

Abstract
Selective or Multiple Reaction monitoring (SRM/MRM) is a liquid-chromatography (LC)/tandem-mass spectrometry (MS/MS) method that enables the quantitation of specific proteins in a sample by analyzing precursor ions and the fragment ions of their selected tryptic peptides. Instrumentation software has advanced to the point that thousands of transitions (pairs of primary and secondary m/z values) can be measured in a triple quadrupole instrument coupled to an LC, by a well-designed scheduling and selection of m/z windows. The design of a good MRM assay relies on the availability of peptide spectra from previous discovery-phase LC-MS/MS studies. The tedious aspect of manually developing and processing MRM assays involving thousands of transitions has spurred to development of software tools to automate this process. Software packages have been developed for project management, assay development, assay validation, data export, peak integration, quality assessment, and biostatistical analysis. No single tool provides a complete end-to-end solution, thus this article reviews the current state and discusses future directions of these software tools in order to enable researchers to combine these tools for a comprehensive targeted proteomics workflow.
Keywords: Targeted proteomics, multiple reaction monitoring, database, bioinformatics, mass spectrometry
1. Introduction
A large body of research suggests a relatively poor correlation between protein and mRNA expression [1]. Since the biological effector molecule is usually the protein and not the mRNA that encodes it, and since mRNA microarray expression analysis is unable to detect differential levels of protein post-translational modification (PTM) (e.g., phosphorylation), there is a very substantial demand for quantitative protein profiling technologies. Proteomics technologies can be broadly divided into two categories: discovery and targeted proteomics. Discovery proteomics experiments (e.g. multidimensional protein identification technology (MudPIT) [2], difference gel electrophoresis (DIGE) [3], isotope-coded affinity tags (ICAT) [4], multiplexed isobaric tagging technology for relative and absolute quantitation (iTRAQ) [5], stable isotope labeling by amino acids in cell culture (SILAC) [6], and LC-MS/MS label-free quantitation [7]) often require large sample quantities and multi-dimensional fractionation, which diminishes throughput. Furthermore, approaches to improve the sensitivity and throughput of protein quantification limit the number of peptides that can be monitored per MS run. For this reason, discovery proteomics optimizes protein identification by spending more time and effort per sample and reducing the number of samples analyzed. In contrast, targeted proteomics strategies limit the number of features that will be monitored and then optimize the chromatography, instrument tuning and acquisition methods in order to achieve the highest sensitivity and throughput for hundreds or thousands of samples. Discovery proteomics relies on stochastic precursor-ion selection, thus making run-to-run peptide identifications variable. In addition, the software and technical expertise needed to run and analyze these methods remains challenging. Discovery proteomics results in MS/MS sequencing of many more peptides (>3) than are needed to identify the parent protein. With complex mixtures this approach also must be coupled with off-line fractionation which results in numerous LC-MS/MS runs that require tens of hours of MS instrument time to detect and quantify hundreds to thousands of proteins in a complex mixture. As an example of the enormous duplication of effort with this approach, since 2007 the MS/Proteomics Resource at Yale University has sequenced and stored 41,938,125 peptides (with an FDR of 0.01) in the Yale Protein Expression Database (YPED) [8, 9] with only 2,895,792 distinct sequences or 6.9% of all YPED data. If we continue to use the same LC-MS/MS approach, then 93% of our instrument time will be wasted by resequencing the same abundant peptides in each experiment. Given the challenges inherent in quantitative analysis of highly complex proteomes, it is not surprising that many proteomic laboratories are moving away from “complete proteome” analysis to a more targeted analysis of protein expression as clinicians seek the biomarkers that may provide valuable insight into the understanding, diagnosis, and personalized treatment of many human diseases.
Targeted Proteomics was just recently selected as Nature Method of the Year and is recognized as the most sensitive and specific way to detect pre-selected components in a complex matrix such as a proteolytic digest of a plasma or tissue extract[10-12]. Multiple Reaction Monitoring (MRM) utilizes a triple quadrupole mass spectrometer carrying out sequential rounds of Selective Reaction Monitoring (SRM), to concurrently quantitate multiple analytes as schematically depicted in Figure 1.
Figure 1.
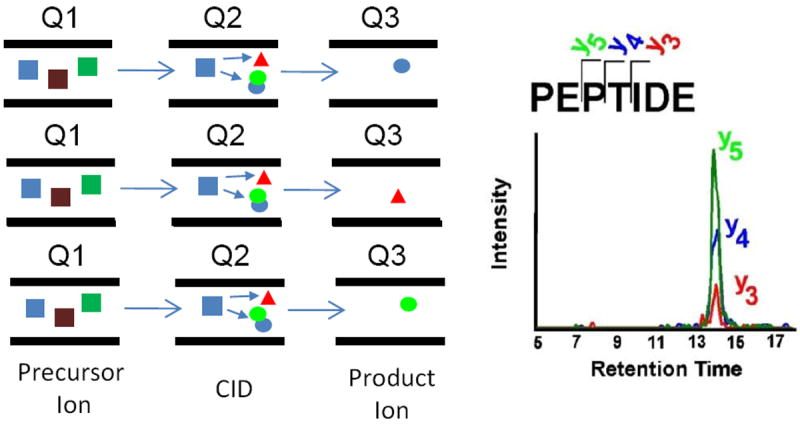
Schematic Diagram of Multiple Reaction Monitoring
A quadrupole mass analyzer is a type of mass filter which typically consists of four parallel metal rods. Ions of a certain mass-to-charge ratio as determined by the applied potential to the rods will travel between the rods and reach the quadrupole’s detector. Triple quadrupole mass spectrometers consist of two quadrupole mass analyzers in series, with a (non mass-resolving) radio frequency (RF)- only quadrupole between them to act as a collision cell for collision-induced dissociation. The first (Q1) and third (Q3) quadrupoles serve as mass filters, whereas the middle (Q2) quadrupole serves as a collision cell. SRM analysis in the triple quadrupole mode is performed by setting the Q1 window to the precursor m/z value (Q1 is not scanning), and Q3 set to the specific m/z value corresponding to a specific fragment of that peptide. In SRM reactions, precursor / fragment ion transitions are monitored while the collision energy is tuned to optimize the intensity of the fragment ions of interest. In the MRM mode, a series of SRM reactions are measured sequentially, and the cycle (typically 1-2 sec) is looped throughout the entire time of the HPLC separation. SRM transitions are determined from the MS/MS spectra of the existing peptides. Typically, doubly (sometimes triply)-charged precursors are selected Multiple transitions per peptide, corresponding to high intensity fragment ions, are then selected and the collision energy optimized to maximize signal strength of MRM transitions by using automation software. The principles upon which the MRM approach rests are based on a very robust MS technology[13] that has been used for many years to quantify a wide range of small molecules in clinical samples ([14]). This high-throughput MS method has a wide linear dynamic range of up to five orders of magnitude and also has a very high sensitivity that allows detection of ng/ml amounts of peptides in biological fluids and cell or tissue protein extracts. [15-17]
For developing a targeted proteomics assay, first the targeted proteins must be chosen, and this is often based on previous experiments such as iTRAQ, DIGE, SILAC, label-free LC-MS, DNA-based microarray technologies (with the caveat that the level of mRNA and protein expression may differ), or on general knowledge of the disease or tissue being studied. Once the protein is selected, a user must select a peptide that is unique to each protein from databases of experimentally identified peptides, or use an algorithm [18] that analyzes the sequence of each targeted protein to predict the best tryptic peptides for analysis. Numerous recent publications outline the advantages and reasons for utilizing MRM analysis. Examples include a recent comparison of MRM analysis to traditional discovery proteomics [19] where they used information-dependent data acquisition (IDA) to monitor 222 peptides from a complex sample, highlighted the improved sensitivity and dynamic range capability of MRM by showing that the number of proteins identified in all samples increased significantly from 34% (IDA) to 88% (MRM). MRM analysis has also been equally applicable to quantifying unmodified as well as modified proteins, such as phosphorylated, ubiquitinylated, methylated, acetylated, etc. proteins. Anderson and Hunter [20] also showed the applicability of this method in plasma when they developed a method to monitor 50 plasma proteins via MRM quantitation. Xiao et. al. [21] have shown that MRM analysis is at least 50 times more sensitive than parent ion monitoring. Carr and researchers from 8 laboratories demonstrated the level of accuracy and precision that MRM assays could achieve by running a multi-site analysis of human plasma [22]. Aebersold and colleagues were able to show the ability of MRM to quantitate proteins over the full dynamic range of yeast. [23]. The results obtained from MRM analysis also have been shown to match antibody-based assays, such as ELISA [24-27]. For absolute quantification of proteins, peptides, and/or their modification states, researchers have utilized stable isotope dilution (SID)[16] as a means to precisely and quantitatively measure the absolute levels of proteins after proteolysis[28]. These stable isotopes can be generated via synthesis of stable isotope peptides (termed AQUA peptides[29]), metabolic labeling of a protein consisting of concatamers of peptides (termed QconCAT[30]), or full length isotope labeled proteins (termed PSAQ [31]).
For detection of low abundance proteins in biological fluids, researchers have employed an approach termed SISCAPA [32]. SISCAPA is a combined assay using a signature peptide and its associated stable isotope-labeled internal standards that are enriched from sample digests by anti-peptide antibodies for a targeted LC-MS readout. Briefly, a synthetic peptide is used to generate a highly specific anti-peptide antibody whose role is to enrich the signature peptide in a complex mixture. The protein digest spiked with a stable isotope-labeled form of this peptide is subjected to affinity chromatography using the anti-peptide antibody immobilized on magnetic beads. After affinity enrichment, both the native peptide and stable isotope-labeled peptide are eluted and analyzed by mass spectrometry.
