

Abstract
The CD28 costimulatory receptor is a critical regulator of T cell function making it an attractive therapeutic target for the treatment of immune mediated diseases. CTLA4Ig, now approved for use in humans, prevents naive T cell activation by binding to B7-proteins and blocking engagement of CD28. However, CTLA4Ig suppresses inflammation even if administered when disease is established, suggesting alternative mechanisms. We identified a novel, CD28-independent mechanism by which CTLA4Ig inhibits activated T cells. We show that in vitro, CTLA4Ig synergizes with nitric oxide from bone marrow derived macrophages to inhibit T cell proliferation. Depletion of Tregs or interference with TGFβ signaling abrogated the inhibitory effect of CTLA4Ig. Parallel in vivo experiments using an allergic airway inflammation model demonstrated that this novel mechanism required both macrophages andTregs. Furthermore, CTLA4Ig was ineffective in SMAD3-deficient mice, supporting a requirement for TGFβ signaling. Thus, in addition to preventing naïve T cells from being fully activated, CTLA4Ig can turn off already activated effector T cells by an NO/Treg/TGFβ-dependent pathway. This mechanism is similar to cell extrinsic effects of endogenous CTLA-4 and may be particularly important in the ability of CTLA4Ig to treat chronic inflammatory disease.
Introduction
Approaches to augment or interfere with immune cell function may be of benefit in many diseases. Members of the CD28 receptor family both activate and inhibit T cell responses, making them attractive therapeutic targets. CD28 is one of the best studied and was the first to be targeted with the development of CTLA4Ig. CTLA4Ig has been shown to be effective both in vitro as well as in numerous animal models of disease (reviewed in (1)). These studies led to the development of the humanized version, abatacept, and the related protein, belatacept, which are approved for use in humans to treat rheumatoid arthritis and prevent renal transplant rejection, respectively (2, 3). Biologics directed against additional members of the CD28 family have also been developed including anti-CTLA-4 antibodies (ipilimumab) to treat malignant melanoma, and promising results have been reported with anti-PD-1 therapy in early cancer trials (4–6).
CTLA4Ig is a fusion protein of the extracellular domain of CTLA-4 and IgG1 that binds to both CD80 and CD86 (also referred to as B7-1 and B7-2, or collectively as B7-proteins) and prevents interaction of B7-proteins with their counter-receptors CD28 and CTLA-4 expressed on T cells (7). In addition, CD80 has been shown to bind PD-L1 and inhibit T cell activation and proliferation through this interaction (8). The primary mechanism of action for CTLA4Ig has been thought to be blockade of CD28 and therefore prevention of initial T cell activation. However, we previously demonstrated that CTLA4Ig was effective if administered after initial antigen activation of T cells and that this was independent of CD28 (9). In this current study, we report the mechanism for this novel mode of action for CTLA4Ig. We demonstrate that the effects of CTLA4Ig are mediated by regulatory T cells (Tregs) and TGFβ and require macrophage derived nitric oxide (NO). These data provide an entirely new insight into how treatment with CTLAA4Ig suppresses inflammation, and may provide information relevant to how endogenous CTLA-4:B7 interactions inhibit T cell responses.
Materials and Methods
Mice
C57Bl/6J and NOS2-deficient mice were purchased from The Jackson Laboratory (Bar Harbor, ME). STAT1-deficient mice were gifts of Dr M. Holtzman and Dr H. Virgin (Washington University School of Medicine, St Louis, MO). CD80/86-deficient mice and FoxP3-DTR mice were provided by Alexander Rudensky (Memorial Sloan Kettering Cancer Center, NY, NY). SMAD-3 deficient mice were provided by Dr. David Beebe (Washington University School of Medicine, St Louis MO). IDO-deficient mice were provided by Dr. Matthew Ciorba (Washington University School of Medicine, St Louis MO). FoxP3-IRES-GFP (B6. Cg-FoxP3tm2Tch/J) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and crossed to OT-II OVA transgenic mice on a RAG 1-deficient background to generate OT-II/FoxP3-GFP/Rag1KO mice. All mice were bred and housed in specific pathogen-free facilities at Washington University School of Medicine. All animal studies have been approved by the Washington University Animal Studies Committee.
Antibodies
α-IFNγ (clone H22, provided by R. Schreiber, Washington University, St Louis, MO) and α-CD4 were purchased from Biolegend (San Diego, CA). α-TGFβ (clone1D11) was purchased from R&D Systems (Minneapolis, MN). Murine CTLA4Ig was provided by Bristol-Myers Squibb (Princeton, NJ.).
Experimental allergic airway inflammation
Mice were immunized and challenged with OVA (Sigma, St Louis, MO) as previously described (10). When indicated, clodronate liposomes were prepared as described (11) and administered (100 μl i.p. and 50 μl i.n.) 1 day prior to inhaled challenge. In some experiments, as indicated, groups of mice were given 100 μg of CTLA4Ig ip on the day of challenge. Neutralizing antibody against IFNγ (250 μg/mouse) was administered 24 hours prior to inhaled challenge. For depletion of Tregs, FoxP3-DTR mice were administered 1 μg diphtheria toxin i.p. (DT, Sigma Chemical Corporation, St Louis, MO) 1 day prior to and again on the day of challenge, and an additional 0.2 μg 2 days after challenge. For bone marrow chimeras, recipient mice were lethally-irradiated with 1000 rad and the following day injected i.v. with bone marrow harvested from femurs of donor mice. The mice were allowed to reconstitute for 8 weeks, then used in the allergic airway inflammation model as described above. Unless otherwise indicated, all in vivo experiments used 4–5 mice per experimental group and have been repeated 3 times, with one representative experiment presented. Cell counts and differentials are presented as the mean ± std. dev.
