g/cm3), and buffer viscosity (η = 1.0204 cP) were estimated using the program SEDNTERP ver.1.09. Sedimentation equilibrium experiments were carried out at 5, 10, 25, 50 and 100 μM and the equilibrium concentration gradients were analyzed by the program, Origin.
Crystallization and data collection
SuperNova crystals were grown at 20°C using hanging-drop vapor diffusion and sitting-drop vapor diffusion methods. The best crystals were obtained with 100 mM sodium acetate, pH 4.2–4.3, 2.2- to 2.3-M NaCl, and 200 mM lithium sulphate. Each drop contained 1 μL of protein solution at a concentration of 25 mg/mL and an equal volume of reservoir solution. The crystals were soaked in solution containing 10% glycerol and 90% of reservoir for cryoprotection and flash-frozen under nitrogen stream. Diffraction data sets were collected at the AR-NW12A beamline of the Photon Factory (Tsukuba, Japan) at 100 K and a wavelength of 1.000 Å and were processed with HKL200023. Supplementary Table 1 shows a summary of the results of the data collection.
Structure determination and refinement
The structure of SuperNova was determined by a molecular replacement method using MOLREP24 and the coordinates of a protomer of the dimeric KillerRed was solved by our group previously (PDB ID: 3A8S), and used as a search model. Structure refinement was performed using CNS25 and REFMAC26. Model inspection was performed manually using COOT27. During refinement, 5% of the reflection data were set aside for the calculation of the free R-factor to monitor refinement. Water molecules were picked with ARP/wARP28. Supplementary Table 1 shows a summary of the results of the structure refinement. The atomic coordinate and structure factor have been deposited in the Protein Data Bank with accession code 3WCK.
Singlet oxygen measurement
Purified proteins were diluted to 100 μM with 50 mM HEPES-KOH (pH 7.4). Singlet oxygen generation was measured using anthracene-9,10-dipropionic acid (ADPA)29 as a singlet oxygen probe. The samples were exposed to intense light provided by a 200 W mercury arc lamp (Hamamatsu Photonics) with a 580AF20 (Omega) filter. Lens and mirrors were adjusted to focus an intense light onto a spot (3 mm in diameter; 1.4 W/cm2) in a sample solution containing 50 μM protein and 10 μM ADPA in a Terasaki dish. A heat-absorbing filter was placed between the mercury arc lamp and the excitation filter to prevent evaporation of the sample. After light exposure, the ADPA fluorescence (430 nm emission; excited at 380 nm) was measured by a F2500 fluorescence spectrophotometer (Hitachi).
Measurement of the phototoxic effect of SuperNova in E. coli and mammalian cells
To measure the phototoxicity of KillerRed, SuperNova, mCherry and EGFP, E. coli [JM109 (DE3)] cells were transformed with a pRSETB vector (Invitrogen) containing a photosensitizing protein and cultured in LB medium for 6.5 days at 18°C. The cells were then diluted to equal concentrations (OD600 = 0.3) and exposed to intense light through a 580AF20 excitation filter (Omega) for 15 min, using a 200-W mercury arc lamp (Hamamatsu Photonics) as a light source. To determine phototoxicity of the KillerRed variants and EGFP in mammalian cells, HeLa cells were transiently transfected with pcDNA3 encoding 2MLS-SuperNova, 2MLS-KillerRed, or 2MLS-EGFP. Eighteen to thirty hours after transfection, the cells were exposed to intense light through a 580AF20 excitation filter (Omega) (8 W/cm2) and examined DIC imaging under the inverted microscope (TE2000, Nikon) equipped with a cooled CCD camera ORCA-R2 (Hamamatsu). The number of surviving E. coli or HeLa cells was counted at various time points after irradiation.
Live imaging of cell division
HeLa cells were transfected with KillerRed-H2B or SuperNova-H2B expressing vectors. One to two days after transfection, cell division were examined by time lapse imaging with an widefield inverted microscope (TE-2000, Nikon) equipped with a × 20 NA 0.75 objective, cooled electron-multiplying charge-coupled device (EM-CCD) camera (iXon Ultra, Andor) and LED based light source (LightEngine SPECTRA, Lumencor). The differential interference contrast images were taken at intervals of 20 min for 24 hrs.
CALI of cofilin in living cells
COS7 cells were transfected with actin-GFP and cofilin-SuperNova- or cofilin-CFP-expressing vectors. Two to 3 days after transfection, actin-GFP fluorescence was examined by time-lapse. CALI was performed on an inverted microscope (FV1000, Olympus) using a × 60 NA1.4 oil immersion objective. The Images were taken using a 488-nm multi-Argon ion laser. Cofilin was inactivated by intense green light irradiation (9.6 W/cm2) through a 540AF30 interference filter (Omega) from a mercury lamp for 1 min. Actin filament motion was quantitatively analyzed by temporal autocorrelation function determination. The time constants of autocorrelation functions for the series of images taken before and after exposing intense green light were compared.