A common feature of the above-mentioned publications is the effort and time required to develop large-scale MRM-based assays. Often these steps include the need to run discovery experiments, generate synthetic peptide standard, and calibrate retention time between samples, thus limiting the throughput of large-scale MRM assays and the time to obtain results. In efforts to make the MRM assay development and instrument usage more efficient, comprehensive software tools and automated workflows have been developed. This manuscript aims to outline these software tools in hopes that future researchers developing large-scale targeted proteomic assays will be able to effectively utilize these tools to reduce the time and effort required for performing large-scale MRM assays on complex proteomic samples.
Targeted proteomic assay development can be divided into three stages: assay development, data collection and data analysis (Figure 2). Often numerous software tools are required to go through all three stages. Given the emerging nature of targeted proteomics, many of these tools are rapidly evolving in efforts to provide a more robust and comprehensive workflow. Recent reviews have covered the MRM software used for both the assay development [33] and downstream data analysis[34]. In this article, we review available commercial and open-source software tools (Table 1) that, when put together, can provide researchers with a complete workflow for developing, collecting, integrating, and analyzing large-scale proteomic MRM assays.
Figure 2.
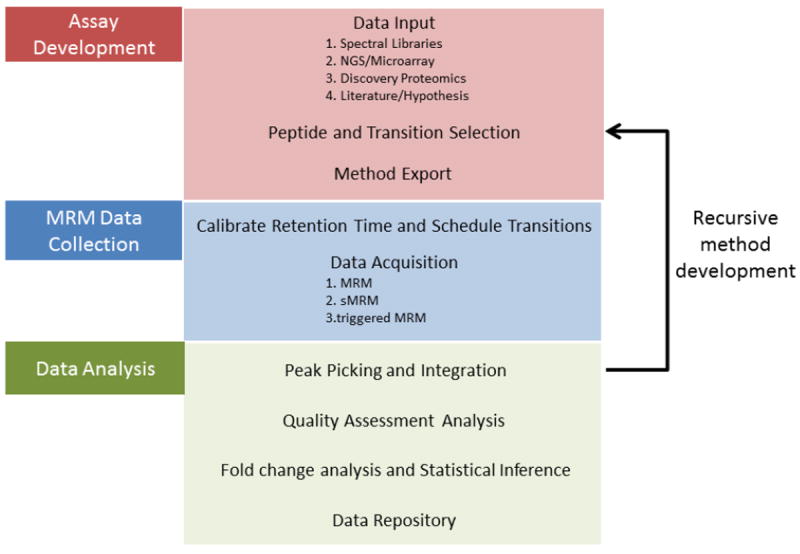
Workflow Diagram for MRM Targeted Proteomics
Table 1.
Current Software Tools for Development of Large Scale Targeted Proteomic Assays
| Software | Vendor/Lab | Publication Date | Platform |
|---|---|---|---|
| 1. Assay Development | |||
|
| |||
|
Databases and Spectral Libraries
| |||
| PeptideAtlas | Institute for Systems Biology (ISB) | 2006 | Web |
| SRMAtlas | ISB | 2009 | Web |
| PRIDE | Martens, EMBL/EBI | 2006 | Web |
| GPMdb | Gpmdb | 2004 | Windows |
| NIST Peptide Mass Spectral Libraries | NIST | 2009 | Web |
| PABST | Seattle Proteome center | 2006 | Web |
|
| |||
|
Peptide and Transition Selection and Method Export
| |||
| Skyline | MacCoss Lab, U Washington | 2010 | Windows |
| MRMPilot | ABSciex | Commercial | Windows |
| Pinpoint | Thermo Scientific | Commercial | Windows |
| MRMaid | Bessant, Cranfield U. | 2009 | Web |
| ATAQS | Aebersold, ISB | 2011 | Web |
|
| |||
|
2. MRM Data Collection
| |||
| iRT calculator | Biognosys | 2012 | Web |
| scheduled MRM (sMRM) | AB Sciex, Waters, Thermo Scientific, and Agilent | Commercial | Windows |
| iSRM (intelligent MRM) | Thermo Scientific | Commercial | Windows |
| xMRM (triggered MRM) | AB Sciex | Commercial | Windows |
| tMRM (triggered MRM) | Agilent | N/A | Windows |
|
| |||
|
3. Data Analysis
| |||
|
Peak Detection and Integration
| |||
| Multiquant | AB Sciex | Commercial | Windows |
| Skyline | MacCoss Lab, U Washington | 2010 | Windows |
| MRMer | McIntosh Lab, ISB | 2008 | Java |
| Pinpoint | Thermo Scientific | Commercial | Windows |
| OpenMS | Kohlbacher & Reinert, FU Berlin & U of Tübingen | 2008 | Windows, Linux, Mac (C++ library) |
| ATAQS | Seattle Proteome Center, ISB | 2011 | Web |
| TIQAM | Seattle Proteome Center, ISB | 2008 | Java (Mac, Win, Linux) |
| mQUEST | Biognosys | 2011 | Linux on a VM; Windows |
|
| |||
|
Quaility Assessment and Data Filtering
| |||
| mProphet | Biognosys | 2011 | Linux on a VM; Windows |
| Audit | Carr, Broad | 2010 | Web |
| Multiquant | AB Sciex | Commercial | Windows |
|
| |||
|
Fold change or statistical analysis
| |||
| SRMstats | Vitek Lab, Purdue | 2012 | Windows, Linux, Mac (R libraries) |
| QuaSAR | Carr, Broad | N/A | Web |
2. Assay development
The goal of MRM assay development is to effectively choose peptides and transitions that serve as proxy for the total protein amount. With this in mind, selection of peptides can be challenging since the process of trypsin digestion can lead to a miscleavages or partial cleavages, thus creating multiple forms for a given peptide sequence. Peptides can also be non-unique, in that their sequence is shared among multiple proteins. Finally, peptides can contain various post-translational modifications such as oxidation, phosphorylation, etc. Those variations split the amount of peptide into multiple forms making detection more difficult; also, all forms of this peptide would need to be monitored in order to accurately correlate the amount of peptide measured with the protein concentration. Due to these reasons, the software tools often contain rules and parameter settings to avoid peptides which suffer these disadvantages. For example, the software will often remove all methionine-containing peptides under consideration due to the effect of oxidation.
Another feature in some of the assay development software is the ability to make inference in the MRM peptide/fragment ions. For example, discovery data is often collected on high resolution mass spectrometers, while Triple Quadrupole Mass Spectrometers often have “unit” m/z resolution (e.g., 0.7 Th full width half max), with the consequence that precursor/product ion pairs can overlap and result in incorrect signal measurements. Therefore, researchers often need to predict or query the fragments of all peptides in a given proteome in order to avoid choosing a Q1/Q3 pair that contains multiple simultaneously detected ions. Given this potential for interference, researchers often choose multiple (2-5) transitions per peptide so that downstream peak integration algorithms can group these for accurate peak integration. An advantage of monitoring multiple MRM transitions is the potential to assess the quality of the peaks based on their relative intensities, thus allowing detection of interference and dropping the problematic transition(s) or the whole peptide from future assays. [35] For MRM assays with only a few transitions, these interference spectra can be determined via visual inspection. For larger-scale assays the relative ordering of peaks can be evaluated quantitatively as a dot-product of the expected peak heights and the observed peak heights, similarly to what has been used in GC-MS[36]. Often the software packages can generate a dot-product score and flag questionable transitions for manual review.
Choosing peptides with the strongest signal is essential for developing high-sensitivity MRM assays, because peptides from the same protein can differ by as much as 100 fold in ionization intensity. [37] Thus, selecting peptides based on the observed signal response maximizes sensitivity and has the added benefit of minimizing the effect of interference. In order to maximize the number of MRM transitions that can be monitored in a liquid chromatography multiple-reaction monitoring assay (LC-MRM), one can schedule the mass spectrometer to collect subsets of peaks based on their retention time (RT) on the column. Thus, many of the software applications have features which predict/calculate RT to generate an MRM method containing, Q1, Q3, and RT values for each peptide. Software such as the sequence-specific retention calculator SSRCalc can calculate a retention time based on peptide sequence and the reversed phase analytical column properties [38-40]. An alternatative method for determining peptide retention times is to run a set of standard peptides or recombinant protein sequences mixed with the sample, thus allowing one to express the RT of the peptides in terms of that of internal standards and generate an empirical retention time. [41] The benefit of using isotopic internal standards in MRM assays is that if the exact concentration of the heavy isotopic peptide is known, the absolute quantitation of that particular peptide can be determined. Another approach to scheduling MRM assays is to use the identical chromatographic setup for discovery MS and targeted MS instrumentation, thus ensuring identical elution times.
2.1 Software for Automating Assay Development
Software and algorithms used to generate MRM assays include MRMaid, MRMpilot, Pinpoint, and Skyline, ATAQS. The initial software packages for MRM assay development were often single-user packages, such as MRMpilot, MRMaid, MaRiMba and they were limited in their scope. Newer software packages such as Skyline and ATAQS aim to integrate the entire targeted proteomic workflow.