Adoptive transfer studies
Splenocytes were isolated from OT-II/FoxP3-GFP/Rag1KO mice (which are CD45.2 positive) and 3 × 106 cells injected i.v. into CD45.1 positive C57Bl/6 recipients. The mice were rested for one day and then primed and challenged with OVA/Alum as described for the allergic airway inflammation studies. Spleen and lung draining lymph nodes were isolated 48 hours after inhaled challenged and isolated cells stained for CD4 and CD45.2. The samples were then analyzed by flow cytometry and the percentage of total CD45.2 cells that are positive for CD4 and FoxP3-GFP expression presented. Each group contains 5 mice and the mean +/− s.e.m presented.
In vitro co-cultures
Bone marrow was isolated from mice of various genotypes as indicated and cultured in non-tissue culture treated Petri dishes in DMEM supplemented with 10% FBS, 15% L-cell supernatant and 5% horse serum (12). Macrophages were harvested 6 days later using cold PBS and 5 × 103 macrophages co-cultured in 96-well plates for 5 days with 1×105 splenocytes labeled with CFSE (Molecular Probes, Invitrogen Corp, Grand Island, NY) and activated with PMA (5ng/ml, Sigma Chemical Co, St Louis, MO) and Ionomycin (0.4 μg/ml, Sigma Chemical Co, St Louis, MO). When indicated, the following reagents were included: CTLA4Ig (10 μg/ml), N6-methyl-l-arginine acetate (L-NMMA, 100 μM. Sigma Chemical Corporation, St Louis, MO), αIFNγ (10 μg/ml), α-TGFβ antibody, or SB431542 (Calbiochem, Billerica, MA). Non adherent cells were harvested, stained with α-CD4-APC and subsequently analyzed on a FacsCalibur flow cytometer (BD Bioscience, San Jose, CA) to measure cell proliferation. Flow cytometry data was analyzed using Winlist 6.0 (Verity Software, Topsham, ME). Cell death was determined by staining with Annexin V and 7-AAD (BD Bioscience, San Jose, CA) and analyzed by flow cytometry. Cytokine concentrations were measured on culture supernatants using the Th1/Th2/Th17 Cytokine Bead Array (BD Biosciences, San Jose, CA). Pooled results from multiple independent experiments are shown. The number of repeats included is indicated in each figure legend. Unless otherwise indicated, all experiments have been repeated at least 3 times and data from one representative experiment presented.
Statistical analysis
For the in vitro coculture experiments, the results from independent experiments were pooled and statistical comparisons performed using a paired T-test using Microsoft Excel (Microsoft Corporation, Redmond, WA). For the BAL cell counts and differentials obtained in the allergic airway inflammation model, p values were obtained using the Kruskall-Wallis one way analysis of variance and Dunns post test comparison or Mann-Whitney U-Test as indicated in the figure legends (GraphPad Prism, GraphPad Corporation, San Diego, CA).
Results
Inhibition of CD28-independent induction of T cell proliferation in vitro requires macrophage derived NO and expression of B7-proteins
Administration of CTLA4Ig at the time of allergen challenge can suppress airway inflammation in wild type mice, but is ineffective in mice lacking NOS2 (9). One of the major sources of NO is the macrophage, and previous work had demonstrated that macrophage derived NO can suppress T cell proliferation (13). Therefore, we tested whether CTLA4Ig synergized with macrophage derived NO in an in vitro co-culture system. Splenocytes were labeled with CFSE, activated with the costimulation independent agonists PMA and ionomycin and cocultured with bone marrow derived macrophages (BMDM). Cell division was measured by flow cytometry. Macrophages partially suppressed T cell proliferation, an effect that was enhanced by addition of CTLA4Ig, while CTLA4Ig alone had no effect on proliferation. Representative CFSE histograms are shown in figure 1A. This was independent of CD28, as splenocytes isolated from CD28-deficient mice were suppressed to the same extent as those from wild type (Figure 1B). Proliferation was restored by the addition of the NOS2 inhibitor N6-methyl-l-arginine acetate (L-NMMA), demonstrating that CTLA4Ig can inhibit T cell proliferation in a CD28-independent, NO-dependent manner.
Figure 1. CD28-independent inhibition of T cell proliferation by CTLA4Ig in vitro.

CFSE labeled splenocytes were co-cultured with bone marrow derived macrophages and activated with PMA and ionomycin. Cells were harvested and stained with α-CD4 and proliferation determined by flow cytometry after 96 hours of culture. Where indicated, CTLA4Ig (10 μg/ml) and/or L-NMMA (100 μM) were included at the start of the culture. A) Representative CFSE dye dilution histograms of wild type splenocytes stimulated with PMA and ionomycin alone or in combination with BMDM and/or CTLA4Ig as indicated. The percentage of cells that have undergone at least one cell division is indicated. B) Splenocytes from wild type or CD28-deficient mice were co-cultured with wild type BMDM. Presented are the combined results from 6 independent experiments C) Splenocytes from wild type or NOS2 deficient mice were co-cultured with BMDM from wild type or NOS2 deficient mice. Presented are the combined results from 3 independent experiments. D) Splenocytes from wild type or CD80/CD86-double deficient mice were co-cultured with BMDM from wild type or CD80/CD86-double deficient mice. Presented are the combined results from 7 independent experiments. * = p<0.05, ** = p<0.01 by 2-tailed paired T-test
To determine the cellular source of NO, we isolated splenocytes and macrophages isolated from wild type and NOS2-deficient mice and tested for T cell suppression with and without CTLA4Ig (Figure 1C). While splenocytes from either genotype were suppressed by wild type macrophages and further suppressed by CTLA4Ig, macrophages derived from NOS2 deficient mice were unable to suppress T cell proliferation either alone or in combination with CTLA4Ig, demonstrating that NO is derived from the macrophage.
We next tested whether expression of CD80 or CD86 was required on the splenocyte or the BMDM (Figure 1D). Macrophages from either wild type or CD80/CD86-double deficient mice were capable of suppressing T cell proliferation and this was further augmented by CTLA4Ig. However, CTLA4Ig was ineffective in co-cultures containing splenocytes from CD80/CD86-deficient mice. Thus, in contrast to the requirement for NOS2, CD80/CD86 expression was dispensable on the macrophage, but required on the splenocyte. These data demonstrate that B7-proteins and NOS2 must be expressed in different cell subsets for CTLA4Ig to be effective. Thus the mechanism cannot be a direct modulation of NOS2 activity by CTLA4Ig binding to B7 on the macrophage.