Autocorrelation function analysis
Region of interests (ROIs; 50 × 50 pixels) were chosen in protrusive lamellipodia for the time trajectory analysis in images of COS7 cells expressing actin-GFP. Then, the temporal autocorrelation function of GFP fluorescence intensity was calculated for the averaged intensity in the ROI by the following equation: 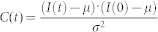 , where I(t) is the fluorescence intensity at time t and μ and σ are the time mean and standard deviations, respectively. The autocorrelation function was averaged for pixels in the ROI in each experimental condition and was fitted by single exponential functions, and time constant values with errors were obtained.
, where I(t) is the fluorescence intensity at time t and μ and σ are the time mean and standard deviations, respectively. The autocorrelation function was averaged for pixels in the ROI in each experimental condition and was fitted by single exponential functions, and time constant values with errors were obtained.
Expression in sensory neurons of C.elegans
For in vivo expression, genes encoding KillerRed and SuperNova were inserted into C. elegans expression vector, L36HS1 (based on the commercial Fire vector) under the control of mec-4 promoter. Transgenic worms were generated by gonadal injection of plasmid constructs (containing no more than 100 ng/μl of DNA). At least 3 extrachromosomal arrays were maintained. The gentle-touch assay was used to examine mechanosensory function14.
Refolding and reoxidation of SuperNova in vitro
Proteins were denatured in 8 M urea and 1 mM dithiothreitol (DTT); and renatured in 35 mM KCl, 2 mM MgCl2, 50 mM Tris pH7.5, 1 mM DTT. Fluorescence recovery experiments in purified SuperNova, KillerRed and mCherry were performed as described30.
Stability of SuperNova in living cell
HEK-293T cells expressing SuperNova, KillerRed or mCherry were exposed to 100 ug/ml cyclohexamide for 0, 24 hr, 48 hr and 72 hrs. Cells were collected by centrifugation. After washing twice in HBSS, cells were suspended by HBSS, and fluorescence was measured using a F-2500 fluorescence spectrophotometer (Hitachi).
Photobleaching assay
HeLa cells expressing SuperNova and KillerRed were exposed to intense laser light (100% laser power, 543 nm) and fluorescence intensity reduction monitored by confocal microscopy (FV1000, Olympus).
Spectroscopy
Fluorescence quantum yields were measured using a Hamamatsu Photonics C9920-01, an absolute photoluminescence quantum-yield measurement device31. To calculate the molar extinction coefficient of SuperNova, absorbance of 100 μM SuperNova protein solution was measured on V630-Bio spectrometer (JASCO) at 579 nm. Molar extinction coefficient at 579 nm ε579 (M−1 cm−1) was defined by following equation. ε579 = A579/c, where A579 and c are absorbance at 579 nm and concentration of protein (M), respectively.
Accession codes
The nucleotide sequence of the SuperNova gene has been deposited with the GenBank under accession code AB522905.
Author Contributions
K.T. performed the gene construction, mutagenesis, protein purification, and CALI experiments and wrote the paper. T.M. predicted the amino acid residues in KillerRed involved in dimer formation, performed the analytical column chromatography, gene construction, live cell imaging and wrote the paper. M.N., S.U., K.F., M.H. and F.I. performed the analytical ultracentrifugation. N.S. and T.A. analyzed the X-ray crystal structure of SuperNova. Y.A. performed the autocorrelation function analysis of the actin filaments. D.F., I.K. and H.S. performed experiments in C. elegans. T.N. contributed to the conceptual development, experimental design, and data analysis and wrote the paper.
Supplementary Material
Supplementary Information
Supplementary Movie 1
Supplementary Movie 2
Acknowledgments
We are grateful to Konstantin Lukyanov, Kazumasa Ohashi, Robert Campbell, and Roger Tsien for providing the gene for KillerRed, the Cofilin-CFP, mTFP1-keratin, and Cx43-mRFP1 expression vectors, respectively. We also thank Matthew J. Daniel for valuable comments. This work was partly supported by grants from the Scientific Research on Advanced Medical Technology of the Ministry of Labor, Health, and Welfare of Japan (T.N.); Precursory Research for Embryonic Science and Technology of the Japan Science and Technology Agency (T.N.); Inamori Foundation; MEXT Grant-in-Aid for Scientific Research on Innovative Areas (23115003, T.N. and 23111502, T.M.) and JSPS Grant-in-Aid for Young Scientists (A) 21687007 (T.M.).