MRMPilot software [42] is commercial software developed by AB Sciex, for developing MRM assays on the company’s instruments. It can devise assays based on existing spectra or can predict spectra de novo, based on chemical properties of the peptides. Assays can be developed for peptides with chemical modifications, PTMs or heavy isotope labels. Once the transitions are defined, MRMPilot Software then builds the Analyst® Software acquisition method. Then it can perform iterative assay development based on experimental results and can store and organize assay files. The software accepts input from ProteinPilot™ Software, Mascot, Sequest, Spectrum Mill, BioML, PeptideAtlas, and MRMAtlas. It also has support for developing MIDAS™ (MRM-triggered MS/MS) acquisition, which utilizes an AB Sciex QTRAP mass spectrometer to trigger a MS/MS experiment once an MRM signal is above a specified baseline, thus enabling researchers to confirm the identification of the peptide.[43]
MRMaid [44, 45] is part of the Proteosuite software suite. It uses spectra from EBI’s PRIDE database and sequence data from UniProt to suggest an MRM assay for a given protein. It is a Web-based tool into which one can enter the name of one or more proteins and obtain a list of transitions based on the entered criteria. Results can be filtered by instrument type, amino acid, transition value, maximum number of peptides per protein and maximum number of transitions per peptide. It considers the probability of observing each peptide and each transition based on data in PRIDE; it calculates the retention time, and the product ion relative intensity to develop an MRM assay. It is not designed to create assays for post translational modifications. It exports the transition list in csv format.
Skyline [46] aims to be a comprehensive pipeline system for developing MRM assays. The user can start by inputting a file of protein sequences for which to develop the assay. The software allows users to build their own spectral library, by inputting data in various formats such as from BiblioSpec, NIST and GPM. The program automatically generates a list of proteolytic peptides and, for each peptide, a list of transitions. One can filter this list by defining the type of protease (trypsin, LysC, etc.), the size and charge of precursor peptides, the types (y, b, etc.) and charge of product ions, the types of modifications of the peptides (static modifications like carbamidomethylation of cysteines, or heavy isotopic modification of specific amino acids). These predicted spectra are used in conjunction with spectral libraries obtained from a local or public repository. There are specific rules and filters that can be selected for generating optimal transitions. Transition calculations can be made for vendor-specific instruments. It is also possible to define retention time standards that are used for setting up scheduled MRM. With suitable settings one can automatically generate an optimal assay. Skyline can export transition lists into various formats, including those specific to particular commercial instruments, as well as open formats. It is also possible to import transition lists from another source and translate them into a different format. Skyline also has features for MRM assay refinement based on the experimental results obtained.
Pinpoint is a commercial package from ThermoScientific which simplifies the creation of targeted quantitative assays and vendor-specific intelligent-SRM (iSRM) assays. The software is designed to automate the preliminary selection of MRM transitions by predicting proteotypic peptides and determining the best MRM transitions. iSRM assays enable you to collect a primary set of transitions for peptide quantitation and trigger secondary transitions at very short dwell times for peptide confirmation without greatly increasing cycle time. Pinpoint methods can then be exported to a Thermo Scientific instrument for data collection. Like MRMPilot, the Pinpoint workflow is iterative in that a preliminary method is used to acquire data, which is in turn used to refine the method. Pinpoint software also provides tools for evaluating this preliminary data and verifying peptide candidates.
ATAQS (Automated and Targeted Analysis with Quantitative SRM) [47] is a software pipeline tool that contains modules to design, manage, analyze and validate an MRM assay. The modular design allows users to begin and end the analysis at any point in the workflow and also to write plug-ins to customize their analysis algorithms. The modules support project management in general, as well as target selection, transition optimization and post-acquisition data analysis. The software is Web-based and allows multiple users to work on a project. ATAQS uses FireGoose [48] to connect to various Web services. Among these Web services are PeptideAtlas (used to select peptide spectra [49]), TIQAM (to generate in-silico peptides for a given protein [50]), PIPE2 (to generate a list of proteins to design an MRM assay as well as for various analysis tasks), and PABST (Peptide Atlas Best SRM Transition) (to generate optimal transitions). The user has the option to generate decoy or heavy-light pairs of data. mProphet[51] is used for validating selected transitions in their ability to correctly identify a protein. An assay document can be generated in TraML format [52] to share with other investigators.
MassHunter Optimizer is part of the MassHunter Workstation, a commercial software package from Agilent for triple quadrupole LC-MS. It allows fast, sensitive compound identification and confirmation that helps design triggered MRM (tMRM) acquisition. Triggered MRM is a more sensitive form of MRM because only the primary quadrupole is set to monitor for a desired peptide until its detection, after which the monitoring of the secondary transitions begins. The software allows for the selection of ten transitions which can be any combination of primary and secondary types. MassHunter also enables the optimization collision energy as well as select the best transitions from empirical data generated on the Agilent platforms using Optimizer-designed methods.
TargetLynx™ Application Manager is available from Waters Corporation automates sample data acquisition, processing and reporting for quantitative results from their instruments. The software can be used to flag peaks that are above a certain threshold, within an m/z window. It can also flag data that does not satisfy certain quality control criteria. The QuantOptimize Application Manager automates MRM method set-up. Up to five MRM transitions can be automatically and individually selected and optimized for collision energy.
Other tools exist that identify peptides that are likely to be suitable for an MRM assay, but the software pipelines described above appear to have greater utility at this time. TIQAM (Targeted Identification for Quantitative Analysis by MRM is a software suite for peptide selection, transition selection and validation [50]. MaRiMba [53] generates and validates a transition list based on input spectra and protein sequences. MRMer [54] calculates transitions and their peak intensities and allows comparison of predicted results with actual results. SRMCollider [55] can calculate ion signatures for a given protein mixture and identify those that can create interference with an MRM assay. Numerous software tools for evaluating the best MRM peptides have emerged such as PeptideSieve[18], ESP predictor[56], and STEPP[57]. These software use different algorithms that consider sequence features and predicted physico-chemical properties of peptides to classify them as “proteotypic”, that is, found in only a single known protein and therefore serves to identify that protein.
2.2 Public Databases
With the emergence of MRM technologies, databases/repositories are needed for capturing MRM data. Current proteomics repositories such as PRIDE[58, 59], GPMdb [60] and PeptideAtlas[61-63] have been based on shotgun proteomics data. These resources do not provide the user with the ability to store, annotate, and query MRM data. While it is possible to develop a new MRM database from scratch, the other alternative is to extend an existing proteomics database/repository to handle MRM data. Databases like GPMdb [60] [64], Peptide Atlas [63], MRMAtlas [65], EBI’s PRIDE[66], NIST (http://peptide.nist.gov/), and YPED [8, 9] are candidates that have the potential for such an extension. The advantage of this approach is that one can leverage existing informatics resources and expertise to integrate targeted and shotgun proteomics data. In addition, many of the existing components, including sample annotation, project-based grouping of samples, and data publication can be reused for MRM purposes. New modules will need to be constructed to allow these databases to accept data from the MRM pipeline as well as to provide new user interfaces to browse and visualize MRM data. It will also be necessary to incorporate the use of standard proteomic data formats such as mzML [67] and TraML [52] for disseminating raw and processed MRM data to the scientific community. For example, Skyline users can now use Panorama[68] Server to be automaticaly upload and sharing Skyline files via a web browser interface.
Another benefit of using existing proteomic databases lies in the selection of transitions for developing MRM assays. Selection of the correct transitions is greatly facilitated if a product ion has been previously observed. Public repositories in which mass spectrometry proteomic data is deposited as described above can be used in the development of MRM assays. The SRMAtlas [37] contains spectrum libraries for a significant number of crude peptides that were synthesized on a large scale. MacCoss and colleagues developed a high-throughput workflow based on in vitro-synthesized proteins [69]. Such MRM assay atlases can be expected to make the measurement of protein levels a more routine undertaking. PABST is a search tool for the best peptides to identify proteins by mass spectrometry [47].
2.3 Standardization and Data formats
Because laboratory instruments and analysis software output data in various and sometimes proprietary formats, it has become important to devise standard data formats that can be produced and recognized by tools used by the proteomics community. For example, mzXML format developed by the Sashimi project (http://sashimi.sourceforge.net/) and the mzData developed by the Human Proteome Organization (http://hupo.org) allow peptide identifications by different software to be combined by integrative programs. Application-specific file formats such as Mascot dat and mgf files, SEQUEST result and dat files, MS2 and DTASelect formats can be converted into XML-based formats such as mzXML [70], mzML [67], pepXML [71], protXML [71], mzIdentML [72], mzQuantML (http://www.psidev.info/mzquantml), X! Tandem XML [73], PRIDE XML (www.ebi.ac.uk/pride/). All these formats are based on the Extensible Markup Language (XML) and can be validated by various standard tools. TraML is the data standard proposed for MRM assay files [52]. It can be used for MRM assays developed for small compound studies as well as for proteomics studies. The format can contain up to 10 types of information sources, ranging from metadata about the file itself to instruments, people, publications, compounds, transition data, and an inclusion and/or exclusion list. Overall, there has been an increasing effort by the ProteomeXchange project to develop a single point of submission to proteomics repositories, and encourage the data exchange and sharing of identifiers between the repositories so that the community may easily find datasets in the participating repositories. [74]
3. MRM data collection
For large scale MRM analysis, the “learned” peptide sequences and their transitions described above are transformed into LC-MRM assays that are run on a triple quadrupole mass spectrometer.