Inhibition of allergic airway inflammation by CTLA4Ig requires macrophages, NOS2 and B7-expression
To establish that the observations made in vitro were relevant to an in vivo response, we tested whether the requirements for CTLA4Ig to modulate allergic airway inflammation paralleled the results obtained with the co-culture system. Mice were primed and challenged with OVA as described (10). CTLA4Ig was injected i.p. at the time of inhaled challenge. In all cases, priming was performed in the absence of administration of CTLA4Ig or any additional drugs, reagents or experimental manipulations. We first tested if macrophages were necessary by depleting them via systemic administration of clodronate liposomes (11). Flow cytometry of whole blood obtained at the time of harvest confirmed an absence of circulating macrophages (data not shown), although there were a few macrophages detected in the BAL fluid (Figure 2B). Mice treated with clodronate or control liposomes developed an eosinophilic inflammatory infiltrate in the lung, as determined by BAL cell counts, differentials and histology (Figure 2A–C). However, CTLA4Ig failed to abrogate the inflammatory response in mice that had been treated with clodronate liposomes, whereas it was effective in control mice, demonstrating a requirement for macrophages and consistent with the in vitro data.
Figure 2. Inhibition of allergic airway inflammation by CTLA4Ig in vivo requires macrophages, and expression of CD80/CD86 and NOS2.

A–C) C57Bl/6J mice were sensitized and challenged with OVA as described. Some mice received nothing, control liposomes or clodronate liposomes to deplete macrophages as indicated. Where indicated, CTLA4Ig was administered at the time of inhaled challenge only. D–F) Bone marrow chimeras established from C57Bl/6J or NOS2 deficient mice reconstituted with either C57Bl/6J or NOS2 deficient marrow mice. The mice were sensitized and challenged with OVA and treated with CTLA4Ig at the time of inhaled challenge. G–I) Bone marrow chimeras established with C57Bl/6J or CD80/CD86-double deficient mice reconstituted with bone marrow cells from C57Bl/6J mice were sensitized and challenged with OVA and treated with CTLA4Ig at the time of inhaled challenge. For all experiments, tissue was collected for analysis 72 hours after inhaled challenge. Each experimental group consisted of 5 mice and each experiment has been repeated 3 times. Shown is data from one representative experiment. * = p< 0.005 as compared to condition without CTLA4Ig as determined by Kruskall-Wallis with Dunns post test comparisons.
We had previously shown that administration of CTLA4Ig did not suppress airway inflammation when administered to NOS2 deficient mice at the time of inhaled challenge (9). However, many cell types express NOS2, including both hematopoietic and non-hematopoietic cells. To determine in which compartment NOS2 expression is required, we established mixed bone marrow chimeras with wild type and NOS2-deficient mice. CTLA4Ig was effective only when NOS2 was expressed in the hematopoietic cell compartment, consistent with the requirement for macrophages as a source of NO (Figure 2D–F).
CD80 and CD86 are expressed primarily on professional APC, however, they are inducible in other cell types (14–16). To test which cell compartment expression was required in we reconstituted wild type mice with bone marrow from CD80/CD86-double deficient mice and vice versa. We found that CD80/CD86 expression in the bone marrow derived compartment is sufficient for CTLA4Ig to suppress airway inflammation, as shown by the results presented in figures 2G–I. In the complementary chimera, in which CD80/CD86-deficient marrow was used to reconstitute wild type or CD80/CD86-deficient mice, we were unable to induce airway inflammation due to the necessity of B7:CD28 interactions for T cell priming (data not shown). Taken together, these data are highly consistent with the results obtained from the in vitro co-culture system.
Regulatory T cells are required for the suppressive effects of CTLA4Ig
The data thus far suggests that a complex interplay of multiple cell types is necessary for the inhibitory effects of CTLA4Ig. Both CD28 and CTLA-4 are expressed on Tregs and have important roles in their development and function leading us to hypothesize that they might be involved in modulation of inflammation by CTLA4Ig (17–20). Using mice in which the diphtheria toxin receptor (DTR) had been knocked into the FoxP3 locus we were able to specifically deplete Tregs by administration of diphtheria toxin (DT) (21). Successful depletion was confirmed in all experiments by flow cytometry (data not shown). As shown in Figure 3D, Treg-depleted splenocytes were not suppressed by CTLA4Ig although proliferation was still inhibited by co-culture with BMDMs in an NO-dependent manner. Thus, while Tregs are required for inhibition by CTLA4Ig, NO can suppress independent of Tregs,
Figure 3. FoxP3+ cells are required for CTLA4Ig mediated inhibition of T cell proliferation and allergic airway inflammation.

A–C) FoxP3-DTR mice were sensitized with OVA and challenged with either OVA or HEL. Mice were either untreated or administered DT to deplete FoxP3+ cells at the time of challenge. CTLA4Ig was administered to the indicated groups at the time of inhaled challenge. Specimens were collected 72 hours after inhaled challenge. Each experimental group consisted of 5 mice and has been repeated 3 times. Shown is data from one representative experiment. * = p< 0.05 as compared to condition without CTLA4Ig D) FoxP3-DTR mice were untreated or administered DT and splenocytes harvested 24 hours later, labeled with CFSE and co-cultured with BMDM alone, with CTLA4Ig or with L-NMMA. Proliferation was determined by flow cytometry after 96 hours. Presented are the combined results from 4 independent experiments. * = p<0.05 by 2-tailed paired T-test. E) splenocytes from OT-II/FoxP3-GFP/Rag1 KO mice (CD45.2+) were adoptively transferred into CD45.1+ C57Bl/6 recipients. The mice were then primed and challenged with OVA. One group received CTLA4Ig at the time of challenge. Spleen and lung draining lymph node harvested 48 hours later. Presented is the percentage of CD45.2+ cells that are FoxP3-GFP positive. Each experimental group consisted of 5 mice and has been performed 2 times. Shown is representative data from one independent experiment. *=p<.05, **= p< 0.01 as determined by Mann-Whitney U-test.