References
- Jay D. G. Selective destruction of protein function by chromophore-assisted laser inactivation. Proc. Natl. Acad. Sci. U. S. A. 85, 5454–5458 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K., Rajfur Z., Vitriol E. & Hahn K. Chromophore-assisted laser inactivation in cell biology. Trends in Cell Biol. 18, 443–450 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck S. et al. Fluorophore-assisted light inactivation: a high-throughput tool for direct target validation of proteins. Proteomics 2, 247–255 (2002). [DOI] [PubMed] [Google Scholar]
- Wong E. V., David S., Jacob M. H. & Jay D. G. Inactivation of myelin-associated glycoprotein enhances optic nerve regeneration. J. Neurosci. 23, 3112–3117 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto K. et al. Chromophore-assisted light inactivation of HaloTag fusion proteins labeled with eosin in living cells. ACS. Chem. Biol. 6, 401–406 (2011). [DOI] [PubMed] [Google Scholar]
- Tour O., Meijer R. M., Zacharias D. A., Adams S. R. & Tsien R. Y. Genetically targeted chromophore-assisted light inactivation. Nat. Biotechnol. 21, 1505–1508 (2003). [DOI] [PubMed] [Google Scholar]
- Bulina M. E. et al. A genetically encoded photosensitizer. Nat. Biotechnol. 24, 95–99 (2006). [DOI] [PubMed] [Google Scholar]
- Bates P. A., Kelley L. A., MacCallum R. M. & Sternberg M. J. E. Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins Suppl 5, 39–46 (2001). [DOI] [PubMed] [Google Scholar]
- Wall M. A., Socolich M. & Ranganathan R. The structural basis for red fluorescence in the tetrameric GFP homolog DsRed. Nat. Struct. Biol. 7, 1133–1138 (2000). [DOI] [PubMed] [Google Scholar]
- Carpentier P., Violot S., Blanchoin L. & Bourgeois D. Structural basis for the phototoxicity of the fluorescent protein KillerRed. FEBS Lett. 583, 2839–2842 (2009). [DOI] [PubMed] [Google Scholar]
- Pletnev S. et al. Structural basis for phototoxicity of the genetically encoded photosensitizer KillerRed. J. Biol. Chem. 284, 32028–32039 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai H. W., Olenych S. G., Wong P., Davidson M. W. & Campbell R. E. Hue-shifted monomeric variants of Clavularia cyan fluorescent protein: identification of the molecular determinants of color and applications in fluorescence imaging. BMC Biol. 6, 13 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell R. E. et al. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 99, 7877–7882 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H. et al. In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron 39, 1005–1017 (2003). [DOI] [PubMed] [Google Scholar]
- Waldeck W. V. et al. Autofluorescent proteins as photosensitizer in eukaryontes. Int J Med Sci. 6, 365–373 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang D. H. et al. Cofilin expression induces cofilin-actin rod formation and disrupts synaptic structure and function in Aplysia synapses. Proc. Natl. Acad. Sci. U S A 102, 16072–16077 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T. Y., DerMardirossian C. & Bokoch G. M. Cofilin phosphatases and regulation of actin dynamics. Curr. Opin. Cell Biol. 18, 26–31 (2006). [DOI] [PubMed] [Google Scholar]
- Qi Y. B., Garren E. J., Shu X., Tsien R. Y. & Jin Y. Photo-inducible cell ablation in Caenorhabditis elegans using the genetically encoded singlet oxygen generating protein miniSOG. Proc. Natl. Acad. Sci. U. S. A. 109, 7499–7504 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawano A. & Miyawaki A. Directed evolution of green fluorescent protein by a new versatile PCR strategy for site-directed and semi-random mutagenesis. Nucleic Acids Res. 28, E78 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T., Miyawaki A. & Nagai T. Direct measurement of protein dynamics inside cells using a rationally designed photoconvertible protein. Nat. Methods 5, 339–345 (2008). [DOI] [PubMed] [Google Scholar]
- Ohashi K. et al. LIM kinase has a dual role in regulating lamellipodium extension by decelerating the rate of actin retrograde flow and the rate of actin polymerization. J. Biol. Chem. 286, 36340–36351 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. & Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- Vagin A. & Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 (2010). [DOI] [PubMed] [Google Scholar]
- Brünger A. et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. 54, 905–921 (1998). [DOI] [PubMed] [Google Scholar]
- Murshudov G. N., Vagin A. A. & Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. Biol. Crystallogr. 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W. G. & Cowtan K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G., Cohen S. X., Lamzin V. S. & Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebeke M., Piette J. & van de Vorst A. Photosensitized production of singlet oxygen by merocyanine 540 bound to liposomes. J. Photochem. Photobiol. B. 9, 281–294 (1991). [DOI] [PubMed] [Google Scholar]
- Reid B. G. & Flynn G. C. Chromophore formation in green fluorescent protein. Biochemistry 36, 6786–6791 (1997). [DOI] [PubMed] [Google Scholar]
- Tomosugi W. et al. An ultramarine fluorescent protein with increased photostability and pH insensitivity. Nat. Methods. 6, 351–353 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Movie 1
Supplementary Movie 2