Quantitation of targeted proteins is often accomplished by quantifying and averaging together the yields of 2-3 tryptic peptides from each targeted protein. The specificity and sensitivity of detection is enhanced by fragmenting each of these tryptic peptides and by then selecting two (or more) of the most intense fragment ions from each tryptic peptide to quantify. Hence, quantitation of each targeted protein is typically based on multiple data points (e.g. 2 peptides × 3 transitions/peptide). This results in an LC-MRM method which collects hundreds to thousands of SRM transitions representing quantitative results for 50-100 proteins in parallel. LC-MRM data is collected by sequentially cycling through hundreds to thousands (depending on the instrument) of transitions at regular intervals, termed cycle time, with each SRM transition requiring a specific amount of data collection time, termed dwell time. As the number of transitions in the assay get larger, the challenge with expanding these assays is that one is forced to reduce both cycle and dwell time, which greatly reduces signal-to-noise ratio and data quality, respectively. For downstream peak detection and integration to be effective, a high-enough peak sampling rate is needed so that the number of points collected represents the signal being measured. Extracting the height of the signal over time generates a Gaussian peak as shown in Figure 1. To define a Gaussian the rule of thumb is that at least 3 points above full-width a half max (FWHM) are needed, but in practice 3 points is not enough to distinguish the peak from noise (in frequency domain). [75] For good statistical practice, collecting at least 5 points above PWHH enables good peak deconvolution and thus produces adequate signal for downstream fold-change analysis. For example, in typical nanoflow chromatography the PWHH is typically 10-15 seconds, thus the implication is that the an LC-MRM analysis must collect signal for each transition every 2-3 seconds in order to obtain enough points to effectively fit and model a peak. Therefore, researchers have built more intelligence into the MRM acquisition logic to improve data quality and assay robustness. Figure 3 outlines the various methods, which have been utilized to collect MRM transitions. The simplest approach is to cycle through the monitoring of all transitions during the entire time of the HPLC elution. For example, Figure 3A illustrates the data collection scheme for a twelve-transition MRM assay based on three peptides with four transitions per peptide. The second approach, often termed “scheduled MRM” (Figure 3B) is based on a peptide retention time and a scheduling window during the LC-MRM run for which each set of transitions from a given peptide a monitored. [76] As more peak detections are multiplexed, these experiments are challenged by the fact that different compounds have different peak widths and different RT stabilities. Having even more intelligence built into the MRM scheduling logic can greatly improve result quality and assay robustness. A third approach, called triggered MRM, combines both scheduling and baseline-triggering (Figure 3C) and is termed xMRM [77] or SRM-triggered SRM [78]. In this method a single MRM transition is scheduled to be collected within a RT window, but when the signal of that particular MRM transition goes above a specified baseline, this triggers additional transitions from that same peptide to be scheduled for monitoring. Utilizing a triggered MRM approach as shown in Figure 3C can effectively maximize both cycle and dwell time while maintaining accurate and quantifiable areas for both primary and secondary transitions. As illustrated in Figure 3, the first approach results in the collection of fewer data points above the PWHH, compared to the second and third approaches.
Figure 3.
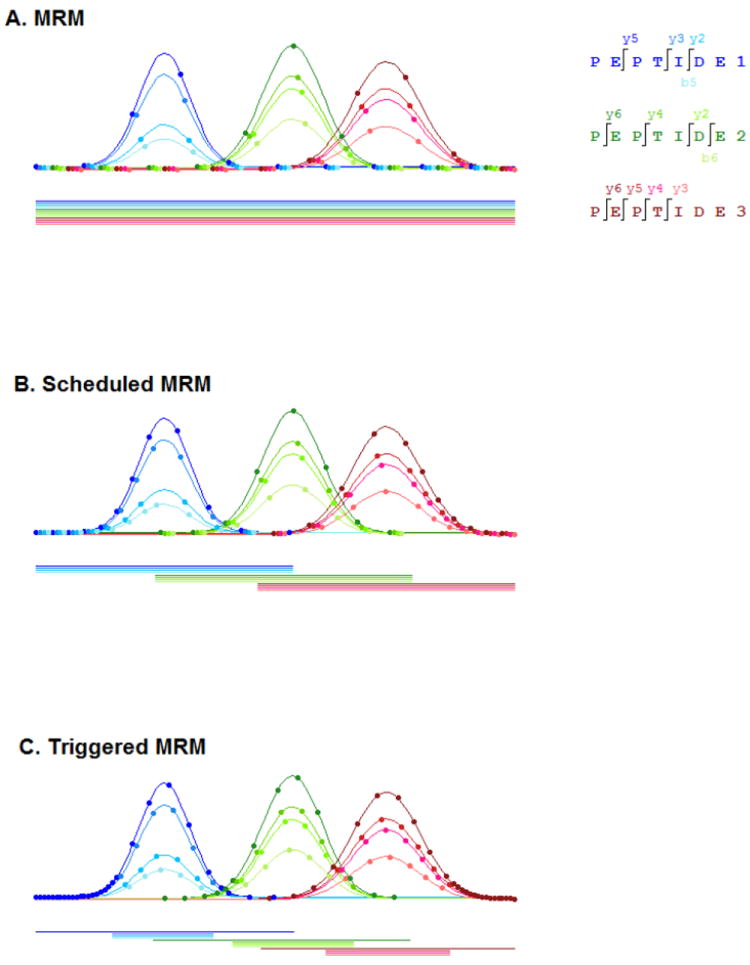
Acquisition methods utilized to collect MRM transitions for a twelve transition MRM assay based on three peptides with four transitions per peptide. (A) MRM is the simplest approach in that all transitions are sequentially looped throughout the entire time of the HPLC separation. (B) Scheduled MRM is based on a peptide retention time and scheduling a window during the LC-MRM run for which each set of transitions from a given peptide a monitored. (C) Triggered MRM combines both scheduling and baseline triggering, where a single MRM transition is scheduled within a RT window and when the signal of that individual MRM transition goes above a specified baseline, the remaining transitions from that same peptide are triggered and collected.
4. Data analysis
4.1 Peak Detection and Integration
The post-acquisition workflow can entail four major steps. The first step is peak detection, integration, and quantification; the second is data quality assessment and data filtering; the third is data visualization and exploratory analysis, and the last is fold-change and/or statistical significance analysis. Currently no individual software package performs all these steps. For peak detection and integration, researchers commonly utilize Skyline, Multiquant, Pinbpoint, MRMer, and ATAQS.
Skyline’s peak integration module can import various types of MRM mass spectrometer files including Agilent, ThermoScientific, AB Sciex, Bruker, and Waters. It also supports mzXMl and mzML open source formats. The windows based application also supports multiple quantitation workflows including label free MS, data-independent, isotope labeled, multiple labeling strategies [uniformly labeled 15N, 13C/15N, etc] for a single peptide, and post-translational modifications. Skyline incorporates peptide identifications from MS/MS spectrum for retention time selection and grouping based on precursor ions. For peak detection and alignment Skyline uses the CRAWDAD [79] for chromatogram retention time alignment and warping. CRAWDAD uses a dynamic time warping (DTW) algorithm to determine retention time shifts and correct for chromatographic variance between replicate runs. The algorithm is applied to the extracted ion chromatograms after signal smoothing by an 11-point second-order Savitsky Golay filter [80] to detect the boundary of the peak. Peak areas are calculated using total integrated area subtracted by background area where the background area is determined by a rectangular area between peak boundaries and background boundary. The software provides a graphical display of chromatogram graphs so manual adjustments can be made and missed integrations can be corrected. The results can then be exported in a comma-separated format for further analysis.
MultiQuant is a commercial software developed by AB Sciex to detect and quantify MRM peak intensities for AB Sciex wiff files. MultiQuant supports both relative and absolute quantitation experiments as well as unlabeled or stable isotope-labeled peptide internal standards. It can process MIDAS Workflow datasets (MRM-triggered MS/MS) [43] [81] in addition to MRM-only datasets. It contains two different peak integration algorithms, MQ4 and SignalFinder, and the user interface has simplified parameter selection to save operator time and to remove operator bias. The MQ4 algorithm has the ability to group transitions for peak detection and integration. MQ4 can perform Gaussian smoothing, baseline subtraction, peak splitting, and calculation of noise percentage. It also contains parameters useful for scheduled MRM experiments such as peak width and retention time window. SignalFinder™ utilizes a confidence-based iterative algorithm that can adapt to varying quantities such as changes in peak shape, baseline and noise. For each transition, a Gaussian curve is fitted to detect baseline level, calculate peak intensity and confidence score. This step is repeated until the confidence score converges. [82] Quantitation methods can be imported or exported as a text file and data results can be exported as either AB Sciex MarkerView™ format or text files. For absolute quantitation experiments, MultiQuant generates standard curve on heavy isotope peptides which can be then applied to unknown samples containing both endogenous and heavy peptides. After calibration is applied by MultiQuant, a concentration and accuracy is computed.
Pinpoint [83] is a commercial software package from Thermo Scientific. It can be used for generating MRM or scheduled MRM assays termed iSRM. It also provides post-acquisition data processing. It supports all Thermo MS platforms including TSQ, Orbitrap, and ion trap. Peptides are quantified by single-point, normal curve, and reversed curve approaches. Single-point quantitation is designed for labeled experiments and provides relative quantitation of endogenous peptide to heavy isotope-labeled peptide. This method is based on the assumption that the same amount of light and heavy-labeled peptides produces an equal signal. A normal curve approach can be applied when a sample has a low amount of endogenous peptides. The reversed-curve approach uses light peptide as internal standards [84].
Waters provides TargetLynx software for sample quantification and confirmation analysis of data collected form Waters Mass Spectrometers. Peak integration is performed by ApexTrack™. ApexTrack™ determines peaks and baselines based on tail, shoulder, and skewness characteristics. TargetLynx develops calibration curves using standard samples. Various types of polynomial curve fitting is available to quantify samples with unknown concentration [85].