To test if Tregs were required for CTLA4Ig to suppress inflammation in vivo, we depleted mice by injecting them with DT only in the peri-challenge period, assuring that priming occurred in the presence of Tregs. As shown in Figure 3A–C, mice depleted of FoxP3-positive cells mounted an inflammatory response that was similar in quality and intensity to mice that did not receive DT. However, depletion of Tregs prevented CTLA4Ig from suppressing inflammation in the OVA challenged mice. Challenge with a different protein antigen, hen egg lysozyme (HEL), did not induce inflammation, confirming that depletion of Tregs did not result in non-specific inflammation. When we analyzed the percentage and number of FoxP3+ cells recovered from the spleen, lung or lymph nodes of mice treated with CTLA4Ig in comparison to controls, we were unable to detect an increase in Treg number (data not shown).
The number of antigen specific Tregs might be too small to detect a change using the above model system. To further examine this, we adoptively transferred cells isolated from OT-II/FoxP3-GFP/Rag1 KO mice into wild type C57Bl/6 mice. The donor cells were CD45.2 positive and the recipient mice expressed CD45.1. In addition, the donor Tregs expressed GFP, allowing for detection and quantitation of the OVA specific Tregs. Following transfer, the mice were primed and challenged and either left untreated or treated with CTLA4Ig. Tissue was harvested 48 hours after challenge and the percentage of transferred cells that express FoxP3 determined by flow cytometry. As shown in figure 3E, we detected a significant increase in FoxP3 positive cells in the spleen and lung draining lymph nodes of mice that were treated with CTLA4Ig. Together, these data establish that FoxP3 positive Tregs are required for the suppressive effect of CTLA4Ig, and suggest that the mechanism may be promoting the expansion or induction of antigen specific Tregs. In addition, CTLA4Ig may also be enhancing Treg function or the susceptibility of the effector T cell to Treg mediated suppression.
CTLA4Ig inhibits by a TGFb-dependent mechanism
Treg cells utilize a number of mechanisms to inhibit effector T cells, including through TGFβ (17, 22). TGFβ itself inhibits T cells directly as well as indirectly by promoting the differentiation of CD4+ T cells into Tregs (22–25). Therefore we tested whether TGFβ might be involved in the inhibitory effects of CTLA4Ig. Inclusion of a neutralizing antibody against TGFβ prevented CTLA4Ig from inhibiting T cell proliferation in vitro (Figure 4A) as did treatment with SB431542, an inhibitor of the TGFβ receptor (Figure 4B). Thus, the effects of CTLA4Ig in vitro require TGFβ/TGFβR signaling. However, inhibition by the BMDMs alone was unaffected by neutralization of TGFβ, indicating that these are separate pathways.
Figure 4. TGFβ is required for CTLA4Ig mediated inhibition of T cell proliferation.
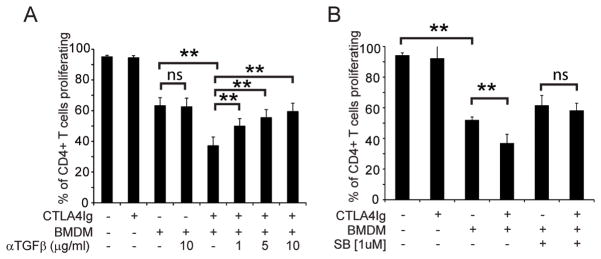
Splenocytes and BMDM were isolated from C57Bl/6J mice and co-cultured either alone or in the presence of A) α-TGFβ antibody or B) an inhibitor of TGFβR signaling. Proliferation was measured by flow cytometry after 96 hours. Presented are the combined results from 5 (panel A) and 3 (panel B) independent experiments. ** = p<0.01 by 2-tailed paired T-test
To confirm a role for TGFβ signaling in vivo, we tested the effect of CTLA4Ig in SMAD3-deficient mice (Figure 5). SMAD3-deficient mice develop spontaneous inflammation, which may complicate the analysis of an in vivo antigen challenge model, however, consistent with a previous report, we found they responded to inhaled allergen challenge comparably to wild type and that the response was antigen specific (26, 27). Following priming and challenge with OVA, the SMAD3-deficient mice developed an eosinophilic inflammatory cell infiltrate in the lung that was indistinguishable from wild type mice. In contrast to wild type, however, this was not blocked by administration of CTLA4Ig, indicating that SMAD3-dependent signaling is required in vivo and consistent with the in vitro findings of a requirement for TGFβ.
Figure 5. CTLA4Ig is ineffective in SMAD3-deficient mice.
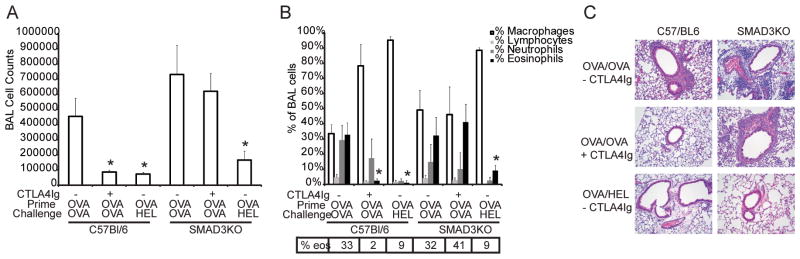
Wild type or SMAD3-deficient mice were sensitized with OVA and given an inhaled challenge with either OVA or HEL. CTLA4Ig was administered to groups of mice as indicated and samples were collected 72 hours after challenge. Each experimental group consisted of 5 mice and has been repeated 3 times. Shown is data from one representative experiment. * = p< 0.05 as compared to OVA primed and challenged without CTLA4Ig as determined by Kruskall-Wallis with Dunns post test comparisons.
