

Abstract
Congenital cystic adenomatoid malformations (CCAM) also known as congenital pulmonary airway malformation is a developmental, non-hereditary, hamartomatous abnormality of lung with unknown etiology. It is a rare disease with an incidence of 1 in 25,000 to 1 in 35,000. It is a disease of infancy with most of the cases diagnosed within first 2 years of life. We report autopsy findings of two fetuses with congenital cystic adenomatoid malformation (Stocker Type II and I) with brief review of literature.
KEY WORDS: Congenital cystic adenomatoid malformation, fetal autopsy, lung
INTRODUCTION
Congenital cystic adenomatoid malformations (CCAM) of lung are rare congenital cystic lung lesions that arise from excessive proliferation of tubular bronchial structures.[1] It is a rare lesion with incidence of 1 in 25,000 to 1 in 35,000 pregnancies and represents 25% of congenital lung malformations and 95% of congenital lung lesions.[2] It was classified into 3 subtypes in 1977,[3] and expanded into five types and renamed as congenital pulmonary airway malformation (CPAM) by Stocker in 2002.[4] Eighty percent of the lesions are recognized in neonatal period; however, there are reports even in adult population.[5] Here, we have described two cases of CCAM, Stocker Type II and I, which were diagnosed by fetal autopsy.
CASE REPORTS
Case 1
A 30-year-old G2 P1 L1, of nonconsanguineous marriage, had a normal antenatal course until 20.5 weeks of gestation, when she presented with history of per vaginal bleeding and abdominal pain since three days. Her past medical history and family history were unremarkable. On examination uterus was of 24 weeks and fetal heart sounds were not localized. Routine blood and biochemical investigations were within normal limits. Subsequently; she delivered a stillborn fetus, clinically it was concluded as inevitable abortion and after informed consent the fetus was sent for pathological examination.
On autopsy, it was a male fetus weighing 1,450 g. External examination did not reveal any anomaly. On internal examination all organs were in situ. The right lung was enlarged (7 × 5 × 3 cm) cut-section showed cysts of varying sizes majority of them were < 2 cm. The left lung was normal (3.5 × 2 × 1 cm). On histology, right lungs showed multiple cysts relatively uniform in size resembling bronchioles, lined by stratified columnar epithelium, and having thin fibromuscular septa. Mucus production was not evident on periodic acid Schiff (PAS) staining [Figure 1a-d]. All other visceral organs were congested, but well-developed and revealed no anomalies. With the above features, a pathological diagnosis of CCAM Stocker type II was offered.
Figure 1.
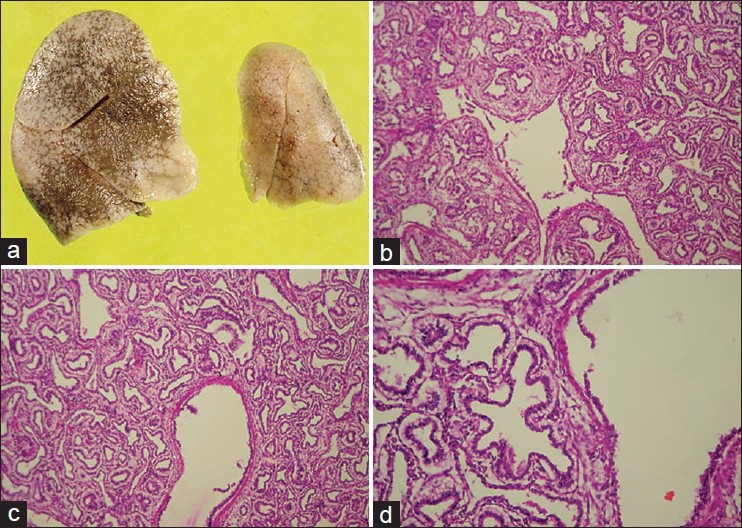
(a) Gross photo of resected lungs showing enlargement of right lung. (b and c) Microscopy showing large cyst (bronchiole like) surrounded by small cysts, lined by stratified columnar epithelium and having thin fibromuscular septa (hematoxylin and eosin (H and E), ×100). (d) High power of the large cyst showing lining epithelium (H and E, ×400)
Case 2
A 29-year-old female G4 P3 L2, with history of 24.6 weeks of amenorrhea sought routine antenatal check-up. She gave history of second degree consanguineous marriage. Her third baby girl died 3 days after birth, details of which was not available. Her blood and biochemical investigations were within normal limits. Routine anomalous scan revealed polyhydramnios along with features of congenital diaphragmatic hernia (CDH). The parents were counseled and they opted for termination. Pregnancy was terminated after informed consent and the fetus was sent for pathological evaluation.