Agilent MassHunter software can integrate and process Agilent LC/MS, GC/MS, and ICP-MS instrument files. Similar to other software described above, MassHunter can perform relative and absolute quantitation experiments as well as unlabeled or stable isotope-labeled peptide internal standards. Final reports are stored in XML format or can be exported to Microsoft Excel.
TIQAM is a stand alone open source software from ISB that has since been superseded by ATAQS. ATAQS utilizes mzXML or mzML files to detect all the peaks above background noise of a targeted peptide. Signals from these peaks are smoothed by discrete Fourier transformation and integrated using mQuest [51]. A peak group is determined by collecting the nearest peaks and validated by examining the peak group characteristics such as the number of transitions, total intensities, and retention time deviation. For the correct peak group, ATAQS further provides peak scores including deviation from expected retention time, the number of matched heavy and light transitions, and the dot product between matched heavy and light intensities. The output of the ATAQS can be sent to the mProphet [51] for peak quality assessment.
MRMer [54] performs both peak integration and heavy/light ratio calculations on mzXML input files [70]. MRMer detects precursor/product groups based on a specified mass tolerance. It then determines group start and stop time, and calculates peak area for each product ion using a trapezoidal approximation. The software enables visual inspection of the result and users can alter the start and stop times if necessary. If samples contains heavy and light transition pairs, a separate input file is required to specify which amino acid residues are isotopically labeled. In this case, the software calculates a heavy/light ratio based on their peak areas. If precursor ion scans are available the MS1 data can be used for peak detection. Finally, data is exported in a tabular format for downstream analysis.
4.2 Data Structure and Peak Quality Assessment
The MRM experiment records various transition-level characteristics, including retention time, signal-to-noise, and peak intensity. For analysis, each characteristic (e.g., intensity) can be structured in the form of a matrix. Figure 4 illustrates examples of data structures obtained by label-free and stable isotope-labeled experiments with three technical runs for each sample. Each peptide contains multiple transitions, with the label-free quantitation having 5 transitions for each endogenous peptide. For a stable isotope-dilution experiment as shown in the figure, the data is represented as each peptide having three transitions matched for both the endogenous and stable isotope-labeled peptides. Each row and column of the data matrix corresponds to a transition and replicate run, respectively. Observation from one transition and a given sample is recorded as an element of the matrix. The rows and columns of the matrix are often grouped according to precursor peptide sequences and biological samples.
Figure 4.
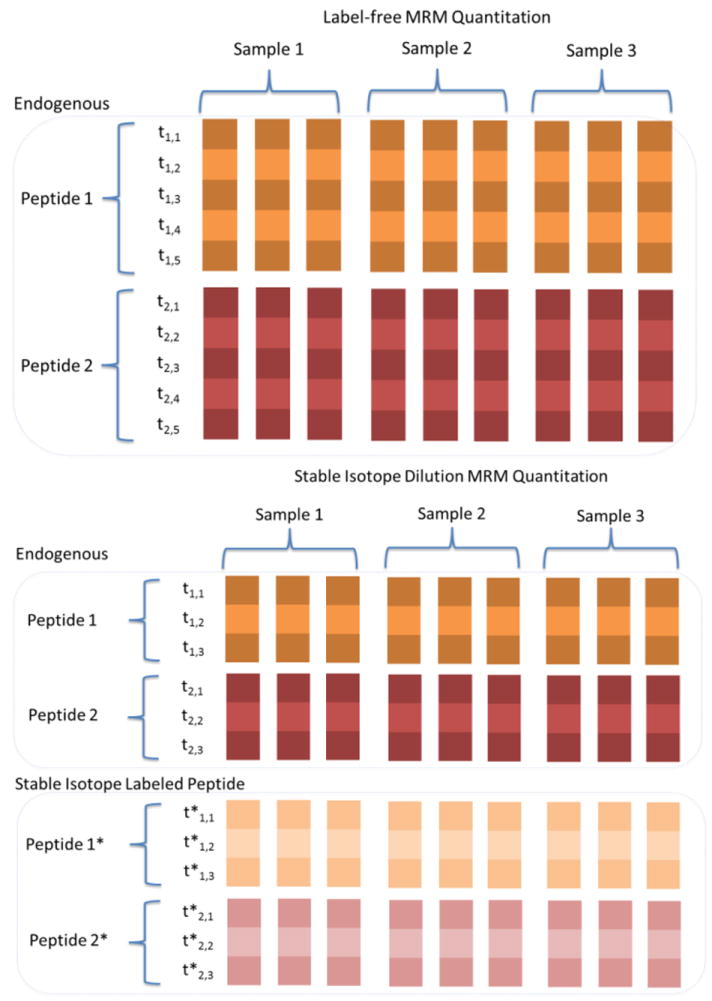
Data structure for label-free and SID MRM quantification with an example of two peptides. In label-free, transition-level peak intensities are grouped according to their peptides. Transition ID is denoted as ti, j, where i indicates a peptide (1,2) and j indicates fragment ions. In the SID experiment, transitions are grouped separately grouped by endogenous and isotopic labeled peptides. Asterisks (*) indicates transitions from the labeled peptides.
Peak quality assessment is essential for large-scale MRM/SRM experiments since the number of transitions in a complete datasets can easily exceed 10,000 individual peak areas or peak area ratios. In efforts to automate this process, software packages include various scoring algorithms that provide data metrics for assessing the quality of each transition. Software packages such as Skyline, TCorr, and Pinpoint evaluate peak integration quality by a utilizing a score based on the dot-product of the observed intensities and a spectral library. Skyline additionally provides a downstream filtering algorithm to eliminate peptides with low intensity and large interferences. Pinpoint calculates percent CV and retention time reproducibility. TargetLynx provides various quality measures based on user-specified thresholds. Peak qualities are assessed by using signal-to-noise ratio, retention time deviation, and the coefficient of determination from the calibration curve. In mProphet, peak quality is determined from various subscores of transition peak groups including the ion intensities or retention time deviation for the sensitive recovery of true peak groups. The algorithm calculates a linear combination of subscores that best separates true and false peak groups by iterative linear discriminant analysis. In particular, mProphet adopts a semi-supervised learning algorithm which requires decoy transitions as negative controls. Decoy transitions are designed to represent false peak groups and can be generated by reversing the peptide sequences or adding a random value to Q1- and Q3-measured masses. They are further used to generate a null distribution to evaluate the false discovery rate [51]. The mProphet algorithm has also been integrated into the ATAQS pipeline. AuDIT (Automated Detection of Inaccurate and imprecise Transitions) detects inaccurate transitions due to signal interference or inconsistency among replicate samples. The algorithm is designed for experiments utilizing stable isotope-labeled internal standards (SIS) with technical replicates. It flags problematic peptides by two criteria. For the first criterion, the algorithm focuses on the relative ratio of the peak areas for any pair of two transitions from the same precursor. Significant difference between the ratios for a pair of analytes (analyte1/analyte2) and the ratios for the SISs (SIS1/SIS2) are determined using the t-test. The second criterion is that, in each transition, the coefficient of variation is evaluated from the ratio of analyte and SIS peak areas peak area ratios (analyte1/SIS1 or analyte2/SIS2) across all replicates [86]. QuaSAR[87] (Quantitative Statistical Analysis of Reaction Monitoring Experiments) is a web-based module associated with Broad Institute GenePattern server which provides coefficient of variation (CV), regression slope and intercept (with confidence intervals), interference detection, and limits of detection (LOD) and quantification (LOQ) for MRM data analysis. The software does require a calibration curve and that both an analyte (light) and an internal standard (IS) (heavy) have been measured for each transition. MultiQuant software calculates a signal-to-noise ratio based on a Relative Noise (RN) value [88]. RN is a single number that expresses the ratio of the actual noise relative to that predicted from the noise model of an instrument. Once determined it can be used to calculate the noise expected at any data point, including under the peak itself, and can be used in a variety of ways, such as to assess whether a data point is real or likely due to noise, and to reproducibly and robustly define the noise in a particular system and, hence, the signal/noise ratio.
In efforts to improve the current peak quality data metrics Colangelo et. al. (unpublished) with AB Sciex research team to develop an additional metric which has been incorporated as a query in MultiQuant, termed Normalized Group Area ratio, which calculates a group area as shown in Figure 5. For each transition the query calculates the ratio of the area of a transition to the area of the first transition for the corresponding group. It then divides by the average of this ratio for all samples (for a given transition). The net result is that the reported value should be close to 1.0 if the ratio of a transition to the first is constant across the samples. If not, one (or both) of the peaks either didn’t integrate well or has an interference (or something odder).
Figure 5.
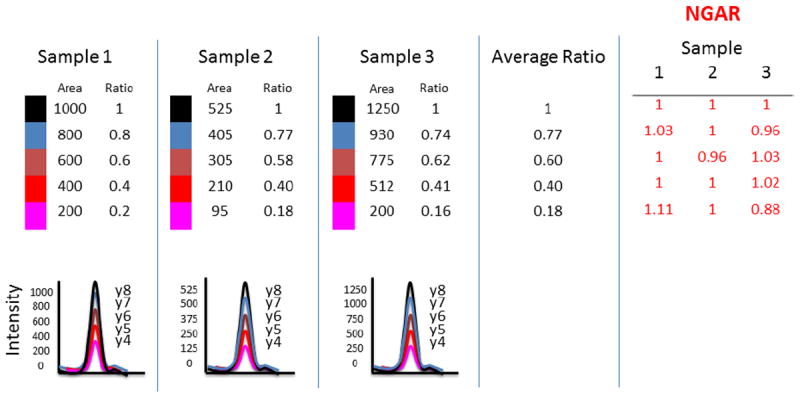
The diagram depicts how Normalized Group Area Ratio (NGAR) is calculated for a peptide with five MRM transitions over three samples three samples. The NGAR algorithm takes the peak areas for each transition within a run and calculates the ratio of the area of a transition to the area of the first transition for the corresponding group. It then divides by the average of this ratio for all samples (for a given transition). The net result is a NGAR that the reported value should be close to 1.0 if the ratio of a transition to the first is constant across the samples.