Discussion
CD28 was initially identified as providing a costimulatory signal required for the activation of naïve T cells, thus fulfilling the role of a second signal in Bretscher and Cohn’s two- signal model of lymphocyte activation (28). Subsequent work has led to an in depth understanding of how CD28 regulates T cell function along with an expansion of the family to include additional activating and inhibitory receptors (reviewed in (29, 30)). The development of reagents that manipulate costimulation has led to important insights into normal T cell function as well as new therapeutics, therefore, an accurate understanding of how these agents work is essential. The data we report in this current study details a novel mechanism of action for CTLA4Ig, with direct implications for therapy as well as for our understanding of immune regulation.
In these studies we have utilized a well-established model of allergic airway inflammation. This model has been shown to be dependent on both B7 and CD28, and administration of CTLA4Ig completely abrogates OVA-induced airway inflammation, although it may be less effective when exposed to a more intense regimen of inhaled challenges (10, 31–35). In addition, the CD28-independent effects of CTLA4Ig were first detected using this model in mice deficient for both CD28 and BTLA (9). These features make it ideal for dissecting the mechanism by which costimulatory pathways regulate inflammation in vivo. Although we recently reported that administration of CTLA4Ig to people with mild atopic asthma did not inhibit the inflammatory response to allergen this does not detract from the utility of the murine model to perform mechanistic studies designed to determine how the CTLA4Ig exerts its anti-inflammatory function (36).
Although independent of CD28, this alternative mechanism remains dependent on expression of CD80/CD86. Others had shown that binding of either CD28 or CTLA-4 to CD80/CD86 could induce secretion of IFNγ (37). However, we did not detect any increase in either IFNγ or NO concentrations in culture supernatants of CTLA4Ig treated cells and abundant IFNγ was present in the co-cultures irrespective of CTLA4Ig (data not shown). Nonetheless, blockade of IFNγ signaling, either by inclusion of a neutralizing antibody or by using STAT-1 mice, prevented NO production (data not shown) and completely abrogated the effect of CTLA4Ig (supplemental figures 1 and 2). Although CTLA4Ig was ineffective in the absence of IFNγ or NOS2, depletion of Tregs or interference with TGFβ did not affect NO-dependent inhibition by macrophages. These data demonstrate that NO and CTLA4Ig act synergistically yet indicate they are through different mechanisms.
In addition to CD28 and CTLA-4, CD80 also binds to PD-L1 (8). This was revealed by examination of cells deficient in both CD28 and CTLA-4 and was shown to inhibit T cell activation and proliferation. While this interaction is also blocked by CTLA4Ig, if interference with CD80:PD-L1 interactions were the primary mechanism operative in our system, we would expect CTLA4Ig to increase T cell proliferation by preventing the inhibitory signal.
Binding of murine CTLA4Ig to CD80 or CD86 on mouse dendritic cells (DC) can induce indoleamine 2,3-dioxygenase (IDO) activity, which can suppress T cell proliferation by several mechanism, including promoting Treg development and function (38, 39). IDO has also been shown to be an important regulator of allergic inflammation in the lung (40, 41). More recently, IDO has been shown to regulate TGFβ production in pDC by a non-enzymatic mechanism that is not inhibited by 1-methyltryptopan (1-MT) (42). Although we had previously shown that treatment of mice with 1-MT had no effect, we tested IDO-deficient mice to determine if this alternative mechanism might be operative in our system (9). However, CTLA4Ig was as effective in inhibiting allergic airway inflammation in IDO-deficient mice as wild type, demonstrating that IDO is not involved in the suppressive effects of CTLA4Ig (supplemental figure 3).
We found that regulatory T cells were absolutely required for CTLA4Ig to inhibit T cell proliferation in vitro as well as in our in vivo model of allergic airway inflammation. Although we did not detect an increase in Treg cell number when wild type mice were treated with CTLA4Ig, adoptive transfer studies revealed an increase in the percentage of antigen specific Tregs suggesting that CTLA4Ig may facilitate the development of inducible Tregs. However, this does not exclude that CTLA4Ig might also augment their functional ability or enhance the sensitivity of the targets of Treg suppression. Treg cells utilize a number of mechanisms to inhibit effector T cells, including through TGFβ (17, 43). In fact, neutralization of TGFβ or inhibition of TGFβ receptor signaling abrogated the effect of CTLA4Ig. Although we did not detect an increase in TGFβ concentration in the culture supernatants, this may be misleading as much of the regulation of TGFβ is post-translational (44).
Several studies have examined the interactions of TGFβ, NO and Tregs, with conflicting results. TGFβ has been shown to down-regulate macrophage production of NO, and this involves both SMAD2 and SMAD3 proteins (45–47). Others have reported that NO interacts with TGFβ or IFNγ to either inhibit or promote Treg differentiation (48, 49). Each of these studies used widely varying systems, likely accounting for the differences in results and conclusions. Nonetheless, it appears that NO can influence Treg development and/or function.
The failure of CTLA4Ig to block inflammation in SMAD3-deficient mice is further support for the importance of a TGFβ-dependent pathway. TGFβ signals through both SMAD-dependent and independent pathways, and these may be differentially involved in T cell suppression by TGFβ and in Treg development and function (23, 26, 50). Non-regulatory CD4 T cells deficient in both SMAD2 and SMAD3 display a phenotype of spontaneous activation and are not suppressed by TGFβ. However, thymic Tregs specifically deleted for SMAD2 and SMAD 3 were essentially normal in both number and function although peripheral conversion of FoxP3- CD4 cells to FoxP3+ cells required SMAD3 (50–52). Thus our data is consistent with either a lack of TGFβ signaling in the effector T cell, or a defect in inducible Treg development or function.