On autopsy, it was a female fetus. External examination revealed low set ears, short neck, and subcutaneous edema. On internal examination all organs were in situ. The left lung was enlarged (8 × 6 × 4 cm), and cystic in consistency. It was occupying most part of the thorax. Right lung, heart, and thymus were pushed posteriolaterally. Right lung was hypoplastic (3 × 2 × 1 cm). The enlarged left lung was pushing the diaphragm inferiomedially; hence, the spleen was found in umbilical region [Figure 2a-c]. Cut-section of the left lung revealed multiple cysts, the larger one was occupying whole of the upper lobe [Figure 2d]. On histology, left lung revealed cysts of varying sizes dominated by larger cysts accompanied by smaller cysts. The cysts were lined by flattened cuboidal epithelial cells having fibromuscular septa [Figure 2e-g]. Focally, few of the cysts showed mucin production which was evident on Periodic acid-Schiff (PAS) staining [Figure 2h and i]. Cartilage was not seen. All other visceral organs were congested, but well developed and revealed no anomalies. With the above features a pathological diagnosis of CCAM Stocker type I was offered.
Figure 2.
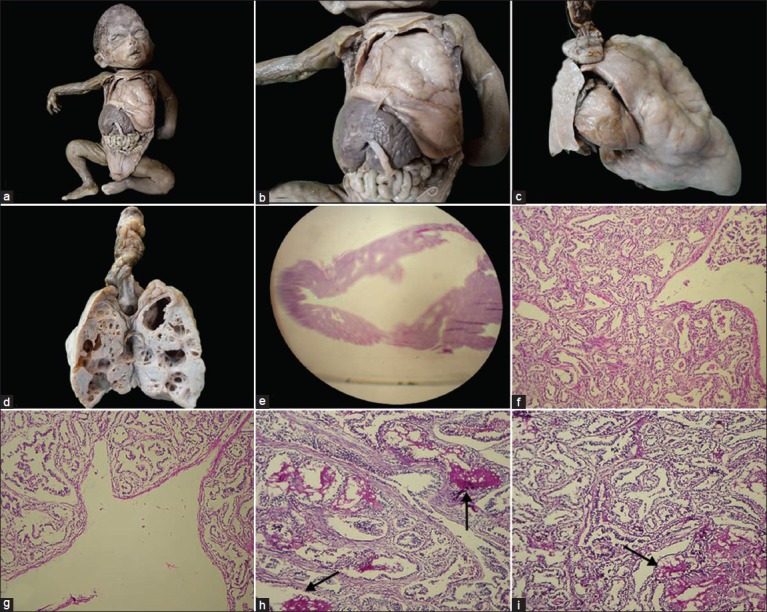
(a and b) Gross photographs showing enlarged left lung occupying most of the thorax compressing heart and right lung. (c) Gross photograph of enlarged left lung with hypoplastic right lung and heart. (d) Cut-section of left lung showing multiple cysts, dominated by large cysts. (e) Whole mount showing large cyst surrounded by small cysts (H and E, ×2). (f and g) Microscopy showing large cysts with surrounded small cysts, lined by flattened epithelium with fibromuscular septa (H and E, ×100). (h and i) Microscopy showing cysts with mucus production (arrow) (periodic acid Schiff staining (PAS), ×100)
DISCUSSION
CCAM is a rare developmental, non-hereditary, hamartomatous abnormality of lung with unknown etiology. This lesion was first described by Chi’n Tang in 1949.[6] Males and females are equally affected. It is usually a unilateral condition and restricted to one lobe, but can involve whole of one lung (as observed in our cases), or both the lungs.[1,7] Based on the anatomical changes, development of human lung is subdivided into embryonal (3-7 weeks), pseudo glandular (7-17 weeks), canalicular (17-29 weeks), saccular (24-36 weeks), and alveolar (36 weeks to maturity). CCAM develops during the pseudo glandular and saccular period (7-35 weeks).[1]
Stocker et al., in 1977,[3] subdivided CCAM into three subtypes. Type I lesion constitute 50-70% and is composed of single or multiple large cysts (> 2 cm) lined by flattened, cuboidal cells frequently producing mediastinal herniation. The walls of the cysts contain prominent smooth muscle and elastic tissue. Occasionally mucus producing cells are seen and presence of cartilage is extremely rare. Mucin production is unique to type I lesion (as observed in our case 2). Type II lesion constitute 15-30% and are composed of multiple small cysts (< 2 cm), lined by ciliated cuboidal to columnar epithelium, structure resembling that of respiratory bronchioles, and distended alveoli are present between the epithelium lined cyst. Mucus cells and cartilage are not seen. This type is usually associated with other systemic anomalies. Type III lesion constitutes 5-10% and are usually large bulky noncystic lesions producing mediastinal shift. Bronchial-like structures are lined by ciliated cuboidal epithelium and separated by masses of alveolus-sized structures by nonciliated cuboidal epithelium.