Most software programs provide a graphical interface for data visualization and manual peak adjustment. For example, Skyline, ATAQS, MultiQuant, PinPoint and TargetLynx all enable one to examine the peak intensities across transition for each given peptide. In addition, most provide an extracted ion chromatogram trace plot for transitions within a given peptide. Users can adjust parameters such as peak start- and end-points, and baseline, and the software will automatically reprocess the peak area and/or peak area ratios. Most software packages have filtering functions to remove poor quality data. Regarding the quality assessment, graphical summary report is available in most packages. For example in mProphet one can generate a histogram of mProphet scores for target and decoy transitions, the FDR and sensitivity curves across mProphet discriminant score cutoffs, and ROC curve for the data. SRMstats provides summary reports on missing data and graphics for the endogenous and reference peak intensities across all MS runs.
4.3 Downstream Statistical analysis
MRM peak integration results can be used for both differential expression analysis as well as estimating either relative or absolute abundance of each protein in any given sample. Differential expression analysis is the most common of these two steps but often proper data normalization is essential to remove technical artifacts before biological ones. The objective of data normalization is to remove intensity-dependent errors and make the distribution of intensities comparable across biological samples and technical runs. Many of the data normalization methods developed for microarray data analysis are applicable to MRM data. For example, median adjustment is utilized in SRMstats[89], this makes the median of log-scaled intensities for heavy isotope-labeled transitions identical. Quantile normalization can be an alternative option but can be very sensitive to strong intensity variables [90]. Another factor to consider before differential expression analysis is to examine any non-biological effect due to experimental setup. For example, sample preparation protocol or date may introduce unwanted variation. This can be particularly critical if the batch effects are correlated with the main effect of interest[91]. During the experiment, it is helpful to record information, such as laboratory conditions, technicians, or reagent lots. When a batch effect is present, it can be adjusted by various approaches including a linear model framework commonly used for microarrays[92].
The second part of downstream statistical analysis of MRM data is to quantitate the estimated amount of each protein in any given sample. SIS peptides have been utilized for absolute quantitation, but to synthesize, purify, and quantitate enough heavy standards for large scale MRM assays makes this approach not cost effective. Thus, in order to estimate the protein abundance without SIS peptides, researchers use the transition level data to infer both the peptide and protein expression abundance. Due to the MRM transitions being surrogates, it is possible to have potential for interference or background in each of these transitions. In order to minimize this effect a common approach is to use the top n most intense signals. The protein level intensities are calculated by summing or averaging the most intense signals. It is based on the empirical discovery that the MS signal for the three most intense tryptic peptides per mole of protein is approximately constant to the protein concentration [93]. Silva et. al. utilized this finding to estimate the absolute protein concentration which allows one to perform quantitative protein comparison within the same sample. The quantification requires a known quantity of intact protein to determine a universal signal response factor. They characterized the absolute quantity by spiking the internal standard proteins into the protein mixture of interest. Extending this model further Ludwig et al. [94] proposed the use of anchor point proteins whose concentrations are known for the MRM framework. This approach requires monitoring the MRM peak intensities of the anchor proteins along with the proteins of interest. They estimate the protein abundance by using the two most intense transitions of the three best flyer peptides for each protein. Absolute abundance is then predicted from the linear relationship between the MRM protein abundance of anchor proteins and their concentration.
For MRM data, two packages have been developed to provide statistical inference of differential expression. The first is an open-source R package, SRMstats [89], that provides normalization for stable isotope-labeled MRM experiments. It normalizes the data based on the median of reference transition intensities across all runs. After filtering out missing transitions, it performs a test for significant difference of a protein expression between multiple experimental conditions or time points. Intensities of endogenous and reference transitions are paired and tested for differential expression using linear mixed-effect model which accounts variations due to experimental conditions, samples, and peptide/transitions. One can specify each effect as fixed or random, based on the experimental design. As a follow-up, the software calculates the minimal number of biological replicates per experimental condition required to detect a desired fold-change and minimal number of peptides per protein and transitions per peptides for future experiments.
The second statistical workflow was outline by Bisson et. al. [95] using a Matlab application to determine confidence levels of differential expression at peptide and protein levels. Transition-level intensities are weighted according to their signal quality and averaged over technical runs. Fold-change between two conditions of a given peptide is calculated by a weighted average of transition-level fold-changes, where the weight is determined by the confidence score for transition-level differential expression. Protein-level fold-change is similarly calculated based on the peptide-level fold-changes. The final protein confidence score is evaluated by combining scores of over- and under-expressed peptides, separately.
5. Interoperability between software tools
Given the numerous software tools outlined above, choosing which programs best work together for the design, collection, and analysis of MRM experiments can seem daunting. There are two main approaches, first if to utilize vendor developed pipeline and second is to utilize open source packages and create a pipeline. For example, if you have a Agilent TripleQuadrupole, utilizing the MassHunter Workstation, which includes MassHunter Optimzer, Spectum Mill, and Mass Profiler Software packages will seamlessly enable you to develop, collect, and process your Agilent instrument data without having to edits method in excel prior to acquisition or convert files to open source format in order to peak integrate. If you have an AB Sciex Triple Quadrupole Mass Spectrometer you can have a similar experience by utilizing AB Sciex’s software suite which includes MRMPilot, Protein Pilot, Analyst, and Multiquant. The downside of these vendor software packages is that they are often limited to processing their own data formats. For example, Multiquant requires a *.wiff for input, thus limiting it to the analysis of AB Sciex mass spectrometer. Also, vendor specific softwares often provide a black box type of analysis in their subroutines and functions (e.g. peak integration). This can be limiting since many of the subroutines are not fully documented (e.g. outlining which algebraic expressions are used) or peer reviewed. The most complete open source package is Skyline, which includes modules for design, method export, peak integration and data quality assessment. Skyline is the only open source program which reads all native vendors file formats. Additionally SRMstats has support for Skyline output, which makes a combined Skyline/SRMstats workflow the most efficient set of tools currently available. Many of the other software programs such as MRMer and ATAQS require you to convert your file to an open source format such as TraML, mzXML or mzML. The downside of file conversion is that it adds an additional step to your analysis, requires you to make a second version of your data file, and often requires additional software such as ProteoWizard [96]. Additionally, vendors are constantly introducing new mass spectrometers, which may or may not be currently supported by existing file converters.
6. Future Development
The development of targeted proteomics assays and the publication of such experiments is growing at an exponential rate [12]. At the same time, the software developed and currently being utilized for MRM assay development and analysis has also greatly improved. The challenge, however, is how to standardize these platforms between laboratories. For instance, while targeted proteomics and discovery proteomics represent two distinct quantitative proteomics approaches, the peptide information produced from shotgun proteomics experiments can help identify representative peptides of the targeted proteins. The problem with using this information is that the software pipeline utilized for discovery phase protein identification often differs between laboratories. For example, one laboratory might utilize MASCOT [97], whereas another might utilize X!tandem [73] or OMSSA [98]. Also, the protein identification results from these data processing algorithms contain inaccuracies that can lead to both incorrect (false positives) and missed (false negatives) protein identifications. Without additional statistical analysis, downstream use of this data can suffer from missed opportunities for biological insight, the pollution of databases with increasing numbers of incorrect identifications, and time spent by biologists investigating false leads. Finally, there are few standardized approaches to assess the quality of peptide identifications. A few public repositories have tried to standardize the large amounts of MS data for different organisms, such as PeptideAtlas [61], GPMdb [64] and PRIDE[58], but often neglect to differentiate between various mass spectrometric fragmentation platforms, which are known to generate different product ions intensities. [99]
As an alternative to generating MRM assays via spectral libraries, recent work on theoretical CID peak heights and fragment pattern calculations for determining transition candidates has been available in software such as MRMPilot (from AB Sciex) and PepNovo+.[100] A more sophisticated spectra predictor has been developed for MALDI-TOF, named PIP[101] and is built on statistical machine learning methods that uses as its input physical attributes of peptides such as amino acid composition, peptide length, mass, and numbers and fractions of acidic, basic, polar, aliphatic and arginine residues. The PIP method shows a good correlation (r around 0.6) between predicted peptide peak heights and actual peptide quantities, but has yet to be applied to electrospray mass spectrometry. Another predictive method based on self organizing maps shows better performance [102]. Nevertheless, the limited accuracy of such predictive methods has been led researchers to prefer empirical data so far. This is still an area of active research and progress in this field should enable selection of peptides for which experimental data is not available.
Recent methods such SWATH-MS [103], Scheduled MRMHR [104], and parallel-reaction monitoring [105] have emerged which utilize high-resolution mass spectrometers to collect MRM-like data. These technologies have the potential to reduce assay development time and increase the number of protein targets. One major challenge going forward is how our existing software tools will support these technologies.