Both CD28 and CTLA-4 have been implicated in regulatory T cell development and function, with particular attention focused on the role of CTLA-4 (17, 18, 53–56). Tregs express high levels of CTLA-4 constitutively, as opposed to effector T cells in which it is expressed only after activation. CTLA-4 deficient mice die of diffuse lymphoproliferation and autoimmunity, in large part due to defective Treg function (53, 54, 57). Recent data supports that CTLA-4 regulates Tregs and conventional T cells by both direct signaling (cell intrinsic mechanism) and indirectly by acting on other cells through its binding to CD80 and CD86 on the APC (cell extrinsic mechanism). Tai et al showed that the intracellular domain of CTLA-4 was not necessary for CTLA-4 to regulate the intrinsic proliferative capacity of Tregs or their ability to suppress the proliferation of naïve effector T cells (58). Others have suggested that CTLA-4 binding induces a loss of CD80 or CD86 expression on the APC as a mechanism of suppression (54, 59). While our data is generated using CTLA4Ig and not CTLA-4 expressed by T cells, it raises the possibility that binding of CTLA4Ig to CD80 and CD86 is at least partially mimicking the cell extrinsic mechanisms of endogenously expressed CTLA-4. While these mechanisms are distinct from our observations in that they ultimately rely upon impaired CD28-mediated costimulation, they are consistent in that they are mediated by signal delivered via the extracellular domain of CTLA-4 to B7 proteins rather than vice versa.
In conclusion, our study has demonstrated that CTLA4Ig can suppress T cell proliferation in vitro and allergen induced airway inflammation in vivo via a mechanism distinct from either IDO or impaired CD28-mediated costimulation. As illustrated in Figure 6, CTLA4Ig can inhibit T cell responses by both CD28-dependent and -independent mechanisms. CTLA4Ig interferes with the activation of naïve T cells by preventing B7 engagement with CD28 (Figure 6, left panel). This pathway is likely most important when the drug is administered at the time of initial sensitization. In addition, CTLA4Ig binding to B7 proteins can also induce a TGFβ- and regulatory T cell dependent inhibition of T cell proliferation and inflammation (Figure 6, right panel). This mechanism of action has not previously been described and may be the predominant mechanism when CTLA4Ig is administered clinically, which is typically at a time well after sensitization has occurred and when inflammation has already been established. Thus, this may very well be the predominant mechanism of action for the drug in the treatment of chronic inflammatory diseases such as rheumatoid arthritis. In addition, the effects we have observed with CTLA4Ig share some similarities with the effects of endogenous CTLA-4 on Tregs. Thus, in addition to illuminating how this important therapeutic agent may work in vivo, it may provide insight into how CTLA-4 regulates effector T cell function.
Figure 6. Proposed model for CTLA4Ig.
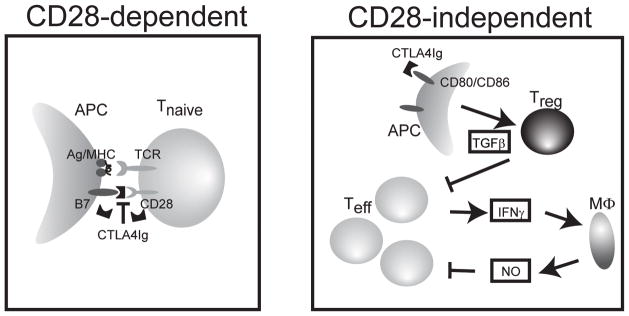
Binding of CTLA4Ig to CD80/CD86 expressed on an antigen presenting cell can prevent naïve T cell activation by preventing CD28 mediated costimulation (left panel). In addition, CTLA4Ig can suppress the proliferation of activated effector T cells (Teff) through a TGFβ and Treg dependent mechanism that is independent of CD28-costimulation (right panel). This pathway requires and is synergistic with macrophage derived NO.
Supplementary Material
Acknowledgments
We would like to thank Dr. David Beebe, Dr. Mike Holtzman, Dr. Herbert Virgin and Dr Matthew Ciorba and Dr. Alexander Rudensky for providing mice used in this study. We thank Drs Jonathan Boomer, Ken Murphy, Andrey Shaw and Robert Schreiber for their critical review of this manuscript and numerous helpful discussions. We thank Dr. John Russell and Julia Sims for assistance with the bone marrow chimeras.
Footnotes
This work was supported by grant HL62683 awarded by the NIH to JMG
There are no conflicts of interest
References
- 1.Linsley PS, Nadler SG. The clinical utility of inhibiting CD28-mediated costimulation. Immunol Rev. 2009;229:307–321. doi: 10.1111/j.1600-065X.2009.00780.x. [DOI] [PubMed] [Google Scholar]
- 2.Genovese MC, Becker JC, Schiff M, Luggen M, Sherrer Y, Kremer J, Birbara C, Box J, Natarajan K, Nuamah I, Li T, Aranda R, Hagerty DT, Dougados M. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Engl J Med. 2005;353:1114–1123. doi: 10.1056/NEJMoa050524. [DOI] [PubMed] [Google Scholar]
- 3.Vincenti F, Larsen C, Durrbach A, Wekerle T, Nashan B, Blancho G, Lang P, Grinyo J, Halloran PF, Solez K, Hagerty D, Levy E, Zhou W, Natarajan K, Charpentier B. Costimulation blockade with belatacept in renal transplantation. N Engl J Med. 2005;353:770–781. doi: 10.1056/NEJMoa050085. [DOI] [PubMed] [Google Scholar]
- 4.Sondak VK, Smalley KS, Kudchadkar R, Grippon S, Kirkpatrick P. Ipilimumab. Nat Rev Drug Discov. 2011;10:411–412. doi: 10.1038/nrd3463. [DOI] [PubMed] [Google Scholar]
- 5.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174:561–569. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deppong CM, Parulekar A, Boomer JS, Bricker TL, Green JM. CTLA4-Ig inhibits allergic airway inflammation by a novel CD28-independent, nitric oxide synthase-dependent mechanism. Eur J Immunol. 2010;40:1985–1994. doi: 10.1002/eji.200940282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimzey SL, Liu P, Green JM. Requirement for CD28 in the effector phase of allergic airway inflammation. J Immunol. 2004;173:632–640. doi: 10.4049/jimmunol.173.1.632. [DOI] [PubMed] [Google Scholar]