[8] Systemic anomalies reported in Type II lesions include; renal anomalies (B/L renal agenesis, abnormalities of ureter, bladder, and urethra), abdominal wall defects, central nervous system defects (hydrocephalus), spinal deformities, gastrointestinal defects (diaphragmatic hernia, jejuna atersia, tracheoesophageal fistula, and imforforate anus), cardiac anomalies (ventriculoseptal defects, tetralogy of fallot, and truncus arteriosis), sirenomelia, and anomalies of reproductive tract.[7]
In 2002 Stocker,[4] modified this classification by adding two more types (type 0 and IV) and renamed the lesion as CPAM, type 0 of tracheobronchial origin has solid appearance with small and firm lungs; and microscopically shows bronchiolar type airway with cartilage, smooth muscle, and glands separated by abundant mesenchymal tissue. Type IV of distal acinar origin, has peripheral cystic type, large cysts (> 10 cm) lined by flattened epithelium and resting on loose mesenchymal tissue.[9] The new classification is not much used as type 0 lesions are very difficult to differentiate from bronchogenic cyst and the similarities between type IV cyst and pleura pulmonary blastoma may cause problem in diagnosis.[5]
The diagnostic sonographic ultrasonographic feature of CCAM is, multicystic lesions at the lung field and common differential diagnosis includes diaphragmatic hernia, bronchogenic cyst, cystic fibrosis, congenital lobar emphysema, and pulmonary sequestration.[2] CCAM will have connections to tracheobronchial tree and derives its blood supply from pulmonary circulation. In contrast, pulmonary sequestration contains immature lung tissue without connection to tracheobronchial tree and derives its blood supply from aberrant systemic blood vessel.[1] In the past, patients whose fetuses presented with presumed diagnosis of CCAM were informed of poor prognosis and advised for termination, but recent studies revealed aggressive obstretric management and termination of pregnancy is not appropriate for patients with affected fetuses. There are reports of even disappearance of lesion on serial USG examination.[10] Termination was offered to our case 2 since it was misdiagnosed as CDH in USG.
For the lung masses, the intrauterine prognosis of the fetus is determined by fetal lung mass size (a single measurement of lung mass at the maximum diameter) or by the CCAM volume ratio (CVR). CCAM volume is calculated by using a formula for prolate ellipse, with measurements of lung mass in three perpendicular planes. The CVR is calculated by dividing the CCAM volume by head circumference (HC, measured in centimeter). Thus CVR = (L × B × W × 0.52/HC).[11]
Cass et al.,[11] in their large study observed that the size of the lung mass or CVR correlated strongly with the fetal outcome. The observed threshold values in their study were 5.2 cm for mass size and 2 for CVR. There was 100% survival of fetuses with mass values less than 5.2 cm and CVR < 2. In the fetuses with lung mass size more than 5.2 cm or CVR more than 2 were associated with marked mediastinal deviation, hydrops, ascites, and heart failure. They also observed that fetal echocardiography will be helpful in management of these fetuses. If there are no signs of heart failure, then the fetuses will be benefited by maternal steroid therapy and serial thoracocentesis. Those fetuses with impending cardiac failure needed open fetal surgeries.