Acknowledgments
We would like to thank Jennifer Colangelo for comments and review of the manuscript. Funding was supported in part by NIH grants UL1 RR024139 (Yale Clinical and Translational Science Award) and P30 DA018343 (NIDA Proteomics Center at Yale University)
Abbreviations
- CV
Coefficient of variation
- ELISA
Enzyme-linked immunosorbent assay
- FDR
False Discovery Rate
- FWHM
Full width of peak at half max peak height
- iTRAQ
isobaric tag for relative and absolute quantitation
- LC-MS/MS
liquid chromatography MS/MS
- MRM
multiple reaction monitoring
- LC-MRM
liquid chromatography multiple reaction monitoring mass spectrometry
- MS/MS
tandem MS
- MudPIT
multidimensional protein identification technology
- NIST
National Institute of Standards and Technology
- PRIDE
proteomics identification database
- SID
stable isotope dilution
- SILAC
stable isotope labeling by amino acids in cell culture
- SIS
stable isotope–labeled internal standard
- SISCAPA
Stable Isotope Standards and Capture by Anti-Peptide Antibodies
- SRM
selective reaction monitoring
- Th
Thomson, unit of mass-to-charge ratio (Da/e)
- YPED
Yale protein expression database
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Greenbaum D, et al. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003;4(9):117. doi: 10.1186/gb-2003-4-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Washburn M, Wolters D, Yates J. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19(3):242–7. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 3.Unlü M, Morgan M, Minden J. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18(11):2071–7. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- 4.Gygi S, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17(10):994–9. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 5.Ross P, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3(12):1154–69. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Ong S, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–8. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 7.Asara J, et al. A label-free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. Proteomics. 2008;8(5):994–9. doi: 10.1002/pmic.200700426. [DOI] [PubMed] [Google Scholar]
- 8.Shifman MA, et al. YPED: a proteomics database for protein expression analysis. AMIA Annu Symp Proc. 2005:1111. [PMC free article] [PubMed] [Google Scholar]
- 9.Shifman M, et al. YPED: a web-accessible database system for protein expression analysis. J Proteome Res. 2007;6(10):4019–24. doi: 10.1021/pr070325f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan S, et al. Mass spectrometry based targeted protein quantification: methods and applications. J Proteome Res. 2009;8(2):787–97. doi: 10.1021/pr800538n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312(5771):212–7. doi: 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- 12.Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods. 2012;9(6):555–66. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- 13.Cooks R. Multiple Reaction Monitoring in Mass Spectrometry/Mass Spectrometry for Direct Analysis of Complex Mixtures. Anal Chem. 1978;50(14):2017–2021. [Google Scholar]
- 14.Shushan B. A review of clinical diagnostic applications of liquid chromatography-tandem mass spectrometry. Mass Spectrom Rev. 2010;29(6):930–44. doi: 10.1002/mas.20295. [DOI] [PubMed] [Google Scholar]
- 15.Gillette MA, Carr SA. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat Methods. 2013;10(1):28–34. doi: 10.1038/nmeth.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keshishian H, et al. Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics. 2007;6(12):2212–29. doi: 10.1074/mcp.M700354-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Onisko B, et al. Mass spectrometric detection of attomole amounts of the prion protein by nanoLC/MS/MS. J Am Soc Mass Spectrom. 2007;18(6):1070–9. doi: 10.1016/j.jasms.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 18.Mallick P, et al. Computational prediction of proteotypic peptides for quantitative proteomics. Nat Biotechnol. 2007;25(1):125–31. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
- 19.Wolf-Yadlin A, et al. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc Natl Acad Sci U S A. 2007;104(14):5860–5. doi: 10.1073/pnas.0608638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5(4):573–88. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Xiao Y, et al. Evaluation of plasma lysophospholipids for diagnostic significance using electrospray ionization mass spectrometry (ESI-MS) analyses. Ann N Y Acad Sci. 2000;905:242–59. doi: 10.1111/j.1749-6632.2000.tb06554.x. [DOI] [PubMed] [Google Scholar]
- 22.Addona TA, et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol. 2009;27(7):633–41. doi: 10.1038/nbt.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Picotti P, et al. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell. 2009;138(4):795–806. doi: 10.1016/j.cell.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, et al. Quantification of beta-catenin signaling components in colon cancer cell lines, tissue sections, and microdissected tumor cells using reaction monitoring mass spectrometry. J Proteome Res. 2010;9(8):4215–27. doi: 10.1021/pr1005197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keshishian H, et al. Quantification of cardiovascular biomarkers in patient plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics. 2009;8(10):2339–49. doi: 10.1074/mcp.M900140-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whiteaker JR, et al. A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat Biotechnol. 2011;29(7):625–34. doi: 10.1038/nbt.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zulak KG, et al. Targeted proteomics using selected reaction monitoring reveals the induction of specific terpene synthases in a multi-level study of methyl jasmonate-treated Norway spruce (Picea abies) Plant J. 2009;60(6):1015–30. doi: 10.1111/j.1365-313X.2009.04020.x. [DOI] [PubMed] [Google Scholar]
- 28.Kirkpatrick DS, Gerber SA, Gygi SP. The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods. 2005;35(3):265–73. doi: 10.1016/j.ymeth.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 29.Gerber SA, et al. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci U S A. 2003;100(12):6940–5. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beynon RJ, et al. Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat Methods. 2005;2(8):587–9. doi: 10.1038/nmeth774. [DOI] [PubMed] [Google Scholar]
- 31.Dupuis A, et al. Protein Standard Absolute Quantification (PSAQ) for improved investigation of staphylococcal food poisoning outbreaks. Proteomics. 2008;8(22):4633–6. doi: 10.1002/pmic.200800326. [DOI] [PubMed] [Google Scholar]
- 32.Anderson NL, et al. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) J Proteome Res. 2004;3(2):235–44. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 33.Cham Mead JA, Bianco L, Bessant C. Free computational resources for designing selected reaction monitoring transitions. Proteomics. 2010;10(6):1106–26. doi: 10.1002/pmic.200900396. [DOI] [PubMed] [Google Scholar]
- 34.Brusniak MY, et al. An assessment of current bioinformatic solutions for analyzing LC-MS data acquired by selected reaction monitoring technology. Proteomics. 2012;12(8):1176–84. doi: 10.1002/pmic.201100571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prakash A, et al. Expediting the development of targeted SRM assays: using data from shotgun proteomics to automate method development. J Proteome Res. 2009;8(6):2733–9. doi: 10.1021/pr801028b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stein SE, Scott DR. Optimization and Testing of Mass-Spectral Library Search Algorithms for Compound Identification. Journal of the American Society for Mass Spectrometry. 1994;5(9):859–866. doi: 10.1016/1044-0305(94)87009-8. [DOI] [PubMed] [Google Scholar]
- 37.Picotti P, Aebersold R, Domon B. The implications of proteolytic background for shotgun proteomics. Mol Cell Proteomics. 2007;6(9):1589–98. doi: 10.1074/mcp.M700029-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Guo DC, et al. Prediction of Peptide Retention Times in Reversed-Phase High-Performance Liquid-Chromatography .1. Determination of Retention Coefficients of Amino-Acid-Residues of Model Synthetic Peptides. Journal of Chromatography. 1986;359:499–517. [Google Scholar]
- 39.Guo DC, et al. Prediction of Peptide Retention Times in Reversed-Phase High-Performance Liquid-Chromatography .2. Correlation of Observed and Predicted Peptide Retention Times and Factors Influencing the Retention Times of Peptides. Journal of Chromatography. 1986;359:519–532. [Google Scholar]
- 40.Parker JM, Guo D, Hodges RS. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry. 1986;25(19):5425–32. doi: 10.1021/bi00367a013. [DOI] [PubMed] [Google Scholar]
- 41.Escher C, et al. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics. 2012;12(8):1111–21. doi: 10.1002/pmic.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cox DM, Tate S, Duchoslav E. Automated Ion Selection and Method Building For MRM Based Protein Validation And Quantification. ASMS; 2007. [Google Scholar]
- 43.Unwin RD, Griffiths JR, Whetton AD. A sensitive mass spectrometric method for hypothesis-driven detection of peptide post-translational modifications: multiple reaction monitoring-initiated detection and sequencing (MIDAS) Nat Protoc. 2009;4(6):870–7. doi: 10.1038/nprot.2009.57. [DOI] [PubMed] [Google Scholar]
- 44.Fan J, et al. MRMaid 2.0: mining PRIDE for evidence-based SRM transitions. OMICS. 2012;16(9):483–8. doi: 10.1089/omi.2011.0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mead JA, et al. MRMaid, the web-based tool for designing multiple reaction monitoring (MRM) transitions. Mol Cell Proteomics. 2009;8(4):696–705. doi: 10.1074/mcp.M800192-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacLean B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26(7):966–8. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brusniak MY, et al. ATAQS: A computational software tool for high throughput transition optimization and validation for selected reaction monitoring mass spectrometry. BMC Bioinformatics. 2011;12:78. doi: 10.1186/1471-2105-12-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shannon PT, et al. The Gaggle: an open-source software system for integrating bioinformatics software and data sources. BMC Bioinformatics. 2006;7:176. doi: 10.1186/1471-2105-7-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deutsch EW, Lam H, Aebersold R. PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 2008;9(5):429–34. doi: 10.1038/embor.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lange V, et al. Targeted quantitative analysis of Streptococcus pyogenes virulence factors by multiple reaction monitoring. Mol Cell Proteomics. 2008;7(8):1489–500. doi: 10.1074/mcp.M800032-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reiter L, et al. mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat Methods. 2011;8(5):430–5. doi: 10.1038/nmeth.1584. [DOI] [PubMed] [Google Scholar]