- 11.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 12.Pan Q, Kravchenko V, Katz A, Huang S, Ii M, Mathison JC, Kobayashi K, Flavell RA, Schreiber RD, Goeddel D, Ulevitch RJ. NF-kappa B-inducing kinase regulates selected gene expression in the Nod2 signaling pathway. Infect Immun. 2006;74:2121–2127. doi: 10.1128/IAI.74.4.2121-2127.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albina JE, Abate JA, Henry WL., Jr Nitric oxide production is required for murine resident peritoneal macrophages to suppress mitogen-stimulated T cell proliferation. Role of IFN-gamma in the induction of the nitric oxide-synthesizing pathway. J Immunol. 1991;147:144–148. [PubMed] [Google Scholar]
- 14.Kim J, Myers AC, Chen L, Pardoll DM, Truong-Tran QA, Lane AP, McDyer JF, Fortuno L, Schleimer RP. Constitutive and inducible expression of B7 family of ligands by human airway epithelial cells. Am J Respir Cell Mol Biol. 2005;33:280–289. doi: 10.1165/rcmb.2004-0129OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elssner A, Jaumann F, Wolf WP, Schwaiblmair M, Behr J, Furst H, Reichenspurner H, Briegel J, Niedermeyer J, Vogelmeier C. Bronchial epithelial cell B7-1 and B7-2 mRNA expression after lung transplantation: a role in allograft rejection? Eur Respir J. 2002;20:165–169. doi: 10.1183/09031936.02.00268102. [DOI] [PubMed] [Google Scholar]
- 16.Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 17.Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lio CW, Dodson LF, Deppong CM, Hsieh CS, Green JM. CD28 facilitates the generation of Foxp3(-) cytokine responsive regulatory T cell precursors. J Immunol. 2010;184:6007–6013. doi: 10.4049/jimmunol.1000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol. 2005;6:152–162. doi: 10.1038/ni1160. [DOI] [PubMed] [Google Scholar]
- 20.Sansom DM, Walker LS. The role of CD28 and cytotoxic T-lymphocyte antigen-4 (CTLA-4) in regulatory T-cell biology. Immunol Rev. 2006;212:131–148. doi: 10.1111/j.0105-2896.2006.00419.x. [DOI] [PubMed] [Google Scholar]
- 21.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 22.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 23.Li MO, Flavell RA. TGF-β: A Master of All T Cell Trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKarns SC, Schwartz RH. Distinct effects of TGF-beta 1 on CD4+ and CD8+ T cell survival, division, and IL-2 production: a role for T cell intrinsic Smad3. J Immunol. 2005;174:2071–2083. doi: 10.4049/jimmunol.174.4.2071. [DOI] [PubMed] [Google Scholar]
- 25.Li MO, Wan YY, Flavell RA. T Cell-Produced Transforming Growth Factor-β1 Controls T Cell Tolerance and Regulates Th1- and Th17-Cell Differentiation. Immunity. 2007;26:579–591. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 26.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anthoni M, Wang G, Leino MS, Lauerma AI, Alenius HT, Wolff HJ. Smad3 -signalling and Th2 cytokines in normal mouse airways and in a mouse model of asthma. Int J Biol Sci. 2007;3:477–485. doi: 10.7150/ijbs.3.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bretscher P, Cohn M. A theory of self-nonself discrimination. Science. 1970;169:1042–1049. doi: 10.1126/science.169.3950.1042. [DOI] [PubMed] [Google Scholar]
- 29.Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229:12–26. doi: 10.1111/j.1600-065X.2009.00770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nurieva RI, Liu X, Dong C. Yin-Yang of costimulation: crucial controls of immune tolerance and function. Immunol Rev. 2009;229:88–100. doi: 10.1111/j.1600-065X.2009.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burr JS, Kimzey SL, Randolph DR, Green JM. CD28 and CTLA4 coordinately regulate airway inflammatory cell recruitment and T-helper cell differentiation after inhaled allergen. Am J Respir Cell Mol Biol. 2001;24:563–568. doi: 10.1165/ajrcmb.24.5.4375. [DOI] [PubMed] [Google Scholar]
- 32.Harris N, Campbell C, Le Gros G, Ronchese F. Blockade of CD28/B7 co-stimulation by mCTLA4-Hγ1 inhibits antigen- induced lung eosinophilia but not Th2 cell development or recruitment in the lung. Eur J Immunol. 1997;27:155–161. doi: 10.1002/eji.1830270123. [DOI] [PubMed] [Google Scholar]
- 33.Harris N, Peach R, Naemura J, Linsley PS, Le Gros G, Ronchese F. CD80 costimulation is essential for the induction of airway eosinophilia. J Exp Med. 1997;185:177–182. doi: 10.1084/jem.185.1.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keane-Myers A, Gause WC, Linsley PS, Chen SJ, Wills-Karp M. B7-CD28/CTLA-4 costimulatory pathways are required for the development of T helper cell 2-mediated allergic airway responses to inhaled antigens. J Immunol. 1997;158:2042–2049. [PubMed] [Google Scholar]
- 35.Deurloo DT, van Esch BC, Hofstra CL, Nijkamp FP, van Oosterhout AJ. CTLA4-IgG reverses asthma manifestations in a mild but not in a more “severe” ongoing murine model. Am J Respir Cell Mol Biol. 2001;25:751–760. doi: 10.1165/ajrcmb.25.6.4607. [DOI] [PubMed] [Google Scholar]
- 36.Parulekar AD, Boomer JS, Patterson BM, Yin-Declue H, Deppong CM, Wilson BS, Jarjour NN, Castro M, Green JM. A randomized controlled trial to evaluate inhibition of T-cell costimulation in allergen-induced airway inflammation. Am J Respir Crit Care Med. 2013;187:494–501. doi: 10.1164/rccm.201207-1205OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orabona C, Grohmann U, Belladonna ML, Fallarino F, Vacca C, Bianchi R, Bozza S, Volpi C, Salomon BL, Fioretti MC, Romani L, Puccetti P. CD28 induces immunostimulatory signals in dendritic cells via CD80 and CD86. Nat Immunol. 2004;5:1134–1142. doi: 10.1038/ni1124. [DOI] [PubMed] [Google Scholar]