The commonest presentation of CCAM in postnatal life is progressive respiratory distress including tachypnea, grunting, retraction, and cyanosis; and in adults it usually presents as repeated chest infections. Chest X-ray, computed tomography (CT), and magnetic resonance imaging (MRI) are helpful in diagnosis.[2]
The main complication of CCAM in neonatal period is compression of the mediastinal structure producing cardiovascular compromise. In adult patient, CCAM is a nidus for pneumonia, abscess formation, fungal infections, spontaneous pneumothorax, hemoptysis, air embolism, intralobar sequestration, and development of bronchogenic carcinoma.[12,13,14] Serial antenatal sonographic evaluation, good obstetric care, and delivery at a tertiary care center are preferred plan of treatment for antenataly detected cases. Postnatal and in adults patients, lobectomy is the treatment of choice for symptomatic cases.[2,5,13,14]
Prognosis also depends on Stocker type, type I lesions carry overall good prognosis. In type II lesion, it is the associated anomalies that determine the prognosis. Type III lesions carry bad prognosis as they are usually large and presents with cardiovascular compromise. Overall bilateral involvement, associated with hydrops and associated congenital anomalies carry poor prognosis.[7]
CONCLUSION
CCAM is a rare developmental malformation of lung. It is usually diagnosed in fetal or neonatal life. Serial ultrasonographic evaluation, fetal lung mass size, CVR, and fetal echocardiography are needed for management of antenatally detected cases. We report these cases for their rarity.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Sood M, Sharma S. Congenital cystic adenomatoid malformation of lung-A case report. Currt Pediatr Res. 2011;15:61–3. [Google Scholar]
- 2.Sahu S, Muthuvel S, Naware SS, Dhavala SS. Congenital cystic adenomatoid malformation of lung. Med J Armed Forces India. 2008;64:268–9. doi: 10.1016/S0377-1237(08)80113-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol. 1977;8:155–71. doi: 10.1016/s0046-8177(77)80078-6. [DOI] [PubMed] [Google Scholar]
- 4.Stocker JT. Congenital pulmonary airway malformation: A new name and an expanded classification of congenital cystic adenomatoid malformation of the lung. Histopathol. 2002;41:424–31. [Google Scholar]
- 5.Feng A, Cai H, Sun Q, Zhang Y, Chen L, Meng F. Congenital cystic adenomatoid malformation of lung in adults: 2 rare cases report and review of the literature. Diagn Pathol. 2012;7:37. doi: 10.1186/1746-1596-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khan NU, Jones MT, Greaves M. Case report: Congenital cystic adenomatoid malformation of an entire lung in a 33-year-old man: A case report and review of the literature. Br J Radiol. 2008;81:e276–8. doi: 10.1259/bjr/23523404. [DOI] [PubMed] [Google Scholar]
- 7.Annam V, Korishetty SI, Yelikar BR, Hippargi SB, Shivalingappa DB. Bilateral congenital cystic adenomatoid malformation, stocker type III with associated findings and review of literature. Indian J Pathol Microbiol. 2010;53:331–3. doi: 10.4103/0377-4929.64324. [DOI] [PubMed] [Google Scholar]
- 8.Sfakianaki AK, Copel JA. Congenital cystic lesions of the lung: Congenital cystic adenomatoid malformation and bronchopulmonary sequestration. Rev Obstet Gynecol. 2012;5:85–93. [PMC free article] [PubMed] [Google Scholar]
- 9.Anand M, Deshmukh SD, Naik A, Gaopande V. Congenital cystic adenomatoid malformation in an adolescent: An unusual presentation with pleural effusion and pneumatocele. Indian J Chest Dis Allied Sci. 2011;53:173–6. [PubMed] [Google Scholar]
- 10.Hsu YR, Lee SY. Prenatal diagnosis of congenital cystic adenomatoid malformation. Chang Gung Med J. 2004;27:61–5. [PubMed] [Google Scholar]
- 11.Cass DL, Olutoye OO, Cassady CI, Moise KJ, Johnson A, Papanna R, et al. Prenatal diagnosis and outcome of fetal lung masses. J Pediatr Surg. 2011;46:292–8. doi: 10.1016/j.jpedsurg.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Zeidan S, Hery G, Lacroix F, Gorincour G, Potier A, Dubus JC, et al. Intralobar sequestration associated with cystic adenomatoid malformation: Diagnostic and thoracoscopic pitfalls. Surg Endosc. 2009;23:1750–3. doi: 10.1007/s00464-008-0183-7. [DOI] [PubMed] [Google Scholar]
- 13.Belcher E, Lawson MH, Nicholson AG, Davison A, Goldstraw P. Congenital cystic adenomatoid malformation presenting as in-flight systemic air embolisation. Eur Respir J. 2007;30:801–4. doi: 10.1183/09031936.00153906. [DOI] [PubMed] [Google Scholar]
- 14.Loachimescu OC, Mehta AC. From cystic pulmonary airway malformation, to bronchioloalveolar carcinoma and adenocarcinoma of the lung. Eur Respir J. 2005;26:1181–7. doi: 10.1183/09031936.05.00011705. [DOI] [PubMed] [Google Scholar]