- 52.Deutsch EW, et al. TraML--a standard format for exchange of selected reaction monitoring transition lists. Mol Cell Proteomics. 2012;11(4) doi: 10.1074/mcp.R111.015040. R111 015040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sherwood CA, et al. MaRiMba: a software application for spectral library-based MRM transition list assembly. J Proteome Res. 2009;8(10):4396–405. doi: 10.1021/pr900010h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin DB, et al. MRMer, an interactive open source and cross-platform system for data extraction and visualization of multiple reaction monitoring experiments. Mol Cell Proteomics. 2008;7(11):2270–8. doi: 10.1074/mcp.M700504-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rost H, Malmstrom L, Aebersold R. A Computational Tool to Detect and Avoid Redundancy in Selected Reaction Monitoring. Molecular & Cellular Proteomics. 2012;11(8):540–549. doi: 10.1074/mcp.M111.013045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fusaro VA, et al. Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat Biotechnol. 2009;27(2):190–8. doi: 10.1038/nbt.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Webb-Robertson BJ, et al. A support vector machine model for the prediction of proteotypic peptides for accurate mass and time proteomics. Bioinformatics. 2010;26(13):1677–83. doi: 10.1093/bioinformatics/btq251. [DOI] [PubMed] [Google Scholar]
- 58.Martens L, et al. PRIDE: the proteomics identifications database. Proteomics. 2005;5(13):3537–45. doi: 10.1002/pmic.200401303. [DOI] [PubMed] [Google Scholar]
- 59.Vizcaino JA, et al. The Proteomics Identifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Craig R, Cortens J, Beavis R. Open source system for analyzing, validating, and storing protein identification data. J Proteome Res. 2004;3:1234–1242. doi: 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- 61.Deutsch EW, et al. Human Plasma PeptideAtlas. Proteomics. 2005;5(13):3497–500. doi: 10.1002/pmic.200500160. [DOI] [PubMed] [Google Scholar]
- 62.Desiere F, et al. The PeptideAtlas project. Nucleic Acids Res. 2006;34(Database issue):D655–8. doi: 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deutsch EW. The PeptideAtlas Project. Methods Mol Biol. 2010;604:285–96. doi: 10.1007/978-1-60761-444-9_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walsh GM, et al. Implementation of a data repository-driven approach for targeted proteomics experiments by multiple reaction monitoring. J Proteomics. 2009;72(5):838–52. doi: 10.1016/j.jprot.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Picotti P, et al. A database of mass spectrometric assays for the yeast proteome. Nat Methods. 2008;5(11):913–4. doi: 10.1038/nmeth1108-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vizcaino JA, et al. PRIDE: Data submission and analysis. Curr Protoc Protein Sci. 2010;Chapter 25(Unit 25):4. doi: 10.1002/0471140864.ps2504s60. [DOI] [PubMed] [Google Scholar]
- 67.Turewicz M, Deutsch EW. Spectra, chromatograms, Metadata: mzML-the standard data format for mass spectrometer output. Methods Mol Biol. 2011;696:179–203. doi: 10.1007/978-1-60761-987-1_11. [DOI] [PubMed] [Google Scholar]
- 68.Sharma V, et al. Panorama: A Private Repository of Targeted Proteomics Assays for Skyline. ASMS; 2012; Vancouver, BC. WP 407. [Google Scholar]
- 69.Stergachis AB, et al. Rapid empirical discovery of optimal peptides for targeted proteomics. Nat Methods. 2011;8(12):1041–3. doi: 10.1038/nmeth.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pedrioli PG, et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol. 2004;22(11):1459–66. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 71.Keller A, et al. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol. 2005;1 doi: 10.1038/msb4100024. 2005 0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eisenacher M. mzIdentML: an open community-built standard format for the results of proteomics spectrum identification algorithms. Methods Mol Biol. 2011;696:161–77. doi: 10.1007/978-1-60761-987-1_10. [DOI] [PubMed] [Google Scholar]
- 73.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20(9):1466–7. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 74.Cote RG, et al. The PRoteomics IDEntification (PRIDE) Converter 2 framework: an improved suite of tools to facilitate data submission to the PRIDE database and the ProteomeXchange consortium. Mol Cell Proteomics. 2012;11(12):1682–9. doi: 10.1074/mcp.O112.021543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.O’Haver T. A Pragmatic* Introduction to Signal Processing. 2013 http://terpconnect.umd.edu/~toh/spectrum/PeakFindingandMeasurement.htm.
- 76.Stahl-Zeng J, et al. High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol Cell Proteomics. 2007;6(10):1809–17. doi: 10.1074/mcp.M700132-MCP200. [DOI] [PubMed] [Google Scholar]
- 77.Cox DM, et al. Improved retention time robustness, dwell time and cycle time in MRM assays. ASMS; 2012; Vancouver, CA. MP26 Poster 625. [Google Scholar]
- 78.Kiyonami R, et al. Increased selectivity, analytical precision, and throughput in targeted proteomics. Mol Cell Proteomics. 2011;10(2) doi: 10.1074/mcp.M110.002931. M110 002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Finney GL, et al. Label-free comparative analysis of proteomics mixtures using chromatographic alignment of high-resolution mu LC-MS data. Analytical Chemistry. 2008;80(4):961–971. doi: 10.1021/ac701649e. [DOI] [PubMed] [Google Scholar]
- 80.Savitzky A, Golay MJE. Smoothing + Differentiation of Data by Simplified Least Squares Procedures. Analytical Chemistry. 1964;36(8):1627. [Google Scholar]
- 81.Cox DM, et al. Multiple reaction monitoring as a method for identifying protein posttranslational modifications. J Biomol Tech. 2005;16(2):83–90. [PMC free article] [PubMed] [Google Scholar]
- 82.Burton L, et al. A novel algorithm for quantitative LC peak integration. ASMS; 2009; Poster I-224. [Google Scholar]
- 83.Peterman S, et al. Streamlining the Process of Biomarker Verification using Pinpoint Software. 2009 Available from: http://www.thermoscientific.com/ecomm/servlet/techresource?storeId=11152&langId=-1&taxonomy=4&resourceId=89932#.
- 84.Rezai T, et al. An Evaluation of Three Peptide Quantitation Strategies for Serum Samples Using Liquid Chromatography-Selected Reaction Monitoring Selected Reaction Monitoring. Available from: https://static.thermoscientific.com/images/D00335~.pdf.
- 85.Waters. TargetLynx Application Manager. 2006 Available from: http://www.waters.com/webassets/cms/library/docs/720001733en.pdf.
- 86.Abbatiello SE, et al. Automated detection of inaccurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin Chem. 2010;56(2):291–305. doi: 10.1373/clinchem.2009.138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mani DR, et al. QuaSAR: A Comprehensive Pipeline for Quantitative and Statistical Analysis of Reaction Monitoring Results. ASMS; 2012; Vancouver, BC. ThP12 Poster 284. [Google Scholar]
- 88.Ivosev G, et al. Relative Noise: A novel method for determining signal to noise ratios for peaks in quantitative LC-MS applications. ASMS; 2010; Salt Lake City, UT. Poster III 374. [Google Scholar]
- 89.Chang CY, et al. Protein significance analysis in selected reaction monitoring (SRM) measurements. Mol Cell Proteomics. 2012;11(4) doi: 10.1074/mcp.M111.014662. M111 014662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bolstad BM, et al. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–93. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 91.Leek JT, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11(10):733–9. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–27. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 93.Silva JC, et al. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol Cell Proteomics. 2006;5(1):144–56. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 94.Ludwig C, et al. Estimation of absolute protein quantities of unlabeled samples by selected reaction monitoring mass spectrometry. Mol Cell Proteomics. 2012;11(3) doi: 10.1074/mcp.M111.013987. M111 013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bisson N, et al. Selected reaction monitoring mass spectrometry reveals the dynamics of signaling through the GRB2 adaptor. Nat Biotechnol. 2011;29(7):653–8. doi: 10.1038/nbt.1905. [DOI] [PubMed] [Google Scholar]
- 96.Chambers MC, et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol. 2012;30(10):918–20. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Perkins DN, et al. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–67. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 98.Geer LY, et al. Open mass spectrometry search algorithm. J Proteome Res. 2004;3(5):958–64. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 99.de Graaf EL, et al. Improving SRM assay development: a global comparison between triple quadrupole, ion trap, and higher energy CID peptide fragmentation spectra. J Proteome Res. 2011;10(9):4334–41. doi: 10.1021/pr200156b. [DOI] [PubMed] [Google Scholar]
- 100.Frank AM. Predicting intensity ranks of peptide fragment ions. J Proteome Res. 2009;8(5):2226–40. doi: 10.1021/pr800677f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Timm W, et al. Peak intensity prediction in MALDI-TOF mass spectrometry: a machine learning study to support quantitative proteomics. BMC Bioinformatics. 2008;9:443. doi: 10.1186/1471-2105-9-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Scherbart A, Nattkemper TW. Looking inside self-organizing map ensembles with resampling and negative correlation learning. Neural Netw. 2011;24(1):130–41. doi: 10.1016/j.neunet.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 103.Gillet LC, et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11(6) doi: 10.1074/mcp.O111.016717. O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Albanese J, Hunter C. Increasing the Multiplexing of High Resolution Targeted Peptide Quantification Assays: Scheduled MRMHR Workflow on the TripleTOF® Systems. 2012 Available from: http://www.absciex.com/Documents/Downloads/Literature/Scheduled%20MRMHR%20Workflow%20TripleTOF%206260212-01.pdf.
- 105.Peterson AC, et al. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics. 2012;11(11):1475–88. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]