- 38.Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, Candeloro P, Belladonna ML, Bianchi R, Fioretti MC, Puccetti P. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3:1097–1101. doi: 10.1038/ni846. [DOI] [PubMed] [Google Scholar]
- 39.Katz JB, Muller AJ, Prendergast GC. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev. 2008;222:206–221. doi: 10.1111/j.1600-065X.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- 40.Grohmann U, Volpi C, Fallarino F, Bozza S, Bianchi R, Vacca C, Orabona C, Belladonna ML, Ayroldi E, Nocentini G, Boon L, Bistoni F, Fioretti MC, Romani L, Riccardi C, Puccetti P. Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat Med. 2007;13:579–586. doi: 10.1038/nm1563. [DOI] [PubMed] [Google Scholar]
- 41.Hayashi T, Beck L, Rossetto C, Gong X, Takikawa O, Takabayashi K, Broide DH, Carson DA, Raz E. Inhibition of experimental asthma by indoleamine 2,3-dioxygenase. The Journal of Clinical Investigation. 2004;114:270–279. doi: 10.1172/JCI21275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, Servillo G, Brunacci C, Calvitti M, Bicciato S, Mazza EM, Boon L, Grassi F, Fioretti MC, Fallarino F, Puccetti P, Grohmann U. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. 2011;12:870–878. doi: 10.1038/ni.2077. [DOI] [PubMed] [Google Scholar]
- 43.Li L, Iwamoto Y, Berezovskaya A, Boussiotis VA. A pathway regulated by cell cycle inhibitor p27Kip1 and checkpoint inhibitor Smad3 is involved in the induction of T cell tolerance. Nat Immunol. 2006;7:1157–1165. doi: 10.1038/ni1398. [DOI] [PubMed] [Google Scholar]
- 44.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 45.Ding A, Nathan CF, Graycar J, Derynck R, Stuehr DJ, Srimal S. Macrophage deactivating factor and transforming growth factors-beta 1 -beta 2 and -beta 3 inhibit induction of macrophage nitrogen oxide synthesis by IFN-gamma. J Immunol. 1990;145:940–944. [PubMed] [Google Scholar]
- 46.Nelson BJ, Ralph P, Green SJ, Nacy CA. Differential susceptibility of activated macrophage cytotoxic effector reactions to the suppressive effects of transforming growth factor-beta 1. J Immunol. 1991;146:1849–1857. [PubMed] [Google Scholar]
- 47.Sugiyama Y, Kakoi K, Kimura A, Takada I, Kashiwagi I, Wakabayashi Y, Morita R, Nomura M, Yoshimura A. Smad2 and Smad3 are redundantly essential for the suppression of iNOS synthesis in macrophages by regulating IRF3 and STAT1 pathways. Int Immunol. 2012;24:253–265. doi: 10.1093/intimm/dxr126. [DOI] [PubMed] [Google Scholar]
- 48.Lee SW, Choi H, Eun SY, Fukuyama S, Croft M. Nitric oxide modulates TGF-beta-directive signals to suppress Foxp3+ regulatory T cell differentiation and potentiate Th1 development. J Immunol. 2011;186:6972–6980. doi: 10.4049/jimmunol.1100485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feng G, Gao W, Strom TB, Oukka M, Francis RS, Wood KJ, Bushell A. Exogenous IFN-gamma ex vivo shapes the alloreactive T-cell repertoire by inhibition of Th17 responses and generation of functional Foxp3+ regulatory T cells. Eur J Immunol. 2008;38:2512–2527. doi: 10.1002/eji.200838411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gu AD, Wang Y, Lin L, Zhang SS, Wan YY. Requirements of transcription factor Smad-dependent and -independent TGF-beta signaling to control discrete T-cell functions. Proc Natl Acad Sci U S A. 2012;109:905–910. doi: 10.1073/pnas.1108352109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martinez GJ, Zhang Z, Chung Y, Reynolds JM, Lin X, Jetten AM, Feng XH, Dong C. Smad3 differentially regulates the induction of regulatory and inflammatory T cell differentiation. J Biol Chem. 2009;284:35283–35286. doi: 10.1074/jbc.C109.078238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takimoto T, Wakabayashi Y, Sekiya T, Inoue N, Morita R, Ichiyama K, Takahashi R, Asakawa M, Muto G, Mori T, Hasegawa E, Saika S, Hara T, Nomura M, Yoshimura A. Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J Immunol. 2010;185:842–855. doi: 10.4049/jimmunol.0904100. [DOI] [PubMed] [Google Scholar]
- 53.Bachmann MF, Kohler G, Ecabert B, Mak TW, Kopf M. Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J Immunol. 1999;163:1128–1131. [PubMed] [Google Scholar]
- 54.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 Control over Foxp3+ Regulatory T Cell Function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 55.Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M, Bluestone JA. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur J Immunol. 2004;34:2996–3005. doi: 10.1002/eji.200425143. [DOI] [PubMed] [Google Scholar]
- 56.Read S, Greenwald R, Izcue A, Robinson N, Mandelbrot D, Francisco L, Sharpe AH, Powrie F. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J Immunol. 2006;177:4376–4383. doi: 10.4049/jimmunol.177.7.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in CTLA-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 58.Tai XG, Van Laethem F, Pobezinsky L, Guinter T, Sharrow SO, Adams A, Granger L, Kruhlak M, Lindsten T, Thompson CB, Feigenbaum L, Singer A. Basis of CTLA-4 function in regulatory and conventional CD4(+) T cells. Blood. 2012;119:5155–5163. doi: 10.1182/blood-2011-11-388918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, Hou TZ, Futter CE, Anderson G, Walker LSK, Sansom DM. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science. 2011;332:600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.